
In Situ Characterization Method to Reveal the Surface Reconstruction Process of an Electrocatalyst

 , ,
, , 
Abstract
1. Introduction
2. Surface Reconstruction
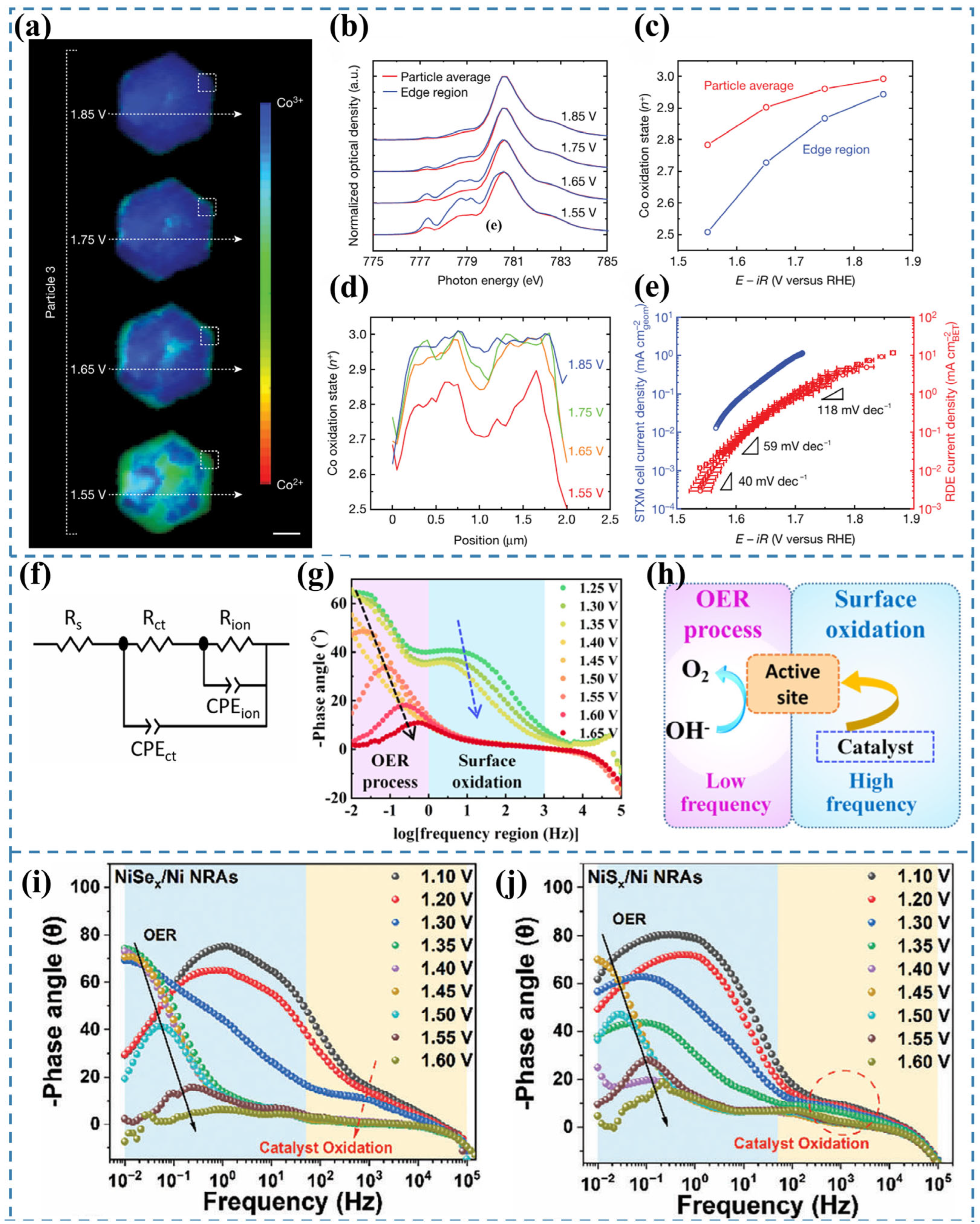
2.1. OER in Alkaline Conditions
2.2. OER in Acidic Conditions
2.3. HER in Alkaline Conditions
2.4. HER in Acidic Conditions
3. In Situ Characterization of Surface Reconstruction
3.1. In Situ Raman

3.2. In Situ XRD

3.3. In Situ Synchrotron GIXRD
3.4. In Situ FTIR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.5. In Situ XPS
3.6. In Situ XAS
3.7. In Situ EELS
3.8. In Situ UV-Vis

3.9. In Situ ATR-IR
3.10. In Situ TEM
3.11. EC-AFM
3.12. STXM
3.13. In Situ EIS
3.14. In Situ DEMS
4. Summary and Outlook
- (1)
- The reliance on a single in situ characterization technique is insufficient to comprehensively elucidate the phase, valence state, structure, and composition of catalysts. Consequently, integrating multiple in situ characterization methods is essential to provide a holistic understanding of catalyst evolution and to accurately identify the catalytically active components.
