
Probing Acidic and Defective Sites in Sulfated UiO-66 and ZrO2 via Adsorptive FTIR Spectroscopy

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Characterization
2.3. In Situ FTIR Experiments
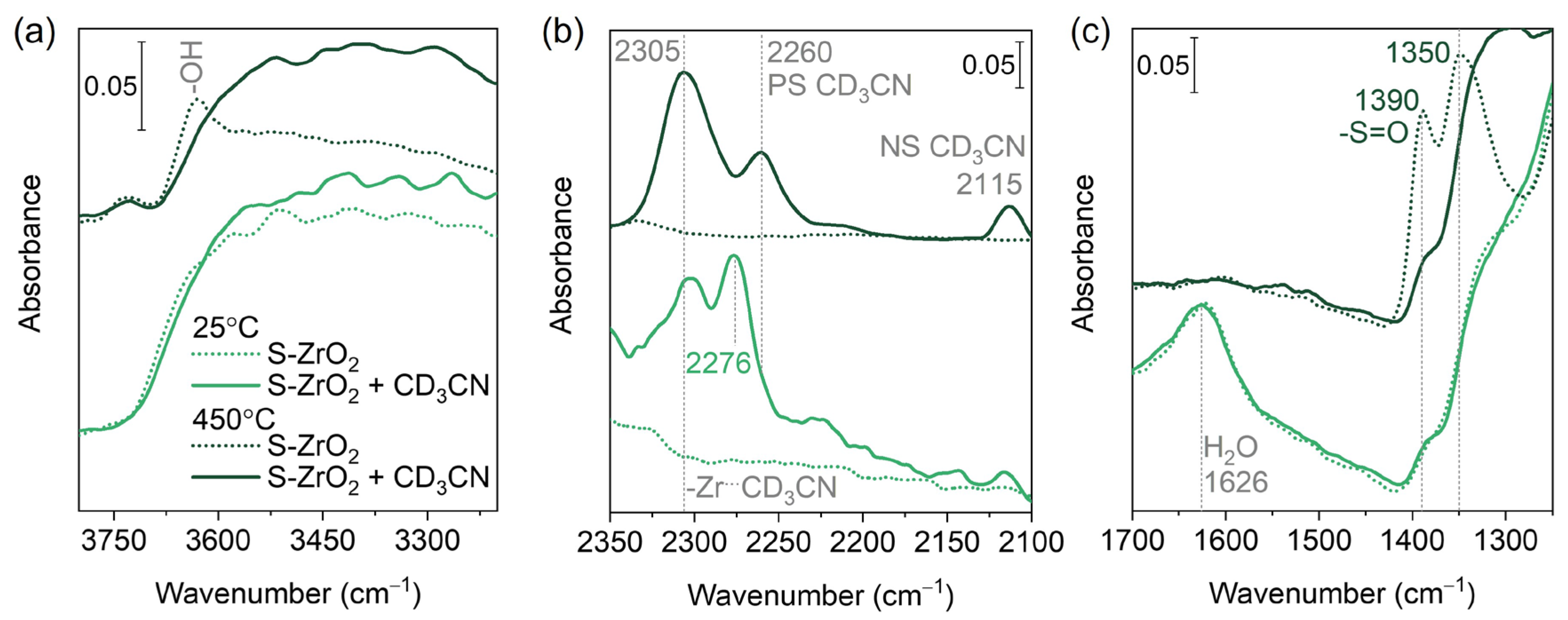
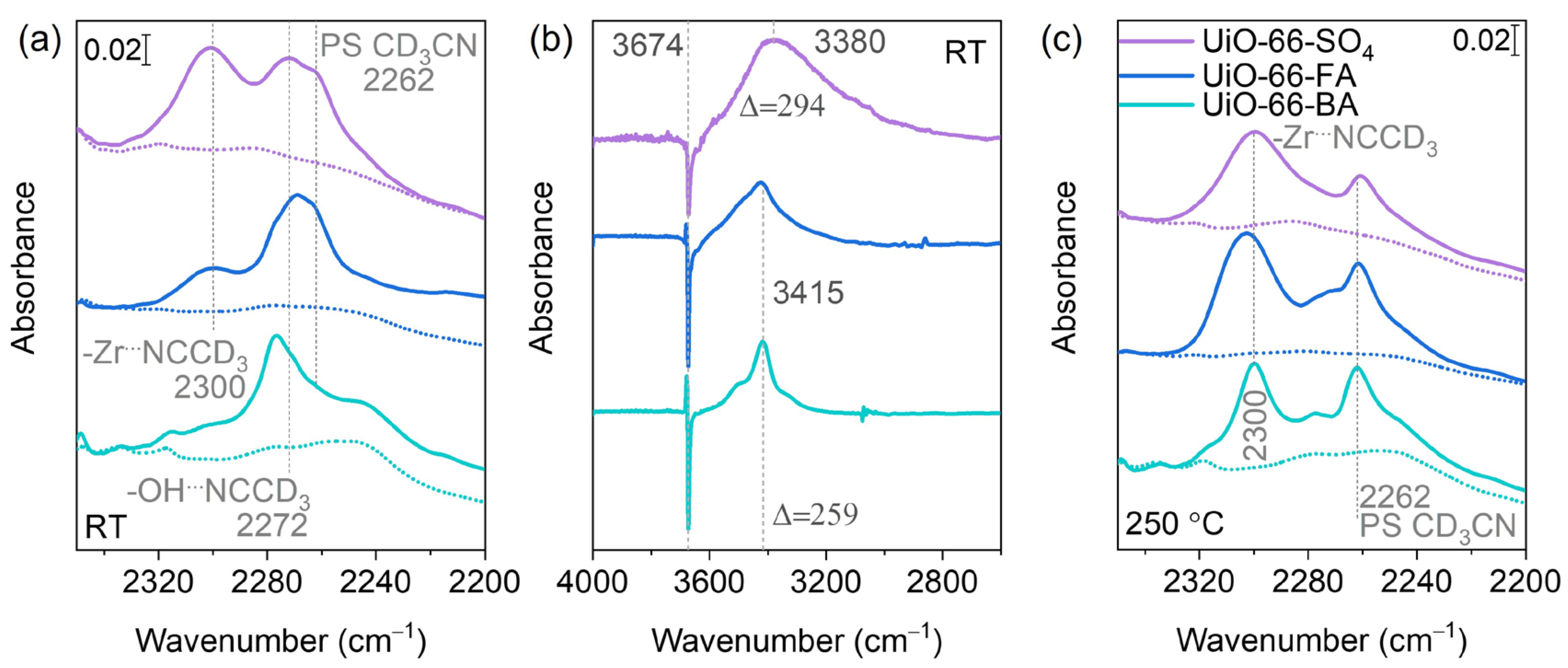
3. Results
