
Ultrahigh Storage Capacity of Alkali Metal Ions in Hexagonal Metal Borides with Orderly Multilayered Growth Mechanism


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Calculation Details
3. Results and Discussion
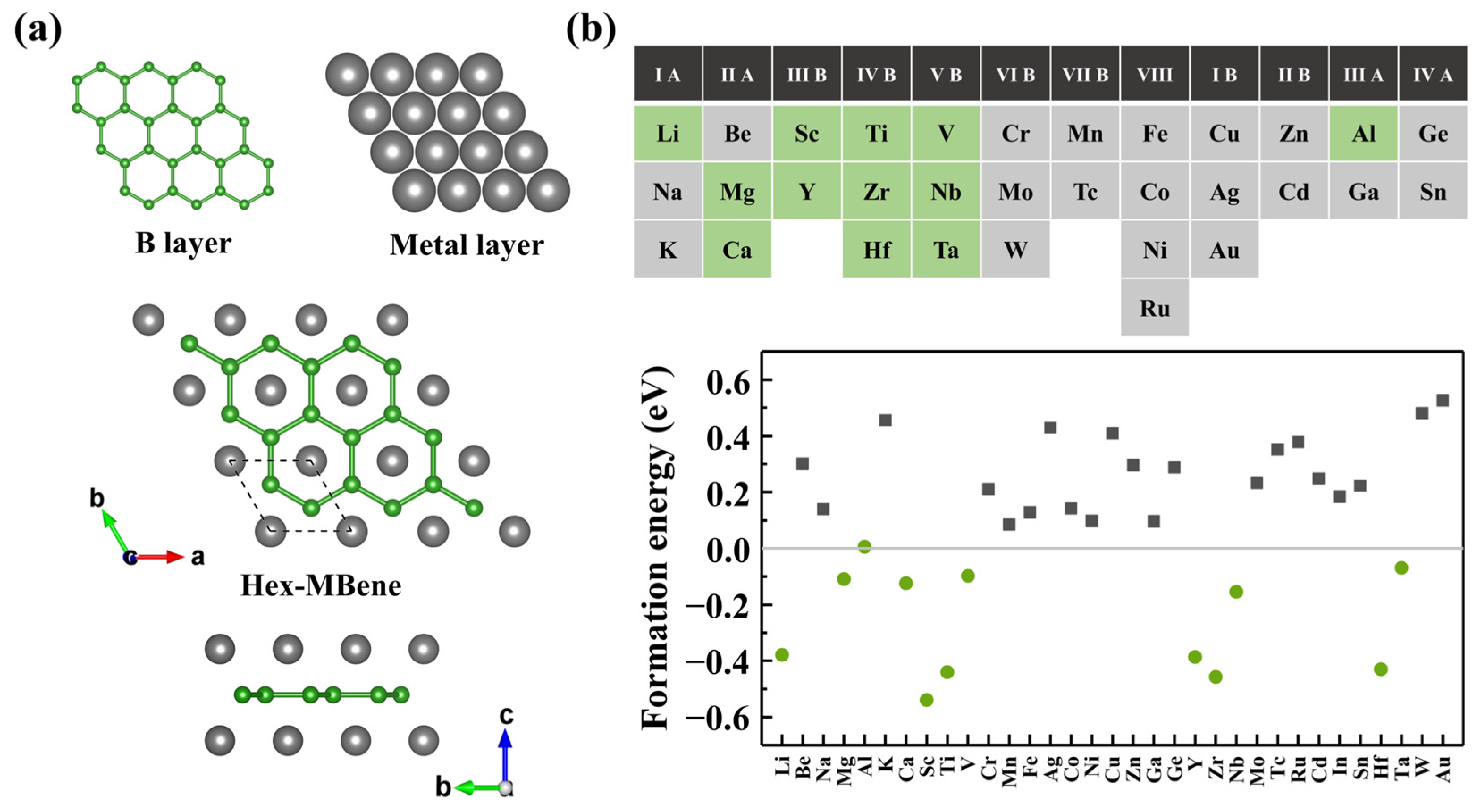
3.1. Structure and Stability of h-MBenes
3.2. Mechanical and Electronic Properties of h-MBenes
3.3. The Storage Capacity and Migration of Alkali Metal on h-MBenes
3.4. Open-Circuit Voltage and Thermal Stability of h-MBene Anodes
3.5. Layer-by-Layer Growth Behavior of Alkali Metals on h-MBene Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Cheng, X.-B.; Jin, Z.; Zhang, R.; Wang, G.; Chen, L.-Q.; Liu, Q.-B.; Huang, J.-Q.; Zhang, Q. Recent advances in understanding dendrite growth on alkali metal anodes. EnergyChem 2019, 1, 100003. [Google Scholar] [CrossRef]
- Manthiram, A.; Yu, X.; Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2, 16103. [Google Scholar] [CrossRef]
- Sandoval, S.E.; Haslam, C.G.; Vishnugopi, B.S.; Liao, D.W.; Yoon, J.S.; Park, S.H.; Wang, Y.; Mitlin, D.; Hatzell, K.B.; Siegel, D.J.; et al. Electro-chemo-mechanics of anode-free solid-state batteries. Nat. Mater. 2025, 24, 673–681. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, H.; Liu, H.; Zhao, C.-Z.; Lu, Y.; Cheng, X.-B.; Huang, J.-Q.; Zhang, Q. Unlocking the Failure Mechanism of Solid State Lithium Metal Batteries. Adv. Energy Mater. 2022, 12, 2100748. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, C.; Lian, R.; Yang, L.; Wang, Y.; Gao, Y.; Xiao, X.; Gogotsi, Y.; Wang, X.; Chen, G.; et al. Mechanisms of the Planar Growth of Lithium Metal Enabled by the 2D Lattice Confinement from a Ti3C2T MXene Intermediate Layer. Adv. Funct. Mater. 2021, 31, 2010987. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, Y.; Shen, S.; Li, X.; Hu, X.; Hu, M.; Su, Y.; Ding, S.; Xiao, C. Directing (110) Oriented Lithium Deposition through High-flux Solid Electrolyte Interphase for Dendrite-free Lithium Metal Batteries. Angew. Chem. Int. Ed. 2023, 62, e202309622. [Google Scholar] [CrossRef]
- Xu, T.; Wang, Y.; Xiong, Z.; Wang, Y.; Zhou, Y.; Li, X. A Rising 2D Star: Novel MBenes with Excellent Performance in Energy Conversion and Storage. Nano-Micro Lett. 2022, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhou, J.; Sun, Z. New two-dimensional transition metal borides for Li ion batteries and electrocatalysis. J. Mater. Chem. A 2017, 5, 23530–23535. [Google Scholar] [CrossRef]
