
Insights into CoFe2O4/Peracetic Acid Catalytic Oxidation Process for Iopamidol Degradation: Performance, Mechanisms, and I-DBP Formation Control




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Methods
2.1. Chemical Reagents and Materials
2.2. Experimental Procedures
2.3. Analysis Methods and Instruments
3. Results and Discussion
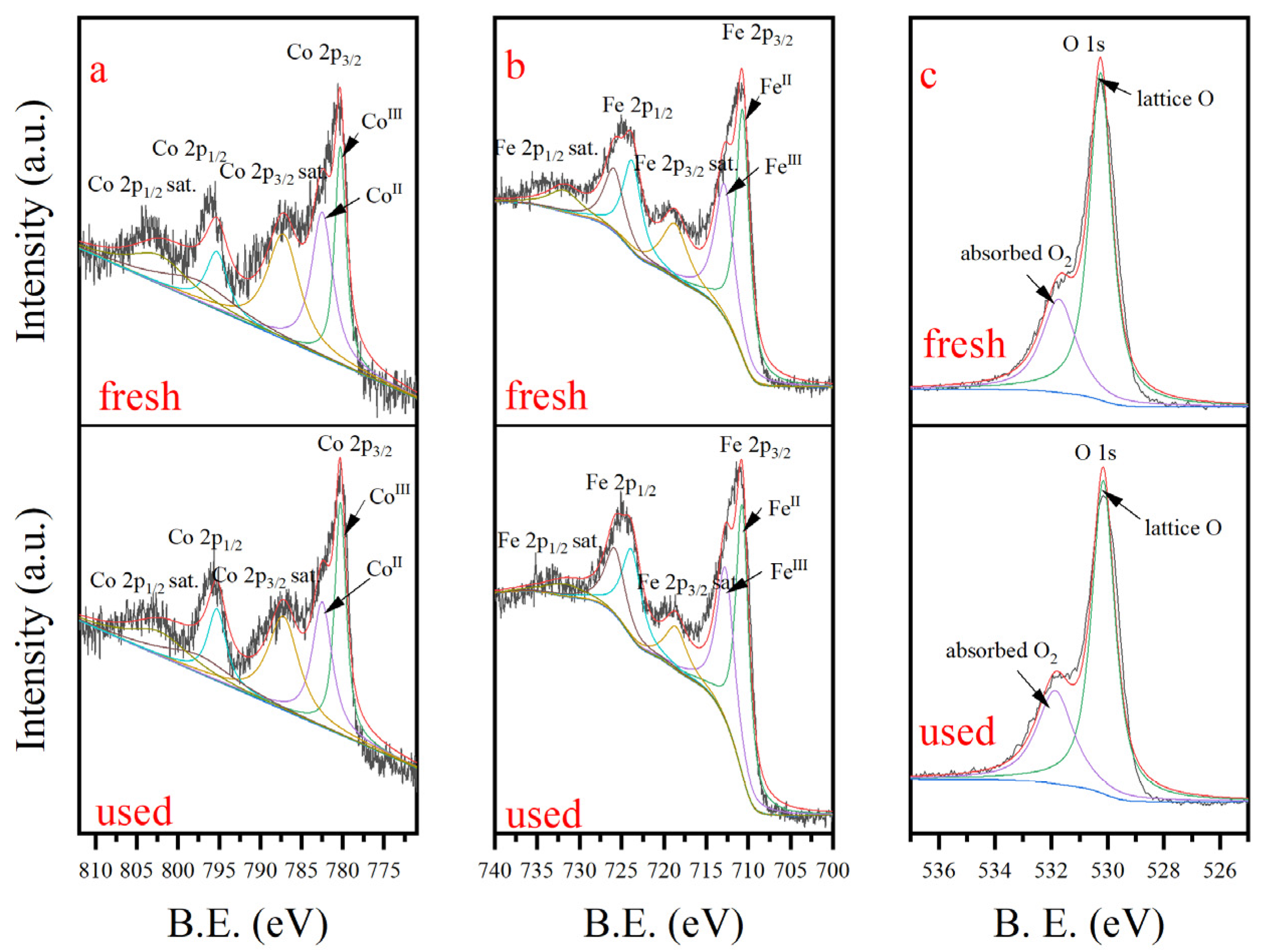
3.1. Characterization of CoFe2O4 Nanoparticles
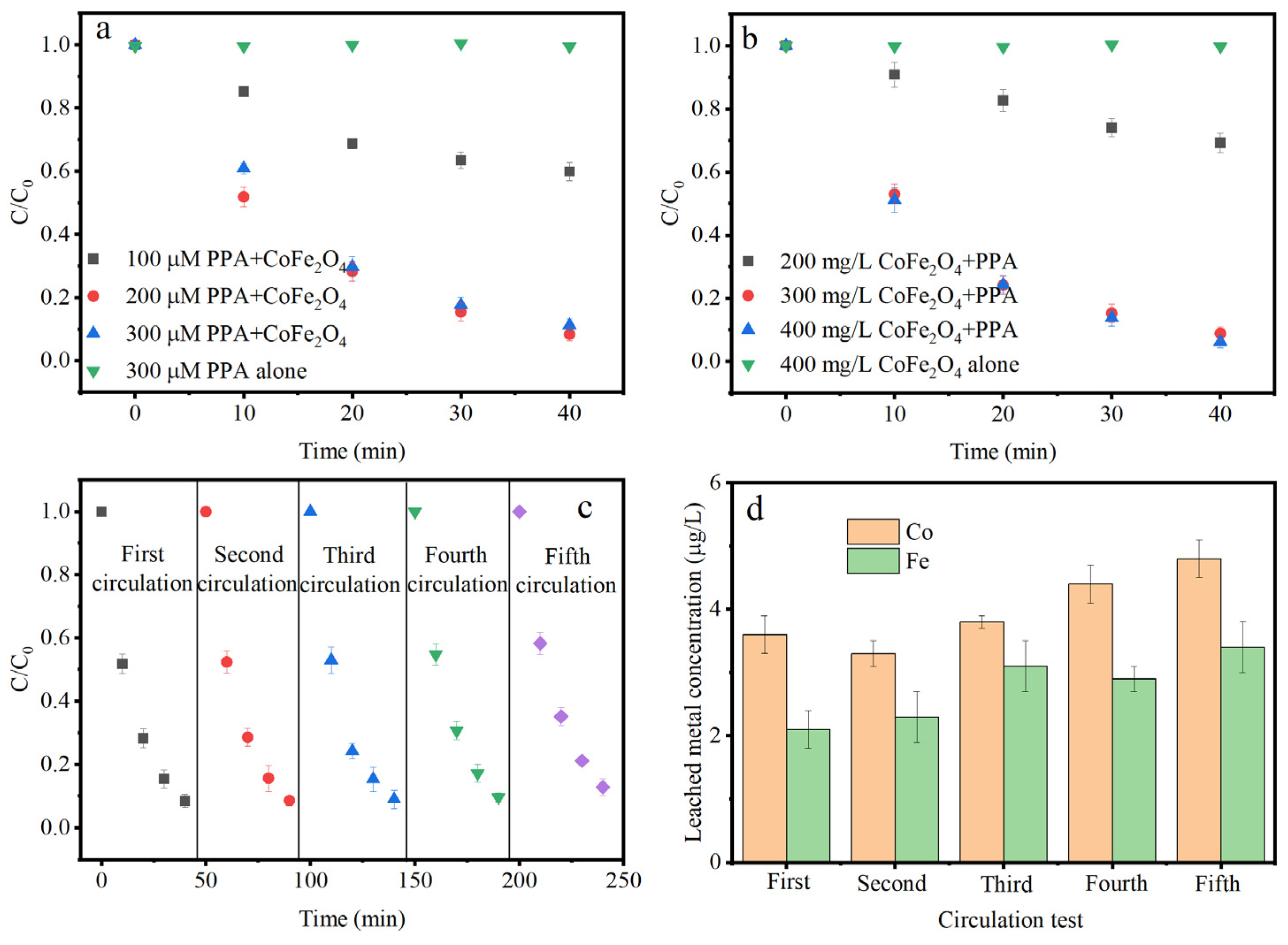
3.2. IPM Degradation Efficiency in CoFe2O4/PAA
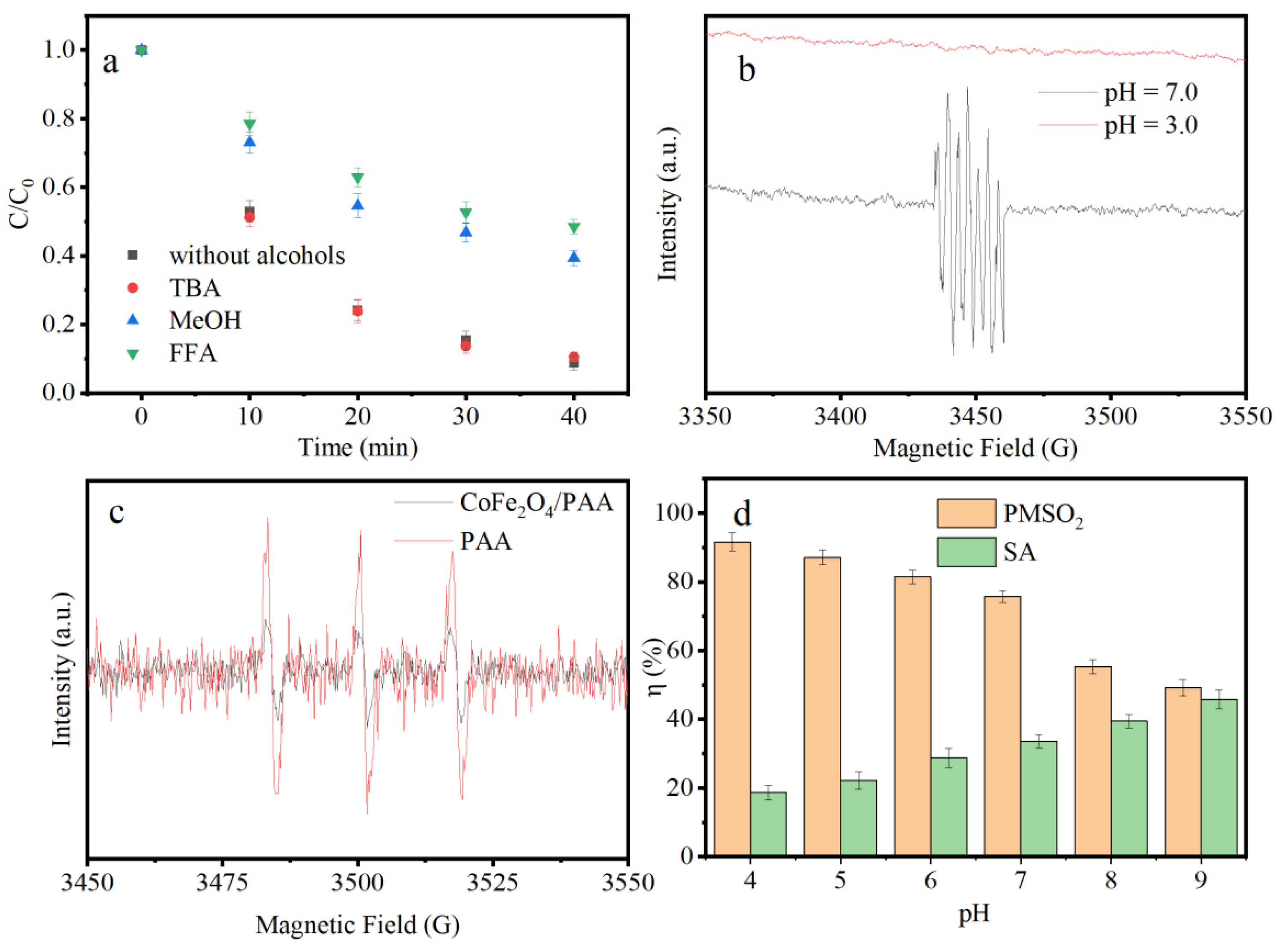
3.3. Determination of ROS in CoFe2O4/PAA
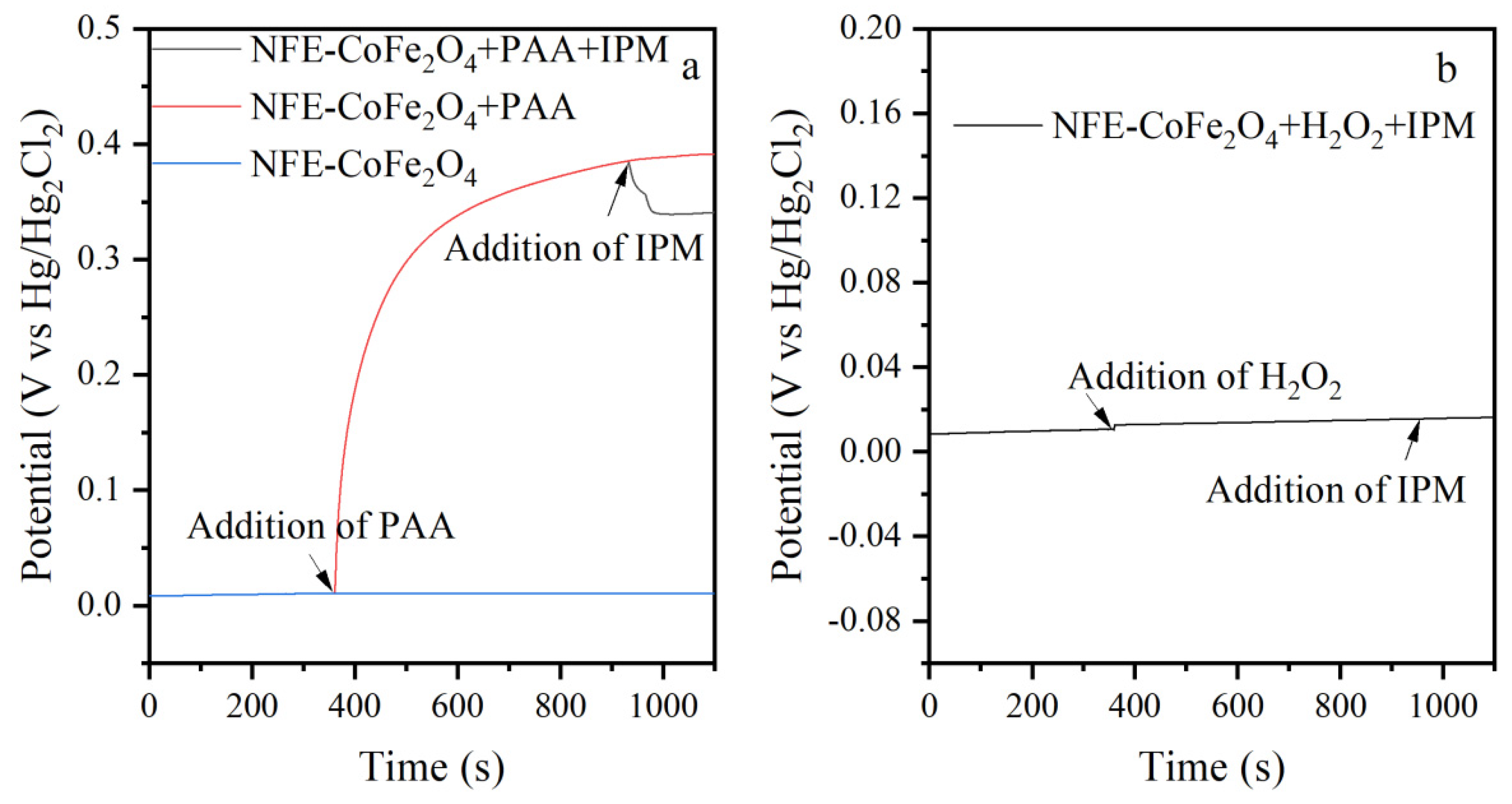
3.4. Electron Transfer Mechanism
3.5. Estimation of MIAA Formation Potential (MIAAFP)
3.6. Proposed IPM Degradation Pathway
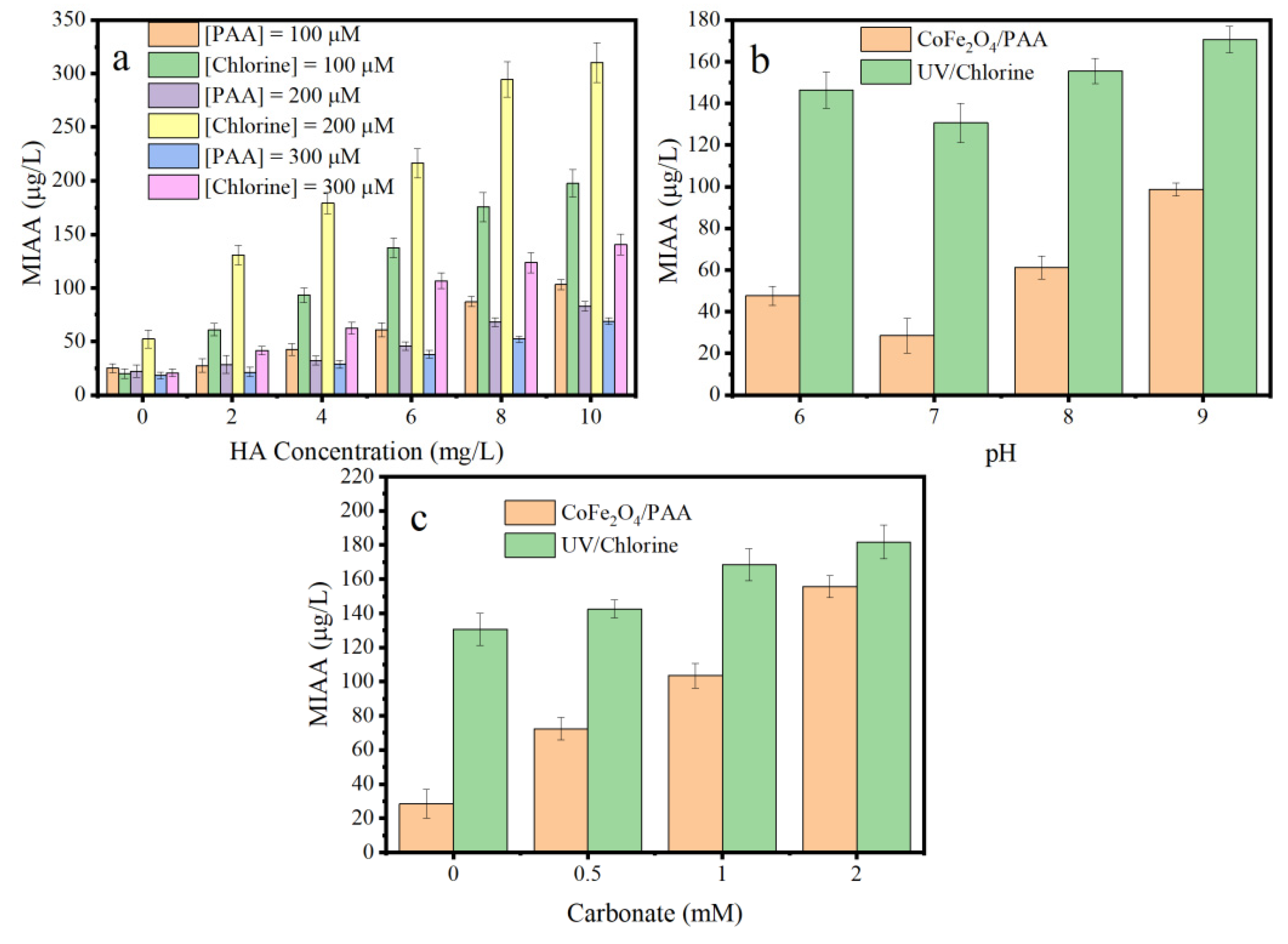
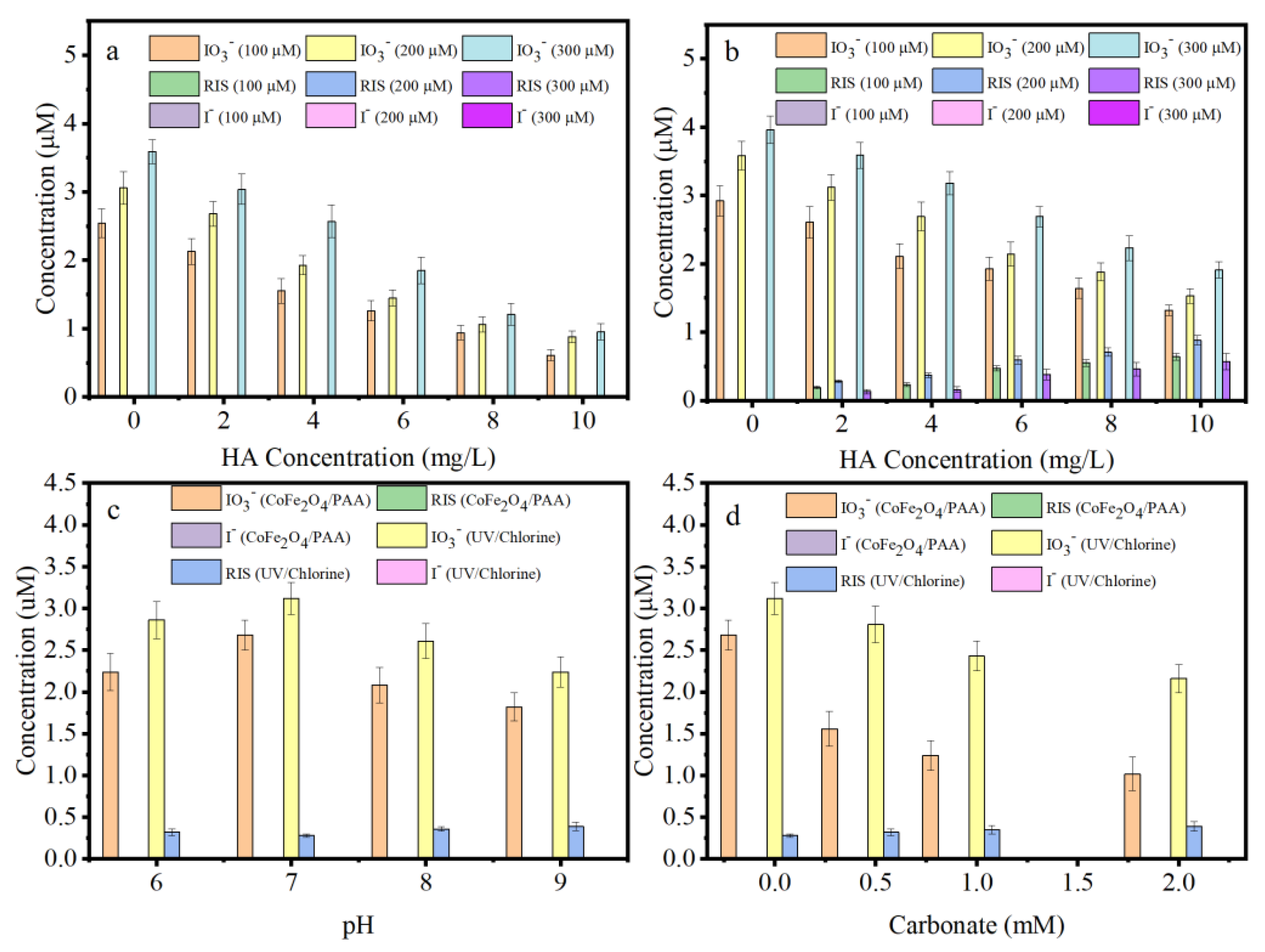
3.7. MIAAFP Control in Real Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Abd El-Rahman, M.K.; Riad, S.M.; Abdel Gawad, S.A.; Fawaz, E.M.; Shehata, M.A. Stability indicating spectrophotometric and spectrodensitometric methods for the determination of diatrizoate sodium in presence of its degradation product. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Bartels, Y.; Jekel, M.; Putschew, A. Impact of the reductive deiodination on the sorption of iodinated X-ray contrast media to filter sand and activated carbon. Water Res. 2024, 258, 121801. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bolton, J.R.; Hofmann, R. Medium pressure UV combined with chlorine advanced oxidation for trichloroethylene destruction in a model water. Water Res. 2012, 46, 4677–4686. [Google Scholar] [CrossRef]
- Hou, M.R.; Li, X.D.; Fu, Y.; Wang, L.L.; Lin, D.G.; Wang, Z.H. Degradation of iodinated X-ray contrast media by advanced oxidation processes: A literature review with a focus on degradation pathways. Chin. Chem. Lett. 2023, 34, 107723. [Google Scholar] [CrossRef]