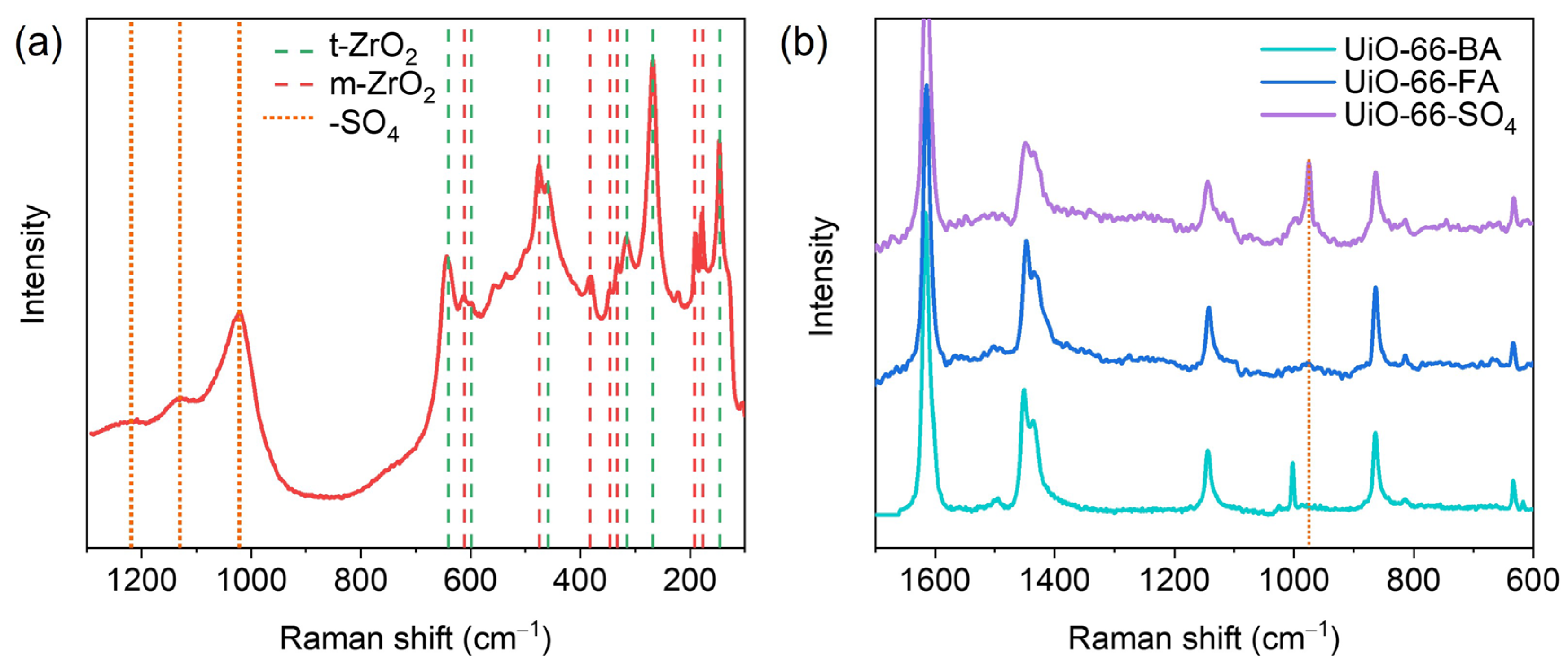
3.1. Basic Characterization
3.2. In Situ FTIR Background