- (2)
- The application of in situ characterization techniques to reveal the surface reconstruction process of electrocatalysts remains a complex endeavor. A significant challenge lies in reducing the technical complexity and operational difficulty associated with in situ testing. Innovations in experimental design, instrumentation, and data analysis are critical to streamline these processes and enhance their accessibility.
- (3)
- Many in situ characterization methods require vacuum conditions, which starkly contrast with the realistic environments where electrocatalyst surface reconstruction occurs. Therefore, advancing techniques that operate under ambient or reaction-relevant conditions represents a crucial development direction. Such advancements will enable a more accurate depiction of surface reconstruction processes under practical scenarios.
- (4)
- Certain characterization methods are limited by their low radiation energies, which restrict their application to materials with strong signal responses. Synchrotron radiation, characterized by its exceptionally high energy, offers a transformative solution by enabling the detection of robust signals even in low-doping materials. This high-energy radiation is particularly advantageous for uncovering the intricate details of surface reconstruction processes, making it a pivotal tool in future research.
- (5)
- The discrepancy between laboratory test conditions and industrial electrolysis environments poses a significant challenge in accurately capturing the surface reconstruction behavior of electrocatalysts. Understanding these processes under realistic operating conditions is imperative for bridging the gap between fundamental research and industrial applications. The development of in situ characterization technologies capable of replicating industrial conditions and capturing real-time surface reconstruction dynamics will be instrumental in advancing the field.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelly, N.A.; Gibson, T.L.; Cai, M.; Spearot, J.A.; Ouwerkerk, D.B. Development of a renewable hydrogen economy: Optimization of existing technologies. Int. J. Hydrogen Energy 2010, 35, 892–899. [Google Scholar] [CrossRef]
- Kuznetsov, D.A.; Han, B.; Yu, Y.; Rao, R.R.; Hwang, J.; Román-Leshkov, Y.; Shao-Horn, Y. Tuning Redox Transitions via Inductive Effect in Metal Oxides and Complexes, and Implications in Oxygen Electrocatalysis. Joule 2018, 2, 225–244. [Google Scholar] [CrossRef]
- Quan, L.; Jiang, H.; Mei, G.; Sun, Y.; You, B. Bifunctional Electrocatalysts for Overall and Hybrid Water Splitting. Chem. Rev. 2024, 124, 3694–3812. [Google Scholar] [CrossRef]
- Chen, J.; Chen, H.; Yu, T.; Li, R.; Wang, Y.; Shao, Z.; Song, S. Recent Advances in the Understanding of the Surface Reconstruction of Oxygen Evolution Electrocatalysts and Materials Development. Electrochem. Energy Rev. 2021, 4, 566–600. [Google Scholar] [CrossRef]
- Liu, X.; Meng, J.; Zhu, J.; Huang, M.; Wen, B.; Guo, R.; Mai, L. Comprehensive Understandings into Complete Reconstruction of Precatalysts: Synthesis, Applications, and Characterizations. Adv. Mater. 2021, 33, 2007344. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, X.; Huang, J.; Wu, C.; Sun, Y.; Zou, G.; Yang, C.; Xiong, J. Recent Progress on Surface Reconstruction of Earth-Abundant Electrocatalysts for Water Oxidation. Small 2019, 15, 1901980. [Google Scholar] [CrossRef]
