
Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles for Enhanced Treatment of Triple-Negative Breast Tumor

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis
2.3. Coating of Iron Oxide Nanoparticles with APU-R848
2.4. In Vitro Study
2.5. Macrophage Polarization
2.6. In Vivo Animal Model
2.7. Biodistribution Study
2.8. Anti-Tumor Efficacy
2.9. Immunohistochemistry
2.10. ELISA Detection
2.11. Statistical Analysis
3. Results and Discussion
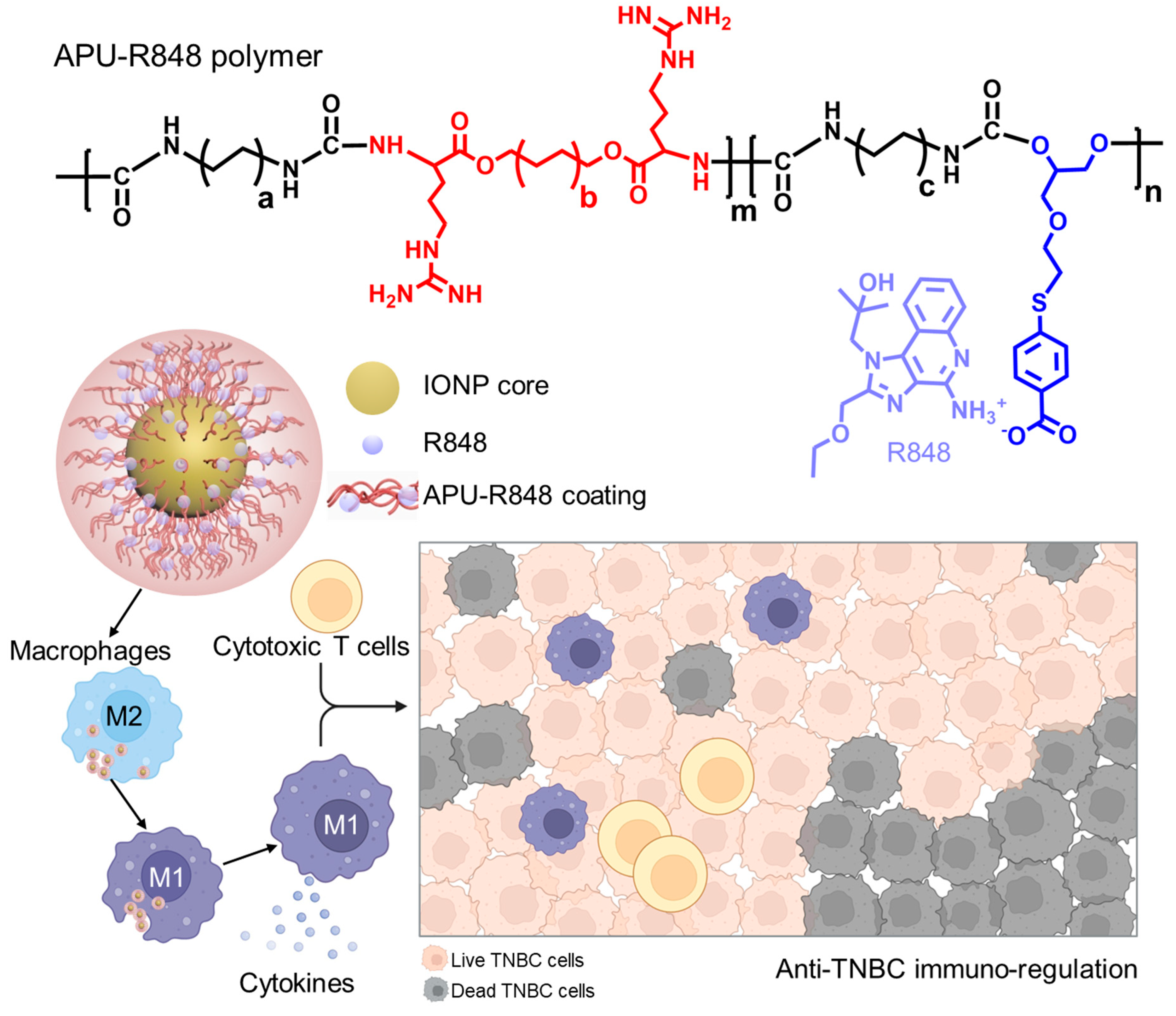
3.1. Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles
3.2. Synthesis and Functionalization of APU-R848 Polymer
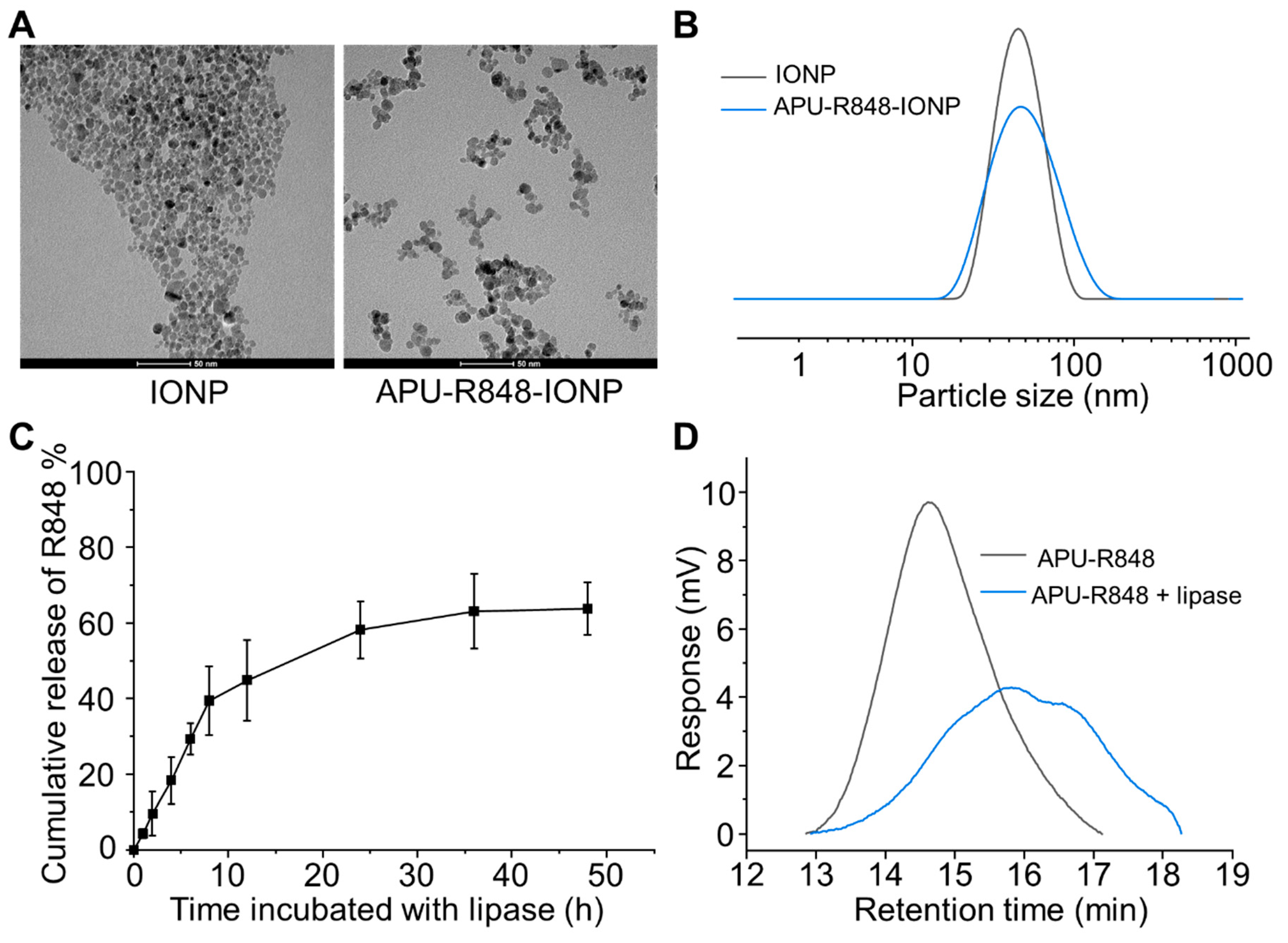
3.3. Coating of APU-R848 Polymers onto Iron Oxide Nanoparticles
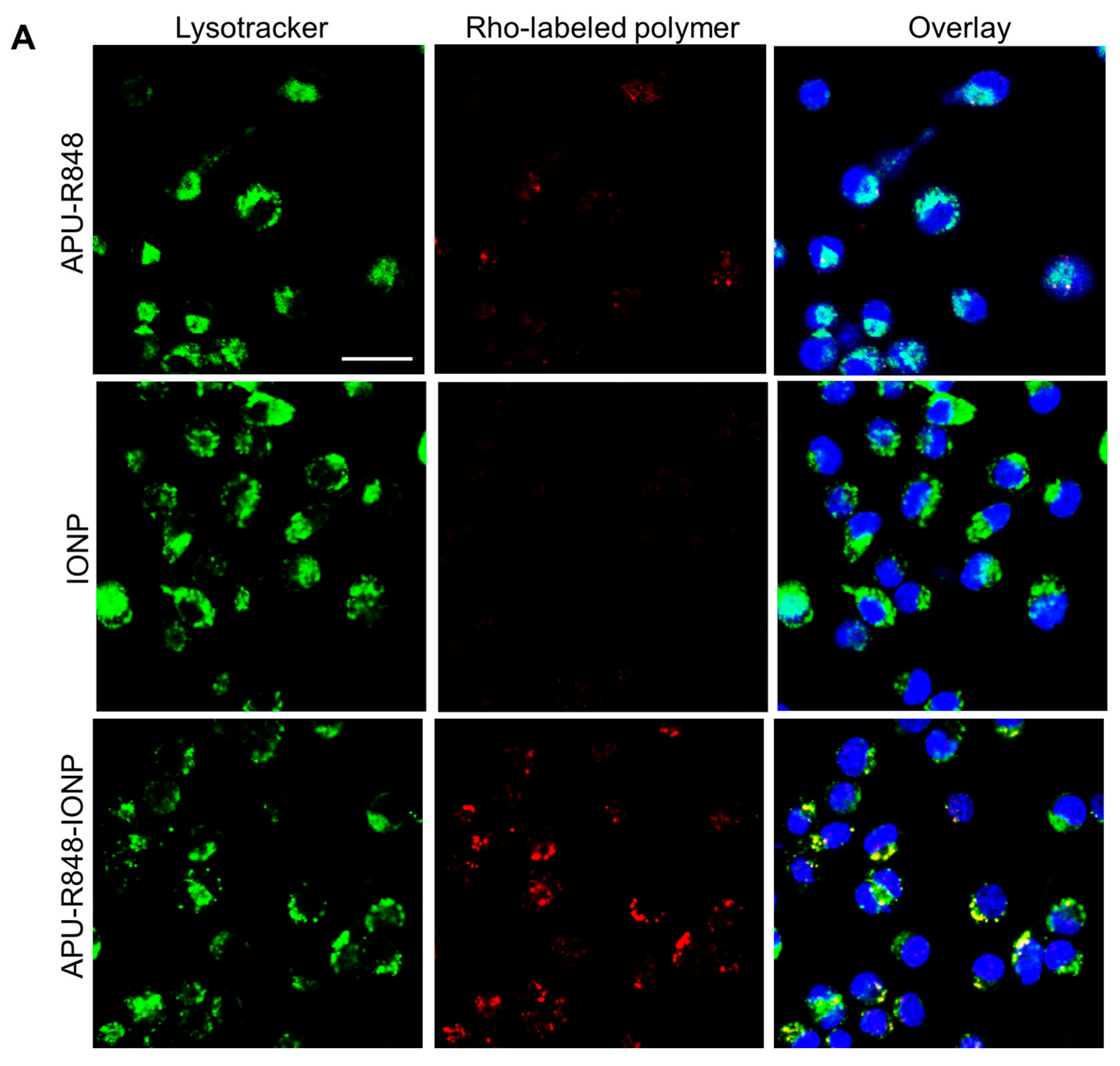
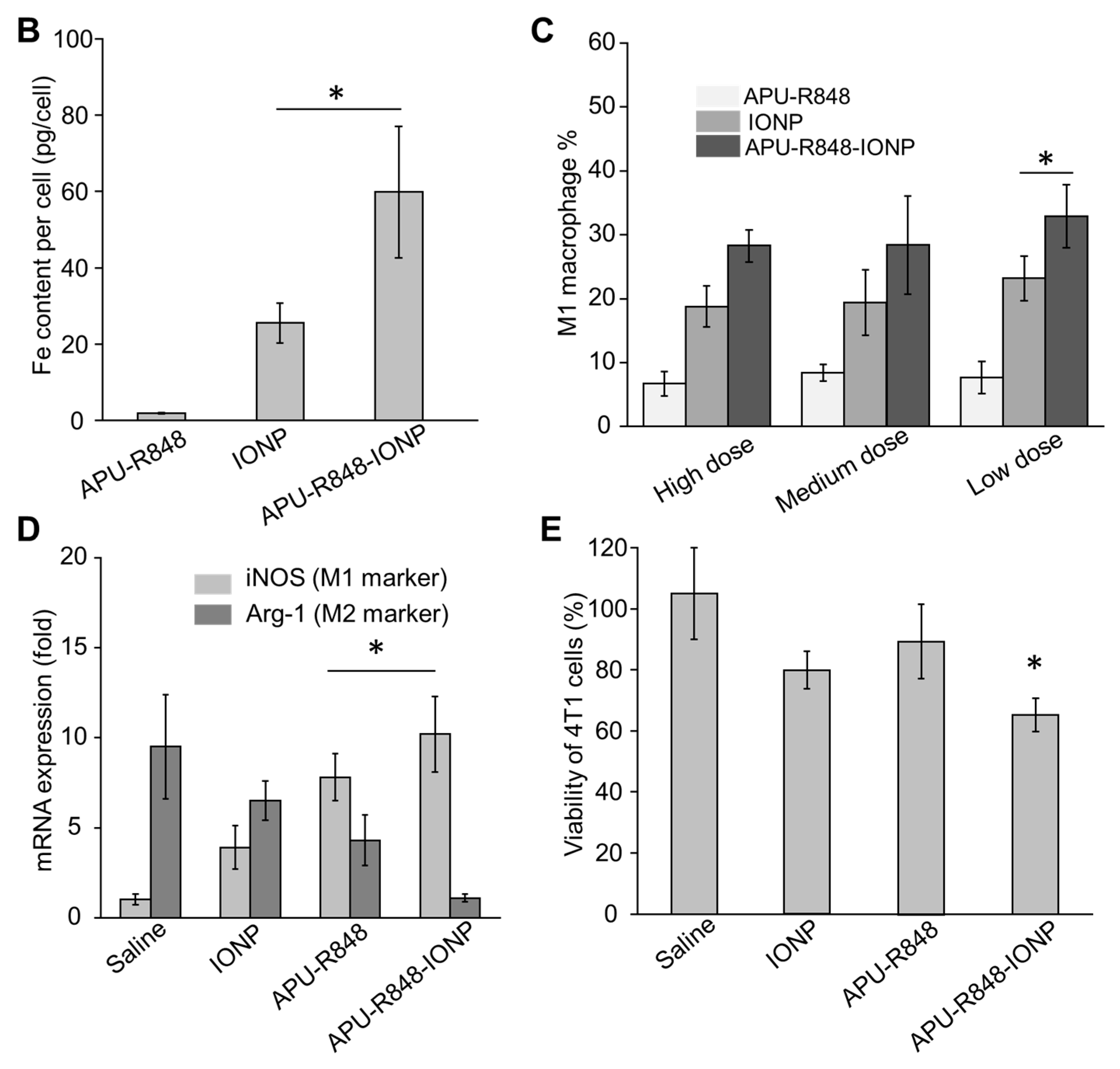
3.4. Internalization of APU-R848-IONPs and the Re-Polarization Effect in Macrophages In Vitro
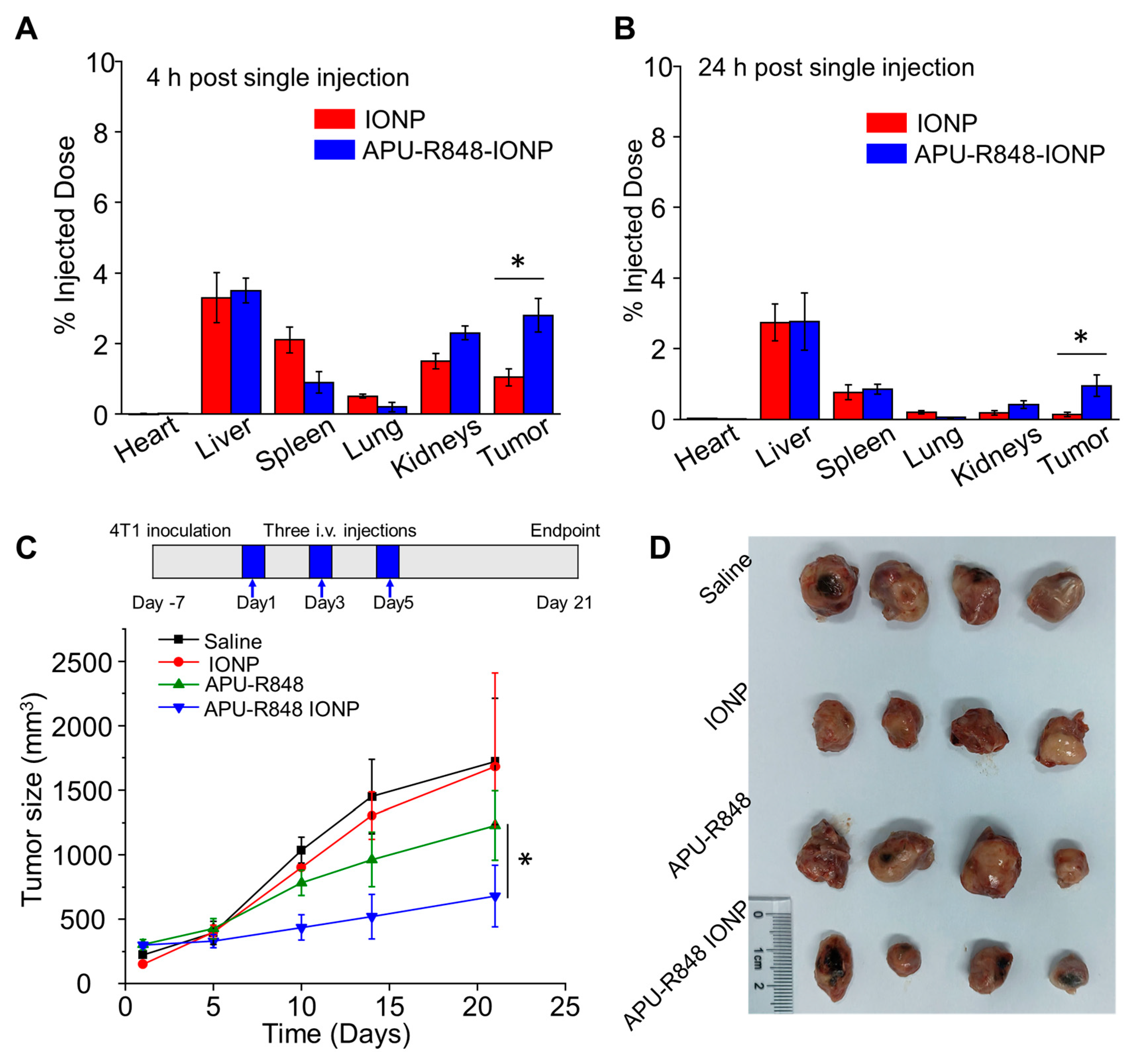
3.5. APU-R848-IONPs Demonstrated Significant Tumor Inhibition in 4T1 Model
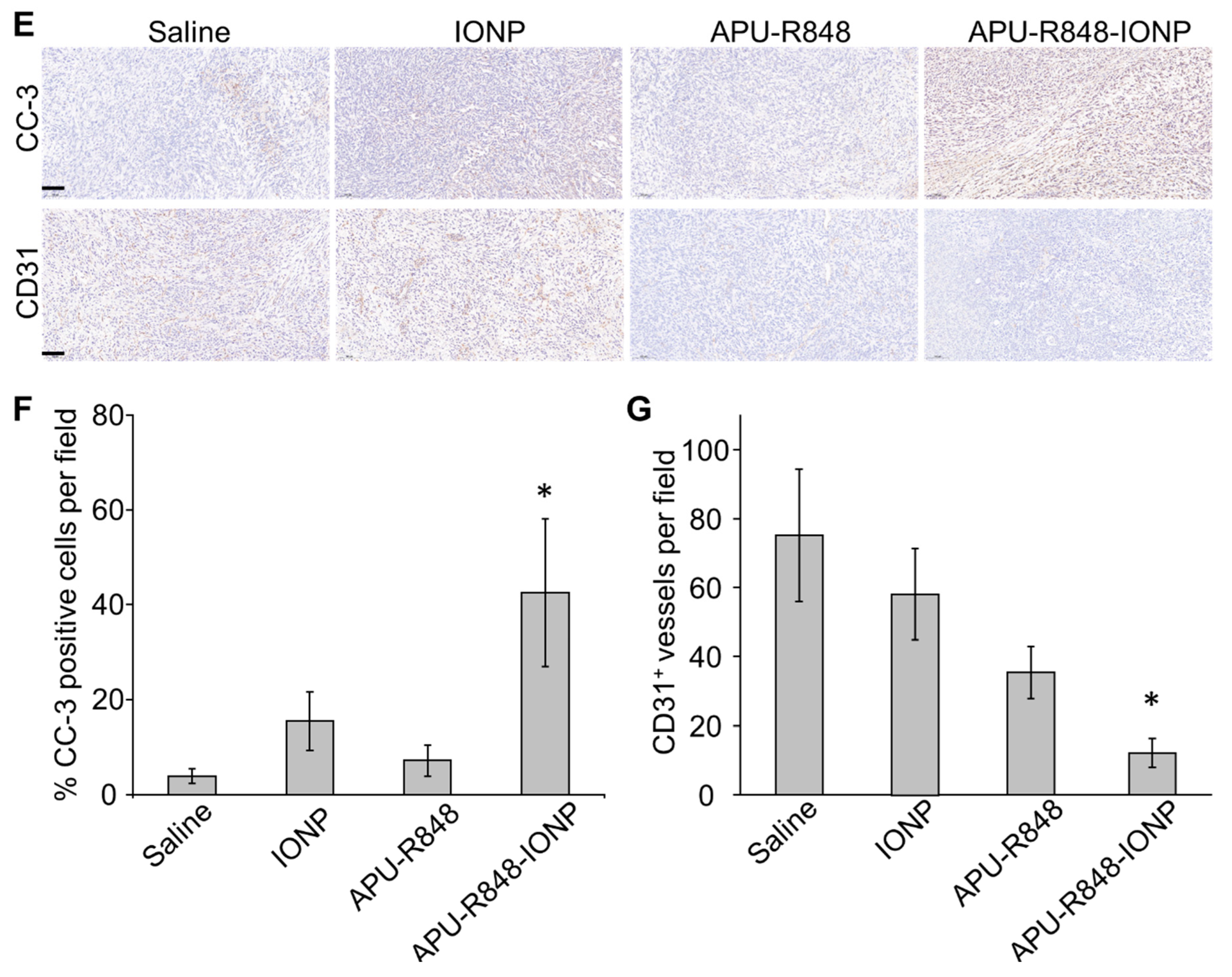
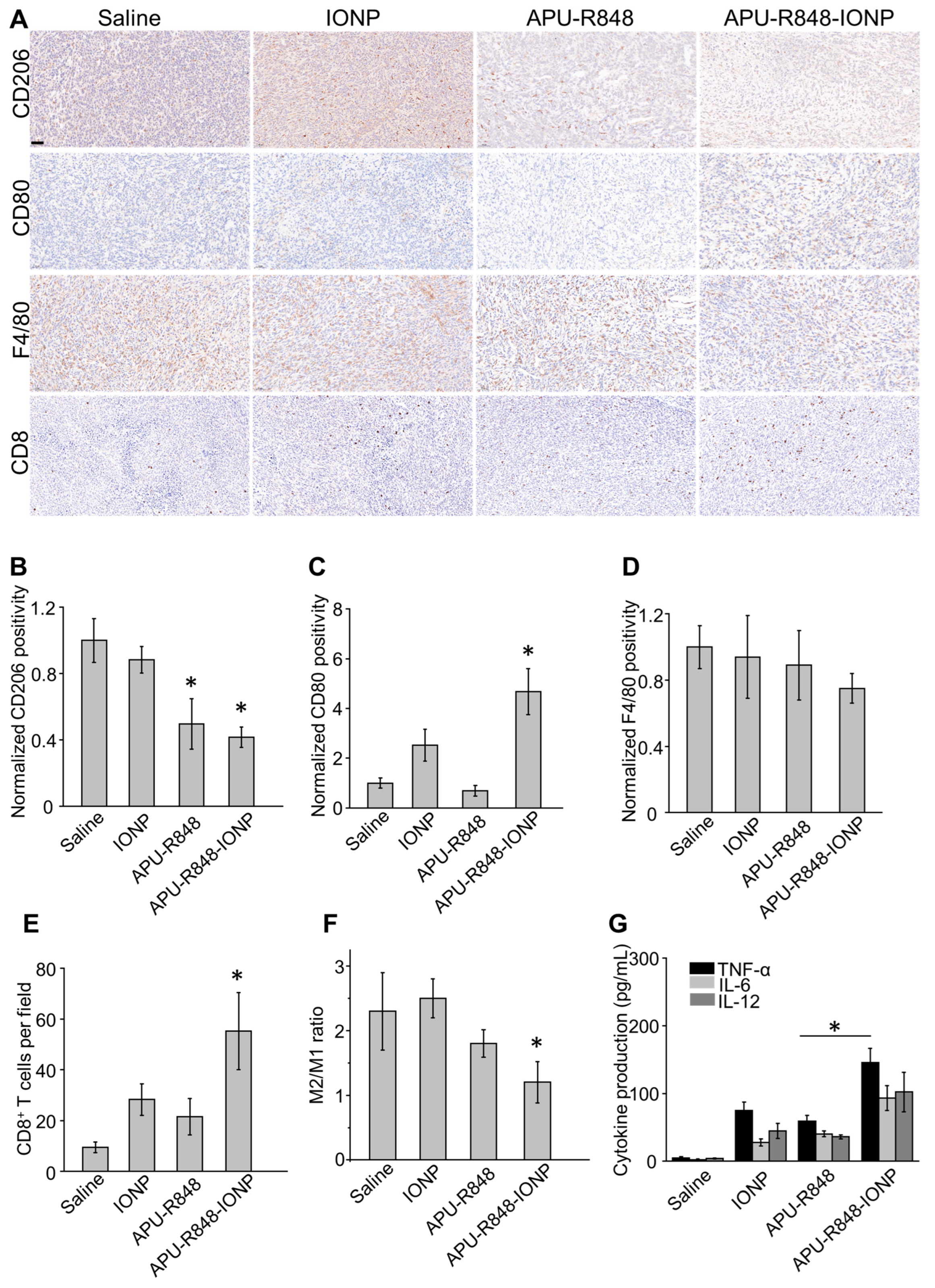
3.6. APU-R848-IONPs Regulated the Tumor Microenvironment and Activated Immune Surveillance in 4T1 Tumors
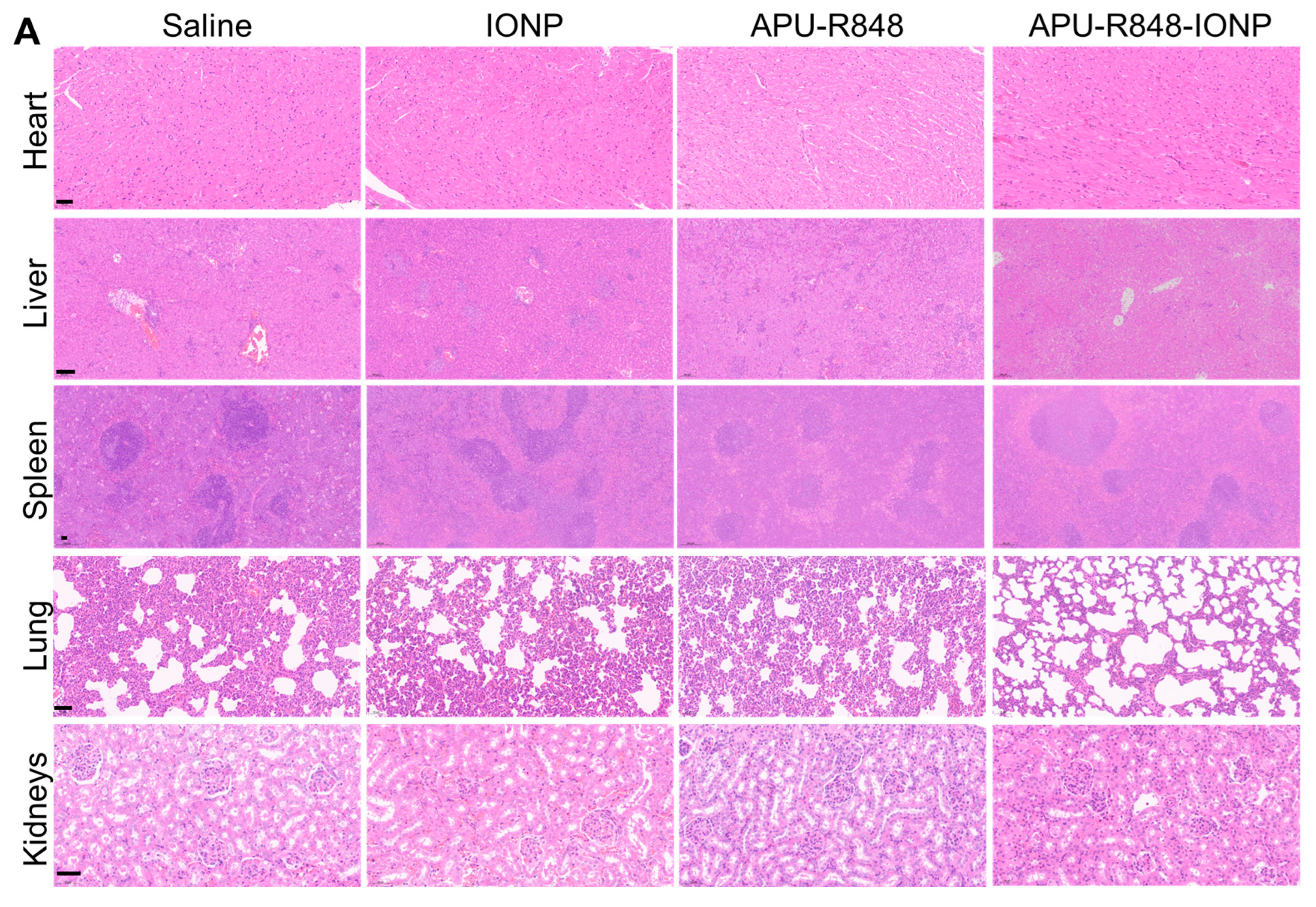
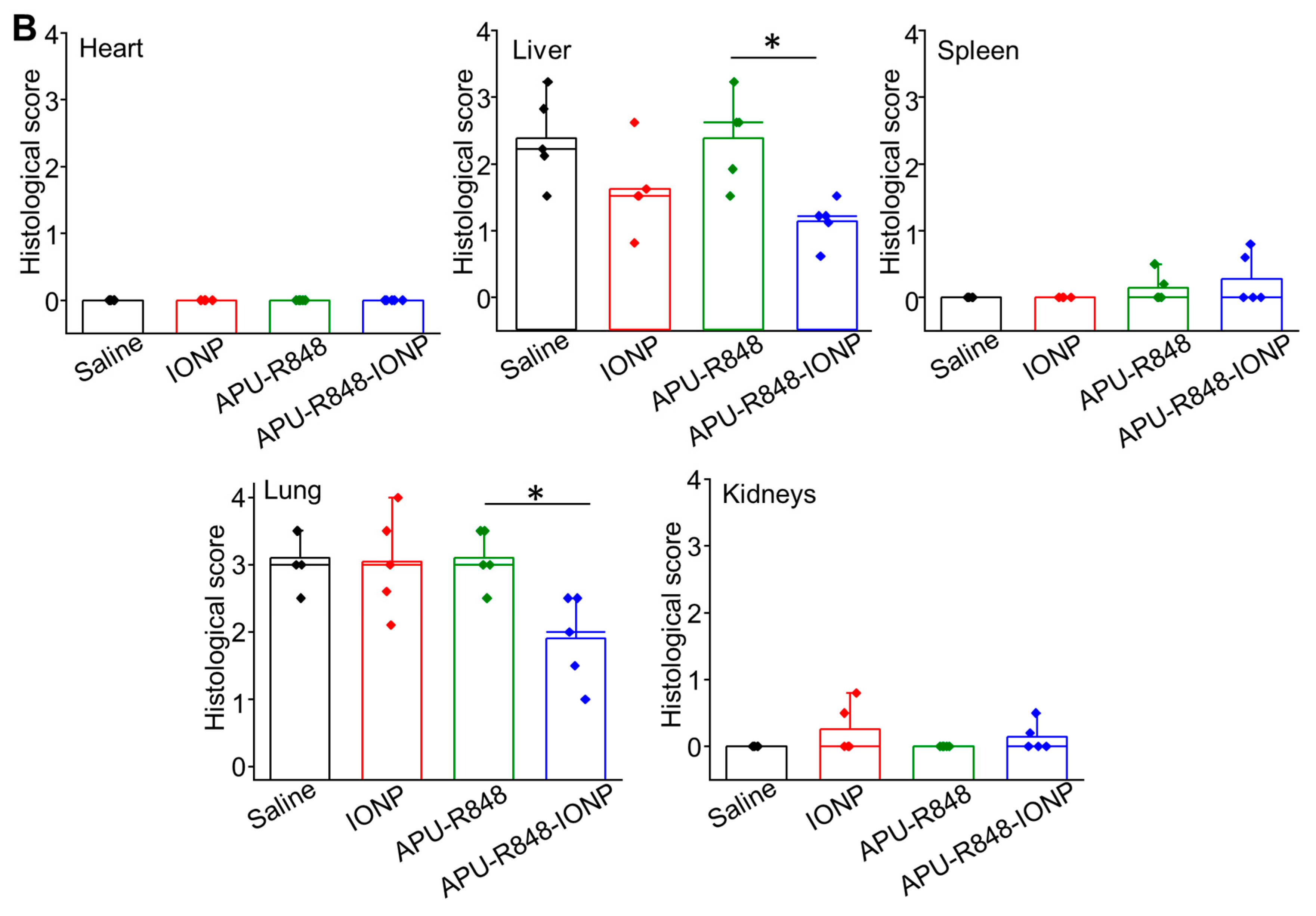
3.7. Biosafety Evaluation of APU-R848-IONP Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Ruijter, T.C.; Veeck, J.; de Hoon, J.P.; van Engeland, M.; Tjan-Heijnen, V.C. Characteristics of triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Keenan, T.E.; Tolaney, S.M. Role of immunotherapy in triple-negative breast cancer. J. Natl. Compr. Cancer Netw. 2020, 18, 479–489. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Miller, S.; Ostrand-Rosenberg, S. Tumor immunity: A balancing act between T cell activation, macrophage activation and tumor-induced immune suppression. Cancer Immunol. Immunother. 2005, 54, 1137–1142. [Google Scholar] [CrossRef]
- Weigert, A.; Brüne, B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide 2008, 19, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Zhao, C.; Pang, X.; Yang, Z.; Wang, S.; Deng, H.; Chen, X. Nanomaterials targeting tumor associated macrophages for cancer immunotherapy. J. Control. Release 2022, 341, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Ovais, M.; Guo, M.; Chen, C. Tailoring nanomaterials for targeting tumor-associated macrophages. Adv. Mater. 2019, 31, 1808303. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Puzzo, M.; De Santo, M.; Morelli, C.; Leggio, A.; Pasqua, L. The advent of molecular targeted therapies against cancer. Toward multi-targeting drugs through materials engineering: A possible future scenario. Small Sci. 2024, 4, 2400113. [Google Scholar] [CrossRef]
- Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics 2019, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Xu, Q.; Gong, R.; Bian, Z.; Chu, C.-C. Targeted Chinese medicine delivery by a new family of biodegradable pseudo-protein nanoparticles for treating triple-negative breast cancer: In vitro and in vivo study. Front. Oncol. 