- Lopez-Prieto, I.J.; Daniels, K.D.; Wu, S.M.; Snyder, S.A. Evaluating surrogate correlation models and iodinated haloacetic acid formation of iodinated contrast media after LPUV/Cl2, LPUV/NH2Cl, and LPUV/H2O2. J. Environ. Chem. Eng. 2021, 9, 105760. [Google Scholar] [CrossRef]
- Redeker, M.; Wick, A.; Meermann, B.; Ternes, T.A. Anaerobic Transformation of the Iodinated X-ray Contrast Medium lopromide, Its Aerobic Transformation Products, and Transfer to Further Iodinated X-ray Contrast Media. Environ. Sci. Technol. 2018, 52, 8309–8320. [Google Scholar] [CrossRef]
- Yeom, Y.; Han, J.; Zhang, X.; Shang, C.; Zhang, T.; Li, X.; Duan, X.; Dionysiou, D.D. A review on the degradation efficiency, DBP formation, and toxicity variation in the UV/chlorine treatment of micropollutants. Chem. Eng. J. 2021, 424, 130053. [Google Scholar] [CrossRef]
- Liu, S.G.; Li, Z.L.; Dong, H.Y.; Goodman, B.A.; Qiang, Z.M. Formation of iodo-trihalomethanes, iodo-acetic acids, and iodo-acetamides during chloramination of iodide-containing waters: Factors influencing formation and reaction pathways. J. Hazard. Mater. 2017, 321, 28–36. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wu, Z.H.; Hua, Z.C.; Guo, K.H.; Zhou, Y.J.; Wang, D.; Xia, B.C.; Fang, J.Y. Mechanistic and kinetic understanding of micropollutant degradation by the UV/NH2Cl process in simulated drinking water. Water Res. 2021, 204, 117569. [Google Scholar] [CrossRef]
- Zhang, W.; Fourcade, F.; Amrane, A.; Geneste, F. Removal of Iodine-Containing X-ray Contrast Media from Environment: The Challenge of a Total Mineralization. Molecules 2023, 28, 341. [Google Scholar] [CrossRef]
- Chávez, A.M.; Quiñones, D.H.; Rey, A.; Beltrán, F.J.; Alvarez, P.M. Simulated solar photocatalytic ozonation of contaminants of emerging concern and effluent organic matter in secondary effluents by a reusable magnetic catalyst. Chem. Eng. J. 2020, 398, 125642. [Google Scholar] [CrossRef]