- Gao, S.; Hao, J.; Zhang, X.; Li, L.; Zhang, C.; Wu, L.; Ma, X.; Lu, P.; Liu, G. Two dimension transition metal boride Y2B2 as a promising anode in Li-ion and Na-ion batteries. Comput. Mater. 2021, 200, 110776. [Google Scholar] [CrossRef]
- Jia, J.; Li, B.; Duan, S.; Cui, Z.; Gao, H. Monolayer MBenes: Prediction of anode materials for high-performance lithium/sodium ion batteries. Nanoscale 2019, 11, 20307–20314. [Google Scholar] [CrossRef]
- Miao, N.; Gong, Y.; Zhang, H.; Shen, Q.; Yang, R.; Zhou, J.; Hosono, H.; Wang, J. Discovery of Two-dimensional Hexagonal MBene HfBO and Exploration on its Potential for Lithium-Ion Storage. Angew. Chem. Int. Ed. 2023, 62, e202308436. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Feng, X.; Huang, T.; Huang, Z.; He, X.; Liu, J.; Xiao, Y.; Wang, X.; Zhang, Q. Rapid synthesis of two-dimensional MoB MBene anodes for high-performance sodium-ion batteries. J. Mater. Sci. Technol. 2025, 212, 67–76. [Google Scholar] [CrossRef]
- Sun, Y.; Li, K.; Wang, B.; Zhang, W.; Wang, E.; Zhou, J.; Sun, Z. Hexagonal Mg2B2 and Ca2B2 monolayers as promising anode materials for Li-ion and Na-ion batteries. Nanoscale 2024, 16, 15699–15712. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Xiao, W.; Zhang, C.; Zhao, Y. Computational investigation of 2D 3d/4d hexagonal transition metal borides for metal-ion batteries. Electrochim. Acta 2021, 384, 138404. [Google Scholar] [CrossRef]
- Zou, R.-F.; Ye, X.-J.; Zheng, X.-H.; Jia, R.; Liu, C.-S. Two-dimensional AlB4/Al2B2: High-performance Dirac anode materials for sodium-ion batteries. Phys. Chem. Chem. Phys. 2023, 25, 28814–28823. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Xu, S.; Li, J.; Yuan, S.; Jia, B.; Gao, S.; Liu, G.; Lu, P. Computational Investigation of Two-Dimensional Vanadium Boride Compounds for Na-Ion Batteries. ACS Omega 2022, 7, 14765–14771. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Zhuo, Z.; Wu, X.; Yang, J. Me-graphene: A graphene allotrope with near zero Poisson’s ratio, sizeable band gap, and high carrier mobility. Nanoscale 2020, 12, 19359–19366. [Google Scholar] [CrossRef]