2021, 10, 600298. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Potuck, A.; Kohn, J.C.; Fung, K.; Reinhart-King, C.A.; Chu, C.-C. Self-assembled cationic biodegradable nanoparticles from pH-responsive amino-acid-based poly (ester urea urethane) s and their application as a drug delivery vehicle. Biomacromolecules 2016, 17, 523–537. [Google Scholar] [CrossRef]
- He, M.; Ro, L.; Liu, J.; Chu, C.C. Folate-decorated arginine-based poly (ester urea urethane) nanoparticles as carriers for gambogic acid and effect on cancer cells. J. Biomed. Mater. Res. Part A 2017, 105, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Potuck, A.N.; Weed, B.L.; Leifer, C.A.; Chu, C. Electrostatically self-assembled biodegradable microparticles from pseudoproteins and polysaccharide: Fabrication, characterization, and biological properties. Biomacromolecules 2015, 16, 564–577. [Google Scholar] [CrossRef]
- He, M.; Sun, L.; Fu, X.; McDonough, S.P.; Chu, C.-C. Biodegradable amino acid-based poly (ester amine) with tunable immunomodulating properties and their in vitro and in vivo wound healing studies in diabetic rats’ wounds. Acta Biomater. 2019, 84, 114–132. [Google Scholar] [CrossRef]
- Spiller, K.L.; Koh, T.J. Macrophage-based therapeutic strategies in regenerative medicine. Adv. Drug Deliv. Rev. 2017, 122, 74–83. [Google Scholar] [CrossRef]
- Kim, S.-H.; Roszik, J.; Grimm, E.A.; Ekmekcioglu, S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front. Oncol. 2018, 8, 67. [Google Scholar] [CrossRef]