- Ye, T.; Zhang, T.Y.; Tian, F.X.; Xu, B. The fate and transformation of iodine species in UV irradiation and UV-based advanced oxidation processes. Water Res. 2021, 206, 117755. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-C.; Lin, Y.-L.; Xu, B.; Xia, Y.; Hu, C.-Y.; Zhang, T.-Y.; Qian, H.; Cao, T.-C.; Gao, N.-Y. Effect of bromide and iodide on halogenated by-product formation from different organic precursors during UV/chlorine processes. Water Res. 2020, 182, 116035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhou, X.; Du, P.; Zhang, T.; Cai, M.; Sun, P.; Huang, C.-H. Oxidation of β-lactam antibiotics by peracetic acid: Reaction kinetics, product and pathway evaluation. Water Res. 2017, 123, 153–161. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Xiong, B.; Bai, F.; Wang, S.; Wan, Y.; Zhang, L.; Xie, P.; Wiesner, M.R. Application of Cobalt/Peracetic Acid to Degrade Sulfamethoxazole at Neutral Condition: Efficiency and Mechanisms. Environ. Sci. Technol. 2020, 54, 464–475. [Google Scholar] [CrossRef]
- Li, S.; Yang, Y.; Zheng, H.; Zheng, Y.; He, C.-S.; Lai, B.; Ma, J.; Nan, J. Introduction of oxygen vacancy to manganese ferrite by Co substitution for enhanced peracetic acid activation and 1O2 dominated tetracycline hydrochloride degradation under microwave irradiation. Water Res. 2022, 225, 119176. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, P.; Ren, Y.; Du, Y.; Geng, C.; Ma, J.; Zhao, F. Treatment of shale gas produced water by magnetic CuFe2O4/TNTs hybrid heterogeneous catalyzed ozone: Efficiency and mechanisms. J. Hazard. Mater. 2022, 423, 127124. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Wang, J. Nano spinel CoFe2O4 deposited diatomite catalytic separation membrane for efficiently cleaning wastewater. J. Membr. Sci. 2020, 615, 118559. [Google Scholar] [CrossRef]