- Jiang, H.; He, Q.; Zhang, Y.; Song, L. Structural Self-Reconstruction of Catalysts in Electrocatalysis. Acc. Chem. Res. 2018, 51, 2968–2977. [Google Scholar] [CrossRef]
- Fan, K.; Zou, H.; Lu, Y.; Chen, H.; Li, F.; Liu, J.; Sun, L.; Tong, L.; Toney, M.F.; Sui, M.; et al. Direct Observation of Structural Evolution of Metal Chalcogenide in Electrocatalytic Water Oxidation. ACS Nano 2018, 12, 12369–12379. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhao, M.; Huang, Z.; Zhu, W.; Zheng, J.; Jiang, Q.; Wang, Z.; Liang, H. Surface Reconstruction of Water Splitting Electrocatalysts. Adv. Energy Mater. 2022, 12, 2201713. [Google Scholar] [CrossRef]
- Liu, X.; Guo, R.; Ni, K.; Xia, F.; Niu, C.; Wen, B.; Meng, J.; Wu, P.; Wu, J.; Wu, X.; et al. Reconstruction-Determined Alkaline Water Electrolysis at Industrial Temperatures. Adv. Mater. 2020, 32, 2001136. [Google Scholar] [CrossRef]
- Abdullah, M.I.; Fang, Y.; Wu, X.; Hu, M.; Shao, J.; Tao, Y.; Wang, H. Tackling activity-stability paradox of reconstructed NiIrOx electrocatalysts by bridged W-O moiety. Nat. Commun. 2024, 15, 10587. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, H.; Wang, J.; Du, Y.; Xi, S.; Sun, Y.; Sherburne, M.; Ager, J.W.; Fisher, A.C.; Xu, Z.J. Exceptionally active iridium evolved from a pseudo-cubic perovskite for oxygen evolution in acid. Nat. Commun. 2019, 10, 572. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, J.; Niu, S.; Li, S.; Du, Y.; Xu, P. Crystalline-Amorphous Ni2P4O12/NiMoOx Nanoarrays for Alkaline Water Electrolysis: Enhanced Catalytic Activity via In Situ Surface Reconstruction. Small 2022, 18, 2105972. [Google Scholar] [CrossRef]
- Fan, K.; Zou, H.; Dharanipragada, N.V.R.A.; Fan, L.; Inge, A.K.; Duan, L.; Zhang, B.; Sun, L. Surface and bulk reconstruction of CoW sulfides during pH-universal electrocatalytic hydrogen evolution. J. Mater. Chem. A 2021, 9, 11359–11369. [Google Scholar] [CrossRef]
- Liu, D.; Ai, H.; Li, J.; Fang, M.; Chen, M.; Liu, D.; Du, X.; Zhou, P.; Li, F.; Lo, K.H.; et al. Surface Reconstruction and Phase Transition on Vanadium–Cobalt–Iron Trimetal Nitrides to Form Active Oxyhydroxide for Enhanced Electrocatalytic Water Oxidation. Adv. Energy Mater. 2020, 10, 2002464. [Google Scholar] [CrossRef]
- Wu, Z.; Gan, Q.; Li, X.; Zhong, Y.; Wang, H. Elucidating Surface Restructuring-Induced Catalytic Reactivity of Cobalt Phosphide Nanoparticles under Electrochemical Conditions. J. Phys. Chem. C 2018, 122, 2848–2853. [Google Scholar] [CrossRef]
- Pathak, I.; Acharya, D.; Chhetri, K.; Rosyara, Y.R.; Muthurasu, A.; Kim, T.; Ko, T.H.; Kim, H.Y. Coengineering of Ni-NDC derived graphitic Ni2P/NiSe2 on a Ti3C2Tx MXene-modified 3D self-supporting electrode: Unraveling 2D–2D multiphases for overall water electrolysis. Compos. Part B Eng. 2025, 296, 112238. [Google Scholar] [CrossRef]
- Zhang, H.; He, X.; Dong, K.; Yao, Y.; Sun, S.; Zhang, M.; Yue, M.; Yang, C.; Zheng, D.; Liu, Q.; et al. Selenate promoted stability improvement of nickel selenide nanosheet array with an amorphous NiOOH layer for seawater oxidation. Mater. Today Phys. 2023, 38, 101249. [Google Scholar] [CrossRef]