- Martí i Líndez, A.-A.; Reith, W. Arginine-dependent immune responses. Cell. Mol. Life Sci. 2021, 78, 5303–5324. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Guo, X.; Lu, Y.; Liu, X.; Jiang, M.; Li, X.; Qin, B.; Luo, Z.; Liu, H. Arginine supplementation targeting tumor-killing immune cells reconstructs the tumor microenvironment and enhances the antitumor immune response. ACS Nano 2022, 16, 12964–12978. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar] [CrossRef] [PubMed]
- Dadfar, S.M.; Roemhild, K.; Drude, N.I.; von Stillfried, S.; Knüchel, R.; Kiessling, F.; Lammers, T. Iron oxide nanoparticles: Diagnostic, therapeutic and theranostic applications. Adv. Drug Deliv. Rev. 2019, 138, 302–325. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Revia, R.A.; Zhang, M. Iron oxide nanoparticles for immune cell labeling and cancer immunotherapy. Nanoscale Horiz. 2021, 6, 696–717. [Google Scholar] [CrossRef]
- Nascimento, C.S.; Alves, É.A.R.; de Melo, C.P.; Corrêa-Oliveira, R.; Calzavara-Silva, C.E. Immunotherapy for cancer: Effects of iron oxide nanoparticles on polarization of tumor-associated macrophages. Nanomedicine 2021, 16, 2633–2650. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Canese, R.; Vurro, F.; Marzola, P. Iron oxide nanoparticles as theranostic agents in cancer immunotherapy. Nanomaterials 2021, 11, 1950. [Google Scholar] [CrossRef]