- Luukkonen, T.; von Gunten, U. Oxidation of organic micropollutant surrogate functional groups with peracetic acid activated by aqueous Co(II), Cu(II), or Ag(I) and geopolymer-supported Co(II). Water Res. 2022, 223, 118984. [Google Scholar] [CrossRef]
- Yang, T.; Wu, S.; Mai, J.; Chen, L.; Huang, C.; Zeng, G.; Wu, Y.; Zhu, M.; Huang, Y.; Mo, Z.; et al. Activation of ferrate(VI) by sulfite for effectively degrading iodinated contrast media and synchronously controlling I-DBPs formation. Chem. Eng. J. 2022, 442, 136011. [Google Scholar] [CrossRef]
- Kong, D.; Zhao, Y.; Fan, X.; Wang, X.; Li, J.; Wang, X.; Nan, J.; Ma, J. Reduced Graphene Oxide Triggers Peracetic Acid Activation for Robust Removal of Micropollutants: The Role of Electron Transfer. Environ. Sci. Technol. 2022, 56, 11707–11717. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.Y.; Wang, M.Y.; Pang, Z.J.; Dai, L.; Lu, J.F.; Zou, J. Simultaneous spectrophotometric determination of peracetic acid and the coexistent hydrogen peroxide using potassium iodide as the indicator. Anal. Methods 2019, 11, 1930–1938. [Google Scholar] [CrossRef]
- Du, Y.; Ma, W.; Liu, P.; Zou, B.; Ma, J. Magnetic CoFe2O4 nanoparticles supported on titanate nanotubes (CoFe2O4/TNTs) as a novel heterogeneous catalyst for peroxymonosulfate activation and degradation of organic pollutants. J. Hazard. Mater. 2016, 308, 58–66. [Google Scholar] [CrossRef]
- Yang, S.J.; Xu, S.S.; Tong, J.Y.; Ding, D.H.; Wang, G.; Chen, R.Z.; Jin, P.K.; Wang, X.C.C. Overlooked role of nitrogen dopant in carbon catalysts for peroxymonosulfate activation: Intrinsic defects or extrinsic defects? Appl. Catal. B-Environ. 2021, 295, 120291. [Google Scholar] [CrossRef]
- Dong, X.B.; Ren, B.X.; Zhang, X.W.; Liu, X.R.; Sun, Z.M.; Li, C.Q.; Tan, Y.; Yang, S.S.; Zheng, S.L.; Dionysiou, D.D. Diatomite supported hierarchical 2D CoNi3O4 nanoribbons as highly efficient peroxymonosulfate catalyst for atrazine degradation. Appl. Catal. B-Environ. 2020, 272, 118971. [Google Scholar] [CrossRef]