- Chang, K.; Tran, D.T.; Wang, J.; Prabhakaran, S.; Kim, D.H.; Kim, N.H.; Lee, J.H. Atomic Heterointerface Engineering of Ni2P-NiSe2 Nanosheets Coupled ZnP-Based Arrays for High-Efficiency Solar-Assisted Water Splitting. Adv. Funct. Mater. 2022, 32, 2113224. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, X.; Li, Y.; Liang, L.; Sasaki, K.; Hao, Q.; Yuan, W.; Li, S.; Liu, H. Constructing reconstruction-inhibited nickel selenide electrocatalysts via incorporating Ag single atom for durable and efficient water oxidation. Appl. Catal. B Environ. Energy 2024, 348, 123830. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, Y.; Tao, H.B.; Liu, Y.; Ma, X.; Li, D.S.; Yang, H.B.; Liu, B. Long-Term Stability Challenges and Opportunities in Acidic Oxygen Evolution Electrocatalysis. Angew. Chem. Int. Ed. 2023, 62, e202216645. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, J.-L.; Li, X.; Yang, S.; Luo, W.; Zhang, Y.; Kim, S.-H.; Kim, D.-H.; Shinde, S.S.; Li, Y.-F.; et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 2021, 4, 1012–1023. [Google Scholar] [CrossRef]
- Feng, J.; Wang, X.; Pan, H. In-situ Reconstruction of Catalyst in Electrocatalysis. Adv. Mater. 2024, 36, 2411688. [Google Scholar] [CrossRef]
- Zhu, K.; Zhu, X.; Yang, W. Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts. Angew. Chem. Int. Ed. 2018, 58, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Tan, X.; Ji, P.; Chen, L.; Yu, J.; Mu, S. Surface reconstruction-derived heterostructures for electrochemical water splitting. EnergyChem 2023, 5, 100091. [Google Scholar] [CrossRef]
- Chen, S.; Ma, L.; Huang, Z.; Liang, G.; Zhi, C. In situ/operando analysis of surface reconstruction of transition metal-based oxygen evolution electrocatalysts. Cell Rep. Phys. Sci. 2022, 3, 100729. [Google Scholar] [CrossRef]
- Wang, C.; Zhai, P.; Xia, M.; Liu, W.; Gao, J.; Sun, L.; Hou, J. Identification of the Origin for Reconstructed Active Sites on Oxyhydroxide for Oxygen Evolution Reaction. Adv. Mater. 2022, 35, 2209307. [Google Scholar] [CrossRef]
- Zhai, Y.; Ren, X.; Zhang, J.; Gan, T.; Yang, N.; Wang, B.; Liu, S. Dynamic Self-Healing of the Reconstructed Phase in Perovskite Oxides for Efficient and Stable Electrocatalytic OER. Small 2024, 21, 2407851. [Google Scholar] [CrossRef]
- Abrashev, M.V.; Ivanov, V.G.; Stefanov, B.S.; Todorov, N.D.; Rosell, J.; Skumryev, V. Raman spectroscopy of alpha-FeOOH (goethite) near antiferromagnetic to paramagnetic phase transition. J. Appl. Phys. 2020, 127, 205108. [Google Scholar] [CrossRef]
- Zhu, K.; Shi, F.; Zhu, X.; Yang, W. The roles of oxygen vacancies in electrocatalytic oxygen evolution reaction. Nano Energy 2020, 73, 104761. [Google Scholar] [CrossRef]
- Liu, X.; Ni, K.; Wen, B.; Guo, R.; Niu, C.; Meng, J.; Li, Q.; Wu, P.; Zhu, Y.; Wu, X.; et al. Deep Reconstruction of Nickel-Based Precatalysts for Water Oxidation Catalysis. ACS Energy Lett. 2019, 4, 2585–2592. [Google Scholar] [CrossRef]
- Gründer, Y.; Lucas, C.A. Surface X-ray diffraction studies of single crystal electrocatalysts. Nano Energy 2016, 29, 378–393. [Google Scholar] [CrossRef]
- Fan, K.; Zong, L.; Liu, J.; Chuang, C.H.; Dong, M.; Zou, Y.; Xu, Y.; Fu, H.Q.; Zhang, L.; Wang, L.; et al. In Situ Reconstruction to Surface Sulfide Adsorbed Metal Scaffold for Enhanced Electrocatalytic Hydrogen Evolution Activity. Adv. Energy Mater. 2024, 14, 2400052. [Google Scholar] [CrossRef]
- Du, J.; You, S.; Li, X.; Tang, B.; Jiang, B.; Yu, Y.; Cai, Z.; Ren, N.; Zou, J. In Situ Crystallization of Active NiOOH/CoOOH Heterostructures with Hydroxide Ion Adsorption Sites on Velutipes-like CoSe/NiSe Nanorods as Catalysts for Oxygen Evolution and Cocatalysts for Methanol Oxidation. ACS Appl. Mater. Interfaces 2019, 12, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-W.; Hsu, Y.-Y.; Shen, Y.-P.; Zheng, Y.; Chan, T.-S.; Sheu, H.-S.; Cheng, Y.-C.; Chen, H.M. Reversible adapting layer produces robust single-crystal electrocatalyst for oxygen evolution. Nat. Commun. 2015, 6, 8106. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, X.; Zhang, J.; Huang, Y.; Liu, B. In Situ/Operando Techniques for Characterization of Single-Atom Catalysts. ACS Catal. 2019, 9, 2521–2531. [Google Scholar] [CrossRef]