- Shah, A.; Dobrovolskaia, M.A. Immunological effects of iron oxide nanoparticles and iron-based complex drug formulations: Therapeutic benefits, toxicity, mechanistic insights, and translational considerations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 977–990. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Barber, D.F. The use of iron oxide nanoparticles to reprogram macrophage responses and the immunological tumor microenvironment. Front. Immunol. 2021, 12, 693709. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, Y.; Mao, Y.; Li, Y.; Shen, Y.; Sheng, J.; Gu, N. Modulation of macrophage polarization by iron-based nanoparticles. Med. Rev. 2023, 3, 105–122. [Google Scholar] [CrossRef]
- Li, J.-X.; Shu, N.; Zhang, Y.-J.; Tong, Q.-S.; Wang, L.; Zhang, J.-Y.; Du, J.-Z. Self-assembled nanoparticles from the amphiphilic prodrug of resiquimod for improved cancer immunotherapy. ACS Appl. Mater. Interfaces 2024, 16, 25665–25675. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; He, H.; Zou, J.; Wang, H.; Chen, Y.; Chen, Y.; Lan, M.; Zhao, Y.; Gao, F. Polydopamine-coated ferric oxide nanoparticles for R848 delivery for photothermal immunotherapy in breast cancer. Int. J. Pharm. 2023, 644, 123249. [Google Scholar] [CrossRef]
- Wu, J.-S.; Li, J.-X.; Shu, N.; Duan, Q.-J.; Tong, Q.-S.; Zhang, J.-Y.; Huang, Y.-C.; Yang, S.-Y.; Zhao, Z.-B.; Du, J.-Z. A polyamidoamine (PAMAM) derivative dendrimer with high loading capacity of TLR7/8 agonist for improved cancer immunotherapy. Nano Res. 2022, 15, 510–518. [Google Scholar] [CrossRef]