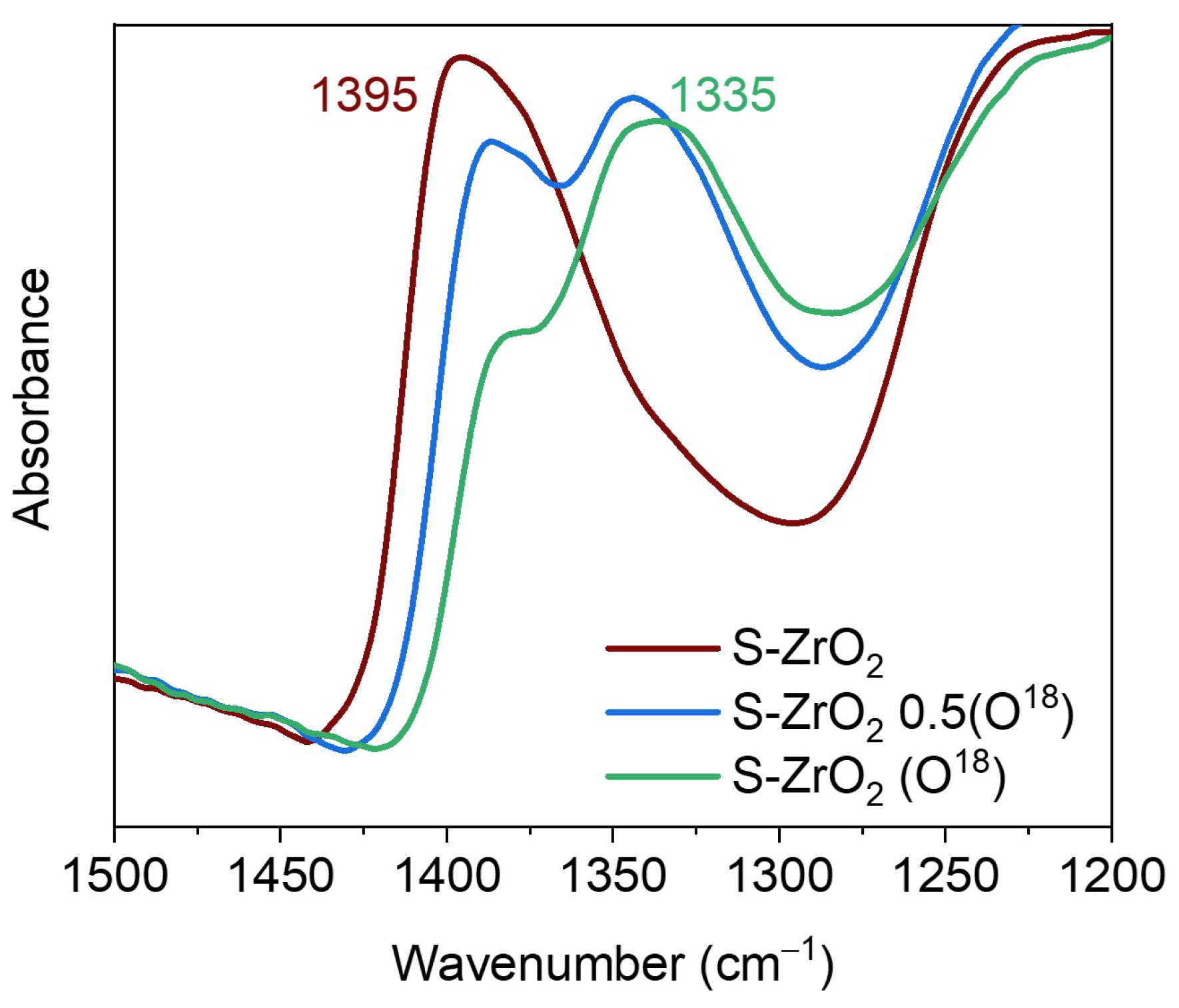
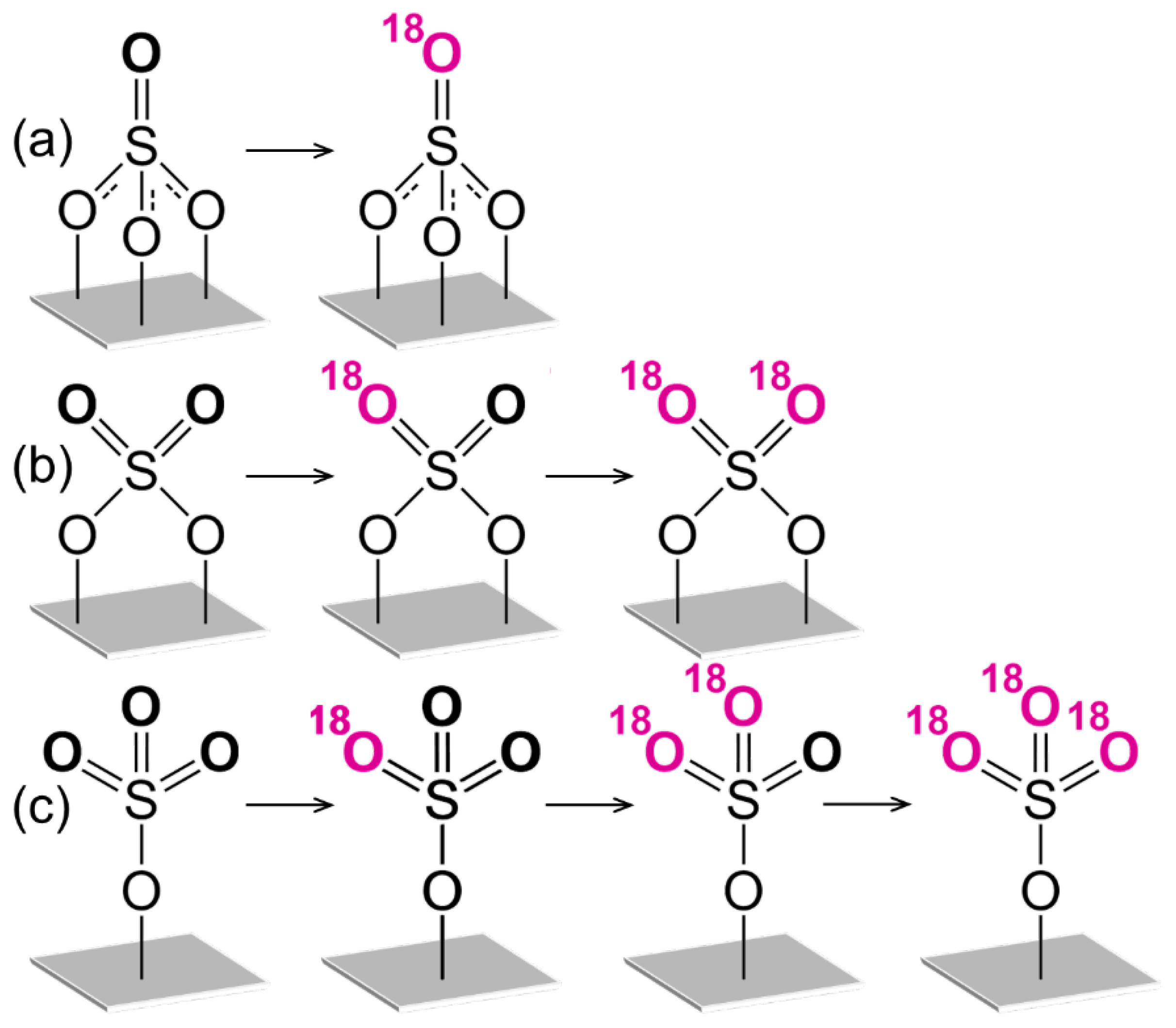
3.3. Oxygen