- Christensen, P.A.; Mashhadani, Z.T.A.W.; Ali, A.H.B.M. In situ FTIR studies on the oxidation of isopropyl alcohol over SnO2 as a function of temperature up to 600 °C and a comparison to the analogous plasma-driven process. Phys. Chem. Chem. Phys. 2018, 20, 9053–9062. [Google Scholar] [CrossRef]
- Lefèvre, G. In situ Fourier-transform infrared spectroscopy studies of inorganic ions adsorption on metal oxides and hydroxides. Adv. Colloid Interface Sci. 2004, 107, 109–123. [Google Scholar] [CrossRef]
- Prinetto, F.; Ghiotti, G.; Nova, I.; Castoldi, L.; Lietti, L.; Tronconi, E.; Forzatti, P. In situ FT-IR and reactivity study of NOx storage over Pt–Ba/Al2O3catalysts. Phys. Chem. Chem. Phys. 2003, 5, 4428–4434. [Google Scholar] [CrossRef]
- Favaro, M.; Yang, J.; Nappini, S.; Magnano, E.; Toma, F.M.; Crumlin, E.J.; Yano, J.; Sharp, I.D. Understanding the Oxygen Evolution Reaction Mechanism on CoOx using Operando Ambient-Pressure X-ray Photoelectron Spectroscopy. J. Am. Chem. Soc. 2017, 139, 8960–8970. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Liu, Z.; Bluhm, H.; Yang, W.; Guo, J.; Hussain, Z. X-ray spectroscopy of energy materials under in situ/operando conditions. J. Electron Spectrosc. Relat. Phenom. 2015, 200, 264–273. [Google Scholar] [CrossRef]
- Tahir, M.; Pan, L.; Idrees, F.; Zhang, X.; Wang, L.; Zou, J.-J.; Wang, Z.L. Electrocatalytic oxygen evolution reaction for energy conversion and storage: A comprehensive review. Nano Energy 2017, 37, 136–157. [Google Scholar] [CrossRef]
- Toparli, C.; Sarfraz, A.; Wieck, A.D.; Rohwerder, M.; Erbe, A. In situ and operando observation of surface oxides during oxygen evolution reaction on copper. Electrochim. Acta 2017, 236, 104–115. [Google Scholar] [CrossRef]
- Song, S.; Zhou, J.; Su, X.; Wang, Y.; Li, J.; Zhang, L.; Xiao, G.; Guan, C.; Liu, R.; Chen, S.; et al. Operando X-ray spectroscopic tracking of self-reconstruction for anchored nanoparticles as high-performance electrocatalysts towards oxygen evolution. Energy Environ. Sci. 2018, 11, 2945–2953. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Jin, J.; Wang, Y.; Peng, Y.; Yin, J.; Shen, W.; Hou, Y.; Zhu, L.; An, L.; et al. Understanding the sulphur-oxygen exchange process of metal sulphides prior to oxygen evolution reaction. Nat. Commun. 2023, 14, 1949. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hong, J.; Lee, D.; Lee, S.-Y.; Zheng, H. In Situ TEM Characterization of Battery Materials. Chem. Rev. 2025, 125, 1840–1896. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wang, C.; Wu, X.; Zhang, J.; Chu, J. In Situ Transmission Electron Microscopy Characterization and Manipulation of Two-Dimensional Layered Materials beyond Graphene. Small 2017, 13, 1604259. [Google Scholar] [CrossRef] [PubMed]
- Klie, R.F.; Zheng, J.C.; Zhu, Y.; Varela, M.; Wu, J.; Leighton, C. Direct Measurement of the Low-Temperature Spin-State Transition in LaCoO3. Phys. Rev. Lett. 2007, 99, 047203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Santhanagopalan, D.; Zhang, W.; Wang, F.; Xin, H.L.; He, K.; Li, J.; Dudney, N.; Meng, Y.S. In Situ STEM-EELS Observation of Nanoscale Interfacial Phenomena in All-Solid-State Batteries. Nano Lett. 2016, 16, 3760–3767. [Google Scholar] [CrossRef]