- Workman, P.; Aboagye, E.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.; Double, J.; Everitt, J.; Farningham, D.; Glennie, M. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.; Chao, P.-H.; Qin, Z.; Böttger, R.; Lee, S.E.; Li, S.-D. Liposomal resiquimod for enhanced immunotherapy of peritoneal metastases of colorectal cancer. Pharmaceutics 2021, 13, 1696. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1747. [Google Scholar] [CrossRef]
- Bahmani, B.; Gong, H.; Luk, B.T.; Haushalter, K.J.; DeTeresa, E.; Previti, M.; Zhou, J.; Gao, W.; Bui, J.D.; Zhang, L. Intratumoral immunotherapy using platelet-cloaked nanoparticles enhances antitumor immunity in solid tumors. Nat. Commun. 2021, 12, 1999. [Google Scholar] [CrossRef]
- Champion, J.A.; Walker, A.; Mitragotri, S. Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res. 2008, 25, 1815–1821. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Su, Y.H.; Wang, X.; Yao, X.; Du, J.Z. Recent progress on nanomedicine-mediated repolarization of tumor-associated macrophages for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2024, 16, e2001. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [PubMed]
- Vermare, A.; Guérin, M.V.; Peranzoni, E.; Bercovici, N. Dynamic CD8+ T cell cooperation with macrophages and monocytes for successful cancer immunotherapy. Cancers 2022, 14, 3546. [Google Scholar] [CrossRef] [PubMed]
- Asami, T.; Ishii, M.; Fujii, H.; Namkoong, H.; Tasaka, S.; Matsushita, K.; Ishii, K.; Yagi, K.; Fujiwara, H.; Funatsu, Y. Modulation of murine macrophage TLR7/8-mediated cytokine expression by mesenchymal stem cell-conditioned medium. Mediat. Inflamm. 