- Yan, T.T.; Ping, Q.; Zhang, A.; Wang, L.; Dou, Y.C.; Li, Y.M. Enhanced removal of oxytetracycline by UV-driven advanced oxidation with peracetic acid: Insight into the degradation intermediates and N-nitrosodimethylamine formation potential. Chemosphere 2021, 274, 129726. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, C.; Guo, K.; Wu, Z.; Wang, L.; Hua, Z.; Fang, J. Kinetics and pathways of the degradation of PPCPs by carbonate radicals in advanced oxidation processes. Water Res. 2020, 185, 116231. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Qian, J.; Pan, B. Are Free Radicals the Primary Reactive Species in Co(II)-Mediated Activation of Peroxymonosulfate? New Evidence for the Role of the Co(II)–Peroxymonosulfate Complex. Environ. Sci. Technol. 2021, 55, 6397–6406. [Google Scholar] [CrossRef]
- Yang, X.; Cai, J.; Wang, X.; Li, Y.; Wu, Z.; Wu, W.D.; Chen, X.D.; Sun, J.; Sun, S.-P.; Wang, Z. A Bimetallic Fe–Mn Oxide-Activated Oxone for In Situ Chemical Oxidation (ISCO) of Trichloroethylene in Groundwater: Efficiency, Sustained Activity, and Mechanism Investigation. Environ. Sci. Technol. 2020, 54, 3714–3724. [Google Scholar] [CrossRef]
- Minh, T.D.; Ncibi, M.C.; Srivastava, V.; Thangaraj, S.K.; Jänis, J.; Sillanpää, M. Gingerbread ingredient-derived carbons-assembled CNT foam for the efficient peroxymonosulfate-mediated degradation of emerging pharmaceutical contaminants. Appl. Catal. B Environ. 2019, 244, 367–384. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, X.; Li, H.; Qian, J.; Pan, B. New Insights into the Activation of Peracetic Acid by Co(II): Role of Co(II)-Peracetic Acid Complex as the Dominant Intermediate Oxidant. ACS EST Eng. 2021, 1, 1432–1440. [Google Scholar] [CrossRef]