- Fang, Z.; Jiang, J.; Guo, H.; Lin, X.; Wu, X.; Zhuo, Z.; Lu, N. Ultrahigh Potassium Storage Capacity of Ca2Si Monolayer with Orderly Multilayered Growth Mechanism. Small 2024, 20, 2401736. [Google Scholar] [CrossRef]
- Jian, Z.; Luo, W.; Ji, X. Carbon electrodes for K-ion batteries. J. Am. Chem. Soc. 2015, 137, 11566–11569. [Google Scholar] [CrossRef]
- Jiang, H.R.; Lu, Z.; Wu, M.C.; Ciucci, F.; Zhao, T.S. Borophene: A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries. Nano Energy 2016, 23, 97–104. [Google Scholar] [CrossRef]
- Zhu, C.; Lin, S.; Zhang, M.; Li, Q.; Su, Z.; Chen, Z. Ultrahigh capacity 2D anode materials for lithium/sodium-ion batteries: An entirely planar B7P2 monolayer with suitable pore size and distribution. J. Mater. Chem. A 2020, 8, 10301–10309. [Google Scholar] [CrossRef]
- Kulish, V.V.; Malyi, O.I.; Persson, C.; Wu, P. Phosphorene as an anode material for Na-ion batteries: A first-principles study. Phys. Chem. Chem. Phys. 2015, 17, 13921–13928. [Google Scholar] [CrossRef]
- Abbas, G.; Alay-e-Abbas, S.M.; Laref, A.; Li, Y.; Zhang, W.X. Two-dimensional B3P monolayer as a superior anode material for Li and Na ion batteries: A first-principles study. Mater. Today Energy 2020, 17, 100486. [Google Scholar] [CrossRef]
- Yang, C.; Sun, X.; Zhang, X.; Li, J.; Ma, J.; Li, Y.; Xu, L.; Liu, S.; Yang, J.; Fang, S.; et al. Is graphite nanomesh a promising anode for the Na/K-Ions batteries? Carbon 2021, 176, 242–252. [Google Scholar] [CrossRef]
- Wang, S.; Si, Y.; Yang, B.; Ruckenstein, E.; Chen, H. Two-Dimensional Carbon-Based Auxetic Materials for Broad-Spectrum Metal-Ion Battery Anodes. J. Phys. Chem. Lett. 2019, 10, 3269–3275. [Google Scholar] [CrossRef]
- Gao, Y.; Li, D.; Cui, T. Hd-Graphene: A Hexagon-Deficient Carbon-Based Anode for Metal-Ion Batteries with High Charge/Discharge Rates. ACS Appl. Electron. Mater. 2021, 3, 5147–5154. [Google Scholar] [CrossRef]
- Shu, H.; Li, F.; Hu, C.; Liang, P.; Cao, D.; Chen, X. The capacity fading mechanism and improvement of cycling stability in MoS2-based anode materials for lithium-ion batteries. Nanoscale 2016, 8, 2918–2926. [Google Scholar] [CrossRef]