2013, 2013, 264260. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yong, L.; Zhu, X.; Feng, Y.; Fu, Y.; Kong, D.; Lu, W.; Zhou, T.-y. Disease progression model of 4T1 metastatic breast cancer. J. Pharmacokinet. Pharmacodyn. 2020, 47, 105–116. [Google Scholar] [CrossRef]
- DuPré, S.A.; Redelman, D.; Hunter Jr, K.W. The mouse mammary carcinoma 4T1: Characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int. J. Exp. Pathol. 2007, 88, 351–360. [Google Scholar] [CrossRef]
- Sun, T.; Kang, Y.; Liu, J.; Zhang, Y.; Ou, L.; Liu, X.; Lai, R.; Shao, L. Nanomaterials and hepatic disease: Toxicokinetics, disease types, intrinsic mechanisms, liver susceptibility, and influencing factors. J. Nanobiotechnology 2021, 19, 108. [Google Scholar] [CrossRef]
- Yarjanli, Z.; Ghaedi, K.; Esmaeili, A.; Rahgozar, S.; Zarrabi, A. Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation. BMC Neurosci. 2017, 18, 51. [Google Scholar] [CrossRef]
- Arami, H.; Khandhar, A.; Liggitt, D.; Krishnan, K.M. In vivo delivery, pharmacokinetics, biodistribution and toxicity of iron oxide nanoparticles. Chem. Soc. Rev. 2015, 44, 8576–8607. [Google Scholar] [CrossRef]
- Cheng, H.W.; Tsao, H.Y.; Chiang, C.S.; Chen, S.Y. Advances in magnetic nanoparticle-mediated cancer immune-theranostics. Adv. Healthc. Mater. 2021, 10, 2001451. [Google Scholar] [CrossRef]
- He, M.; Chu, C.-C. A new family of functional biodegradable arginine-based polyester urea urethanes: Synthesis, chracterization and biodegradation. Polymer 2013, 54, 4112–4125. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zeta Potential in Saline | DLS Size | TEM Size | |