- Gong, Y.; Chen, Z.; Wu, Y.; Wang, A.; Zhao, S. Revisiting the Iron(II)/Cobalt(II)-Based Homogenous Fenton-like Processes from the Standpoint of Diverse Metal–Oxygen Complexes. Environ. Sci. Technol. 2024, 58, 16589–16599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, J.; Pang, S.; Zhou, Y.; Guan, C.; Gao, Y.; Li, J.; Yang, Y.; Qiu, W.; Jiang, C. Is Sulfate Radical Really Generated from Peroxydisulfate Activated by Iron(II) for Environmental Decontamination? Environ. Sci. Technol. 2018, 52, 11276–11284. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.; Zhang, Z.; Feng, J.; Wei, T.; Ren, Y.; Zhu, Y.; Ma, J. CuO with (001)-plane exposure efficiently induces peroxymonosulfate to form ≡Cu-OOSO3-intermediates directly oxidizing organic contaminants in water. Chem. Eng. J. 2022, 441, 136100. [Google Scholar] [CrossRef]
- Huang, G.-X.; Si, J.-Y.; Qian, C.; Wang, W.-K.; Mei, S.-C.; Wang, C.-Y.; Yu, H.-Q. Ultrasensitive Fluorescence Detection of Peroxymonosulfate Based on a Sulfate Radical-Mediated Aromatic Hydroxylation. Anal. Chem. 2018, 90, 14439–14446. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, Y.; Pang, S.-Y.; Jiang, J.; Yang, Z.; Shen, Y.; Wang, Z.; Wang, P.-X.; Wang, L.-H. New Insights into the Combination of Permanganate and Bisulfite as a Novel Advanced Oxidation Process: Importance of High Valent Manganese-Oxo Species and Sulfate Radical. Environ. Sci. Technol. 2019, 53, 3689–3696. [Google Scholar] [CrossRef]
- Zong, Y.; Guan, X.; Xu, J.; Feng, Y.; Mao, Y.; Xu, L.; Chu, H.; Wu, D. Unraveling the Overlooked Involvement of High-Valent Cobalt-Oxo Species Generated from the Cobalt(II)-Activated Peroxymonosulfate Process. Environ. Sci. Technol. 2020, 54, 16231–16239. [Google Scholar] [CrossRef]
- Wu, W.; Tian, D.; Liu, T.; Chen, J.; Huang, T.; Zhou, X.; Zhang, Y. Degradation of organic compounds by peracetic acid activated with Co3O4: A novel advanced oxidation process and organic radical contribution. Chem. Eng. J. 2020, 394, 124938. [Google Scholar] [CrossRef]