- Wen, T.; Qu, B.; Tan, S.; Huang, G.; Song, J.; Wang, Z.; Wang, J.; Tang, A.; Pan, F. Rational design of artificial interphase buffer layer with 3D porous channel for uniform deposition in magnesium metal anodes. Energy Storage Mater. 2023, 55, 816–825. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, Y.; Guo, H.; Wu, X.; Lu, N.; Zhuo, Z. Two-Dimensional Biphenylene-Based Carbon Allotrope Family with High Potassium Storage Ability. J. Phys. Chem. Lett. 2023, 14, 9655–9664. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, M.; Lin, S.; Hao, J.; Wang, Y.; Zhao, H.J.; Li, Y. Be2C5 Monolayer with Quasiplanar Pentacoordinate Carbon Atoms and Ultrahigh Energy Density as a Dirac Anode for Potassium-Ion Batteries. PRX Energy 2023, 2, 033012. [Google Scholar] [CrossRef]
- Ni, S.; Jiang, J.; Wang, W.; Wu, X.; Zhuo, Z.; Wang, Z. Beryllium dinitride monolayer: A multifunctional direct bandgap anisotropic semiconductor containing polymeric nitrogen with oxygen reduction catalysis and potassium-ion storage capability. J. Mater. Chem. A 2025, 13, 10214–10223. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AM@M2B2 | Eads (eV) | Δe (e) | LAM-M (Å) | Eb (eV) |
|---|---|---|---|---|
| Li@Mg2B2 | −0.215 | −0.84 | 3.02 | 0.018 |
| Li@Al2B2 | −0.488 | −0.86 | 2.74 | 0.048 |
| Li@V2B2 | −0.985 | −0.85 | 2.89 | 0.010 |
| Na@Mg2B2 | −0.488 | −0.70 | 3.32 | 0.014 |
| Na@Y2B2 | −0.520 | −0.58 | 3.70 | 0.019 |
| K@Al2B2 | −1.111 | −0.84 | 3.46 | 0.014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, J.; Guo, H.; Lu, N. Ultrahigh Storage Capacity of Alkali Metal Ions in Hexagonal Metal Borides with Orderly Multilayered Growth Mechanism. Nanomaterials 2025, 15, 886. https://doi.org/10.3390/nano15120886
Jiang J, Guo H, Lu N. Ultrahigh Storage Capacity of Alkali Metal Ions in Hexagonal Metal Borides with Orderly Multilayered Growth Mechanism. Nanomaterials. 2025; 15(12):886. https://doi.org/10.3390/nano15120886
Chicago/Turabian StyleJiang, Jiaxin, Hongyan Guo, and Ning Lu. 2025. "Ultrahigh Storage Capacity of Alkali Metal Ions in Hexagonal Metal Borides with Orderly Multilayered Growth Mechanism" Nanomaterials 15, no. 12: 886. https://doi.org/10.3390/nano15120886
APA StyleJiang, J., Guo, H., & Lu, N. (2025). Ultrahigh Storage Capacity of Alkali Metal Ions in Hexagonal Metal Borides with Orderly Multilayered Growth Mechanism. Nanomaterials, 15(12), 886. https://doi.org/10.3390/nano15120886