- Gong, M.; Zhou, W.; Kenney, M.J.; Kapusta, R.; Cowley, S.; Wu, Y.; Lu, B.; Lin, M.C.; Wang, D.Y.; Yang, J.; et al. Blending Cr2O3 into a NiO–Ni Electrocatalyst for Sustained Water Splitting. Angew. Chem. Int. Ed. 2015, 54, 11989–11993. [Google Scholar] [CrossRef]
- Li, X.; Ren, Z.; Banis, M.N.; Deng, S.; Zhao, Y.; Sun, Q.; Wang, C.; Yang, X.; Li, W.; Liang, J.; et al. Unravelling the Chemistry and Microstructure Evolution of a Cathodic Interface in Sulfide-Based All-Solid-State Li-Ion Batteries. ACS Energy Lett. 2019, 4, 2480–2488. [Google Scholar] [CrossRef]
- Gazquez, J.; Luo, W.; Oxley, M.P.; Prange, M.; Torija, M.A.; Sharma, M.; Leighton, C.; Pantelides, S.T.; Pennycook, S.J.; Varela, M. Atomic-Resolution Imaging of Spin-State Superlattices in Nanopockets within Cobaltite Thin Films. Nano Lett. 2011, 11, 973–976. [Google Scholar] [CrossRef]
- Zhou, S.; Miao, X.; Zhao, X.; Ma, C.; Qiu, Y.; Hu, Z.; Zhao, J.; Shi, L.; Zeng, J. Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition. Nat. Commun. 2016, 7, 11510. [Google Scholar] [CrossRef] [PubMed]
- Wijten, J.H.J.; Mandemaker, L.D.B.; van Eeden, T.C.; Dubbeld, J.E.; Weckhuysen, B.M. In Situ Study on Ni–Mo Stability in a Water-Splitting Device: Effect of Catalyst Substrate and Electric Potential. ChemSusChem 2020, 13, 3172–3179. [Google Scholar] [CrossRef]
- Lu, J.; Bravosuarez, J.; Takahashi, A.; Haruta, M.; Oyama, S. In situ UV–vis studies of the effect of particle size on the epoxidation of ethylene and propylene on supported silver catalysts with molecular oxygen. J. Catal. 2005, 232, 85–95. [Google Scholar] [CrossRef]
- Pishgar, S.; Strain, J.M.; Gulati, S.; Sumanasekera, G.; Gupta, G.; Spurgeon, J.M. Investigation of the photocorrosion of n-GaP photoanodes in acid with in situ UV-Vis spectroscopy. J. Mater. Chem. A 2019, 7, 25377–25388. [Google Scholar] [CrossRef]
- Lee, S.; Moysiadou, A.; Chu, Y.-C.; Chen, H.M.; Hu, X. Tracking high-valent surface iron species in the oxygen evolution reaction on cobalt iron (oxy)hydroxides. Energy Environ. Sci. 2022, 15, 206–214. [Google Scholar] [CrossRef]
- Zhu, J.; Zi, S.; Zhang, N.; Hu, Y.; An, L.; Xi, P. Surface Reconstruction of Covellite CuS Nanocrystals for Enhanced OER Catalytic Performance in Alkaline Solution. Small 2023, 19, 2301762. [Google Scholar] [CrossRef]
- Gao, L.; Cui, X.; Sewell, C.D.; Li, J.; Lin, Z. Recent advances in activating surface reconstruction for the high-efficiency oxygen evolution reaction. Chem. Soc. Rev. 2021, 50, 8428–8469. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.; Huang, B.; Hu, Y.; An, L.; Yao, Y.; Lu, M.; Jin, J.; Zhang, N.; Xi, P.; et al. Activated Ni-O-Ir Enhanced Electron Transfer for Boosting Oxygen Evolution Reaction Activity of LaNi1-xIrxO3. Small 2022, 18, 2204723. [Google Scholar] [CrossRef]
- Wang, L.; Meng, Q.; Xiao, M.; Liu, C.; Xing, W.; Zhu, J. Insights into the Dynamic Surface Reconstruction of Electrocatalysts in Oxygen Evolution Reaction. Renewables 2024, 2, 272–296. [Google Scholar] [CrossRef]
- Liu, D.; Shadike, Z.; Lin, R.; Qian, K.; Li, H.; Li, K.; Wang, S.; Yu, Q.; Liu, M.; Ganapathy, S.; et al. Review of Recent Development of In Situ/Operando Characterization Techniques for Lithium Battery Research. Adv. Mater. 2019, 31, 1806620. [Google Scholar] [CrossRef]