- Zhang, H.; Li, C.; Lyu, L.; Hu, C. Surface oxygen vacancy inducing peroxymonosulfate activation through electron donation of pollutants over cobalt-zinc ferrite for water purification. Appl. Catal. B Environ. 2020, 270, 118874. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J. Magnetic Nanoscaled Fe3O4/CeO2 Composite as an Efficient Fenton-Like Heterogeneous Catalyst for Degradation of 4-Chlorophenol. Environ. Sci. Technol. 2012, 46, 10145–10153. [Google Scholar] [CrossRef]
- Li, J.; Xu, M.; Yao, G.; Lai, B. Enhancement of the degradation of atrazine through CoFe2O4 activated peroxymonosulfate (PMS) process: Kinetic, degradation intermediates, and toxicity evaluation. Chem. Eng. J. 2018, 348, 1012–1024. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Y.; Wang, Z.; Wei, W.; Tang, W.; Shi, J.; Xiong, R. Electronic structure studies of the spinel CoFe2O4 by X-ray photoelectron spectroscopy. Appl. Surf. Sci. 2008, 254, 6972–6975. [Google Scholar] [CrossRef]
- Zhao, Y.; An, H.; Dong, G.; Feng, J.; Wei, T.; Ren, Y.; Ma, J. Oxygen vacancies induced heterogeneous catalysis of peroxymonosulfate by Ni-doped AgFeO2 materials: Evolution of reactive oxygen species and mechanism. Chem. Eng. J. 2020, 388, 124371. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, J.; Zhao, L.; Geng, C.; Zhao, F.; Yang, T.; Ma, J. Insights into Co3O4 nano-rod/peroxymonosulfate catalytic oxidation system for chemical cleaning ultrafiltration membrane: Performance, mechanisms, and effects on the membrane stability. Sep. Purif. Technol. 2024, 330, 125375. [Google Scholar] [CrossRef]
- Ren, W.; Nie, G.; Zhou, P.; Zhang, H.; Duan, X.; Wang, S. The Intrinsic Nature of Persulfate Activation and N-Doping in Carbocatalysis. Environ. Sci. Technol. 2020, 54, 6438–6447. [Google Scholar] [CrossRef]
- Kong, X.; Wu, Z.; Ren, Z.; Guo, K.; Hou, S.; Hua, Z.; Li, X.; Fang, J. Degradation of lipid regulators by the UV/chlorine process: Radical mechanisms, chlorine oxide radical (ClO•)-mediated transformation pathways and toxicity changes. Water Res. 2018, 137, 242–250. [Google Scholar] [CrossRef]