|---|---|---|---|
| IONP (without polymer coating) | −30.5 ± 7.2 mV | 37.5 ± 8.2 nm | 12.5 ± 3.6 nm |
| APU-IONP (IONP coated with APU) | 15.2 ± 5.5 mV | 50.6 ± 7.5 nm | n.a. |
| APU-R848-IONP (IONP coated with APU-R848) | 5.6 ± 2.3 mV | 58.5 ± 6.1 nm | 13.2 ± 3.0 nm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Li, J.; Ma, L.; Wang, Z.; Du, B.; Kwan, H.Y.; Bian, Z.; Chu, C.-C. Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles for Enhanced Treatment of Triple-Negative Breast Tumor. Nanomaterials 2025, 15, 1006. https://doi.org/10.3390/nano15131006
Ji Y, Li J, Ma L, Wang Z, Du B, Kwan HY, Bian Z, Chu C-C. Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles for Enhanced Treatment of Triple-Negative Breast Tumor. Nanomaterials. 2025; 15(13):1006. https://doi.org/10.3390/nano15131006
Chicago/Turabian StyleJi, Ying, Juan Li, Li Ma, Zhijie Wang, Bochu Du, Hiu Yee Kwan, Zhaoxiang Bian, and Chih-Chang Chu. 2025. "Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles for Enhanced Treatment of Triple-Negative Breast Tumor" Nanomaterials 15, no. 13: 1006. https://doi.org/10.3390/nano15131006
APA StyleJi, Y., Li, J., Ma, L., Wang, Z., Du, B., Kwan, H. Y., Bian, Z., & Chu, C.-C. (2025). Development of Immune-Regulatory Pseudo-Protein-Coated Iron Oxide Nanoparticles for Enhanced Treatment of Triple-Negative Breast Tumor. Nanomaterials, 15(13), 1006. https://doi.org/10.3390/nano15131006