- Sun, H.; Tung, C.-W.; Qiu, Y.; Zhang, W.; Wang, Q.; Li, Z.; Tang, J.; Chen, H.-C.; Wang, C.; Chen, H.M. Atomic Metal–Support Interaction Enables Reconstruction-Free Dual-Site Electrocatalyst. J. Am. Chem. Soc. 2021, 144, 1174–1186. [Google Scholar] [CrossRef]
- Shin, D.; Park, J.B.; Kim, Y.-J.; Kim, S.J.; Kang, J.H.; Lee, B.; Cho, S.-P.; Hong, B.H.; Novoselov, K.S. Growth dynamics and gas transport mechanism of nanobubbles in graphene liquid cells. Nat. Commun. 2015, 6, 6068. [Google Scholar] [CrossRef]
- Mefford, J.T.; Akbashev, A.R.; Kang, M.; Bentley, C.L.; Gent, W.E.; Deng, H.D.; Alsem, D.H.; Yu, Y.-S.; Salmon, N.J.; Shapiro, D.A.; et al. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 2021, 593, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Dette, C.; Hurst, M.R.; Deng, J.; Nellist, M.R.; Boettcher, S.W. Structural Evolution of Metal (Oxy)hydroxide Nanosheets during the Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2018, 11, 5590–5594. [Google Scholar] [CrossRef]
- Toma, F.M.; Cooper, J.K.; Kunzelmann, V.; McDowell, M.T.; Yu, J.; Larson, D.M.; Borys, N.J.; Abelyan, C.; Beeman, J.W.; Yu, K.M.; et al. Mechanistic insights into chemical and photochemical transformations of bismuth vanadate photoanodes. Nat. Commun. 2016, 7, 12012. [Google Scholar] [CrossRef] [PubMed]
- Mudali, U.K.; Padhy, N. Electrochemical scanning probe microscope (EC-SPM) for the in situ corrosion study of materials: An overview with examples. Corros. Rev. 2011, 29, 73–103. [Google Scholar] [CrossRef]
- Deng, J.; Nellist, M.R.; Stevens, M.B.; Dette, C.; Wang, Y.; Boettcher, S.W. Morphology Dynamics of Single-Layered Ni(OH)2/NiOOH Nanosheets and Subsequent Fe Incorporation Studied by in Situ Electrochemical Atomic Force Microscopy. Nano Lett. 2017, 17, 6922–6926. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Kong, H.; Kim, J.; Choi, S.; Ciucci, F.; Hao, Y.; Yang, S.; Shao, Z.; Lim, J. Non-precious-metal catalysts for alkaline water electrolysis: Operando characterizations, theoretical calculations, and recent advances. Chem. Soc. Rev. 2020, 49, 9154–9196. [Google Scholar] [CrossRef]
- Wang, J.; Kim, H.; Hyun, H.; Jo, S.; Han, J.; Ko, D.; Seo, S.; Kim, J.; Kong, H.; Lim, J. Probing and Resolving the Heterogeneous Degradation of Nickel-Rich Layered Oxide Cathodes across Multi-Length Scales. Small Methods 2020, 4, 2000551. [Google Scholar] [CrossRef]
- Lim, J.; Li, Y.; Alsem, D.H.; So, H.; Lee, S.C.; Bai, P.; Cogswell, D.A.; Liu, X.; Jin, N.; Yu, Y.S.; et al. Origin and hysteresis of lithium compositional spatiodynamics within battery primary particles. Science 2016, 353, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Mefford, J.T.; Zhao, Z.; Bajdich, M.; Chueh, W.C. Interpreting Tafel behavior of consecutive electrochemical reactions through combined thermodynamic and steady state microkinetic approaches. Energy Environ. Sci. 2020, 13, 622–634. [Google Scholar] [CrossRef]
- Bergmann, A.; Jones, T.E.; Moreno, E.M.; Teschner, D.; Chernev, P.; Gliech, M.; Reier, T.; Dau, H.; Strasser, P. Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction. Nat. Catal. 2018, 1, 711–719. [Google Scholar] [CrossRef]
- Wu, J.; Wang, X.; Zheng, W.; Sun, Y.; Xie, Y.; Ma, K.; Zhang, Z.; Liao, Q.; Tian, Z.; Kang, Z.; et al. Identifying and Interpreting Geometric Configuration-Dependent Activity of Spinel Catalysts for Water Reduction. J. Am. Chem. Soc. 2022, 144, 19163–19172. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, L.; Yang, T.; Wang, E.; Yu, X.; Hou, Y.; Du, Z.; Cao, S.; Chou, K.-C.; Hou, X. Enhanced overall water splitting by morphology and electronic structure engineering on pristine ultrathin metal-organic frameworks. J. Mater. Sci. Technol. 2025, 220, 92–103. [Google Scholar] [CrossRef]