- Gao, J.; Duan, X.; O’Shea, K.; Dionysiou, D.D. Degradation and transformation of bisphenol A in UV/Sodium percarbonate: Dual role of carbonate radical anion. Water Res. 2020, 171, 115394. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Larson, R.A.; Zepp, R.G. Reactivity of the carbonate radical with aniline derivatives. Environ. Toxicol. Chem. 1988, 7, 265–274. [Google Scholar] [CrossRef]
- Moore, J.S.; Phillips, G.O.; Sosnowski, A. Reaction of the Carbonate Radical Anion with Substituted Phenols. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1977, 31, 603–605. [Google Scholar] [CrossRef]
- Hu, C.-Y.; Hou, Y.-Z.; Lin, Y.-L.; Li, A.-P.; Deng, Y.-G. Degradation kinetics of diatrizoate during UV photolysis and UV/chlorination. Chem. Eng. J. 2019, 360, 1003–1010. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Zhang, J.; Zhao, F.; Fan, W.; Yang, S.; Ma, J. Insights into CoFe2O4/Peracetic Acid Catalytic Oxidation Process for Iopamidol Degradation: Performance, Mechanisms, and I-DBP Formation Control. Nanomaterials 2025, 15, 897. https://doi.org/10.3390/nano15120897
Wu H, Zhang J, Zhao F, Fan W, Yang S, Ma J. Insights into CoFe2O4/Peracetic Acid Catalytic Oxidation Process for Iopamidol Degradation: Performance, Mechanisms, and I-DBP Formation Control. Nanomaterials. 2025; 15(12):897. https://doi.org/10.3390/nano15120897
Chicago/Turabian StyleWu, Haiwei, Jiaming Zhang, Fangbo Zhao, Wei Fan, Song Yang, and Jun Ma. 2025. "Insights into CoFe2O4/Peracetic Acid Catalytic Oxidation Process for Iopamidol Degradation: Performance, Mechanisms, and I-DBP Formation Control" Nanomaterials 15, no. 12: 897. https://doi.org/10.3390/nano15120897
APA StyleWu, H., Zhang, J., Zhao, F., Fan, W., Yang, S., & Ma, J. (2025). Insights into CoFe2O4/Peracetic Acid Catalytic Oxidation Process for Iopamidol Degradation: Performance, Mechanisms, and I-DBP Formation Control. Nanomaterials, 15(12), 897. https://doi.org/10.3390/nano15120897