- Wang, S.; Yan, Y.; Du, Y.; Zhao, Y.; Li, T.; Wang, D.; Schaaf, P.; Wang, X. Inhibiting the Deep Reconstruction of Ni-Based Interface by Coordination of Chalcogen Anions for Efficient and Stable Glycerol Electrooxidation. Adv. Funct. Mater. 2024, 34, 2404290. [Google Scholar] [CrossRef]
- Deng, L.; Hung, S.F.; Lin, Z.Y.; Zhang, Y.; Zhang, C.; Hao, Y.; Liu, S.; Kuo, C.H.; Chen, H.Y.; Peng, J.; et al. Valence Oscillation of Ru Active Sites for Efficient and Robust Acidic Water Oxidation. Adv. Mater. 2023, 35, 2305939. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Hung, S.-F.; Chen, H.-Y.; Chan, T.-S.; Chen, H.M.; Liu, B. In Operando Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4. J. Am. Chem. Soc. 2015, 138, 36–39. [Google Scholar] [CrossRef]
- Doyle, R.L.; Lyons, M.E.G. Kinetics and Mechanistic Aspects of the Oxygen Evolution Reaction at Hydrous Iron Oxide Films in Base. J. Electrochem. Soc. 2013, 160, H142–H154. [Google Scholar] [CrossRef]
- Xie, C.; Chen, W.; Du, S.; Yan, D.; Zhang, Y.; Chen, J.; Liu, B.; Wang, S. In-situ phase transition of WO3 boosting electron and hydrogen transfer for enhancing hydrogen evolution on Pt. Nano Energy 2020, 71, 104653. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.-X.; Gou, W.; Zhang, M.; Xia, Z.; Zhang, S.; Chang, C.-R.; Ma, Y.; Qu, Y. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover. Energy Environ. Sci. 2019, 12, 2298–2304. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, Y.-C.; Dong, C.-L.; Xie, C.; Liu, Z.; Du, S.; Chen, W.; Yan, D.; Tao, L.; Shu, Z.; et al. Operando Identification of the Dynamic Behavior of Oxygen Vacancy-Rich Co3O4 for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2020, 142, 12087–12095. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, H.; Xi, S.; Lee, W.S.V.; Xue, J. Understanding of Oxygen Redox in the Oxygen Evolution Reaction. Adv. Mater. 2022, 34, 2107956. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.W.; Yue, K.; Wu, L.; Yang, J.; Luan, D.; Zhang, X.; Lou, X.W. Surface Transformation in Lanthanum Nickelate for Enhanced Oxygen Evolution Catalysis. Angew. Chem. Int. Ed. 2025; early view. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, Y.; Yang, T.; Liu, S.; Yang, L.; Wang, E.; Yu, X.; Wang, H.; Chou, K.-C.; Hou, X. In Situ Characterization Method to Reveal the Surface Reconstruction Process of an Electrocatalyst. Nanomaterials 2025, 15, 917. https://doi.org/10.3390/nano15120917
Zhan Y, Yang T, Liu S, Yang L, Wang E, Yu X, Wang H, Chou K-C, Hou X. In Situ Characterization Method to Reveal the Surface Reconstruction Process of an Electrocatalyst. Nanomaterials. 2025; 15(12):917. https://doi.org/10.3390/nano15120917
Chicago/Turabian StyleZhan, Yiqin, Tao Yang, Shuang Liu, Liming Yang, Enhui Wang, Xiangtao Yu, Hongyang Wang, Kuo-Chih Chou, and Xinmei Hou. 2025. "In Situ Characterization Method to Reveal the Surface Reconstruction Process of an Electrocatalyst" Nanomaterials 15, no. 12: 917. https://doi.org/10.3390/nano15120917
APA StyleZhan, Y., Yang, T., Liu, S., Yang, L., Wang, E., Yu, X., Wang, H., Chou, K.-C., & Hou, X. (2025). In Situ Characterization Method to Reveal the Surface Reconstruction Process of an Electrocatalyst. Nanomaterials, 15(12), 917. https://doi.org/10.3390/nano15120917