Isotopic Exchange
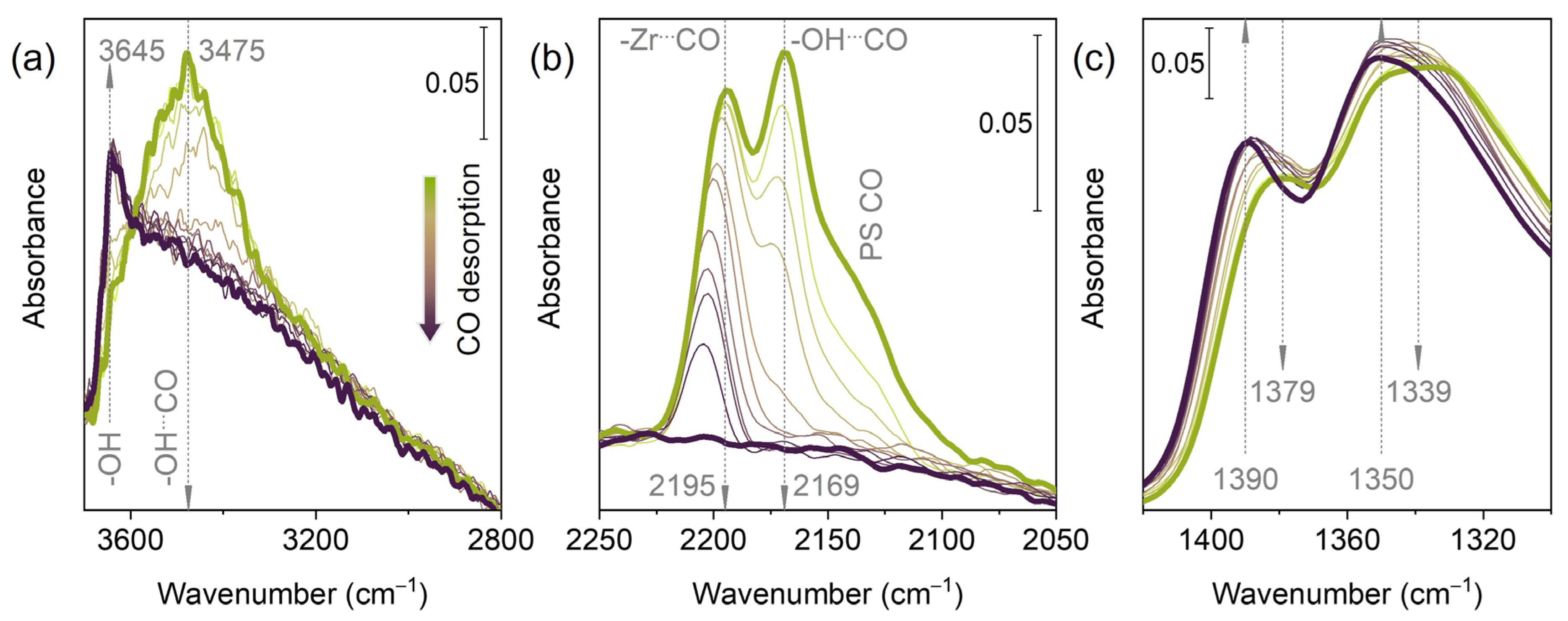
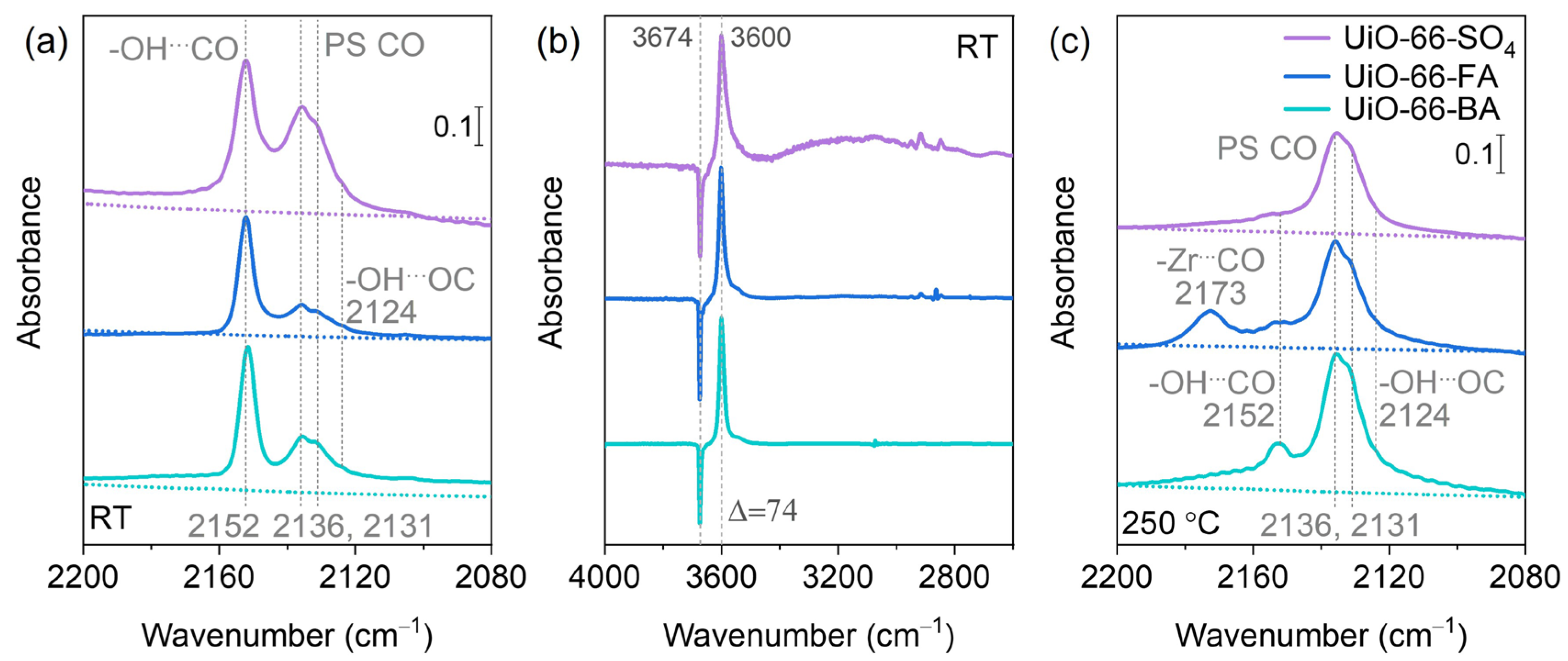
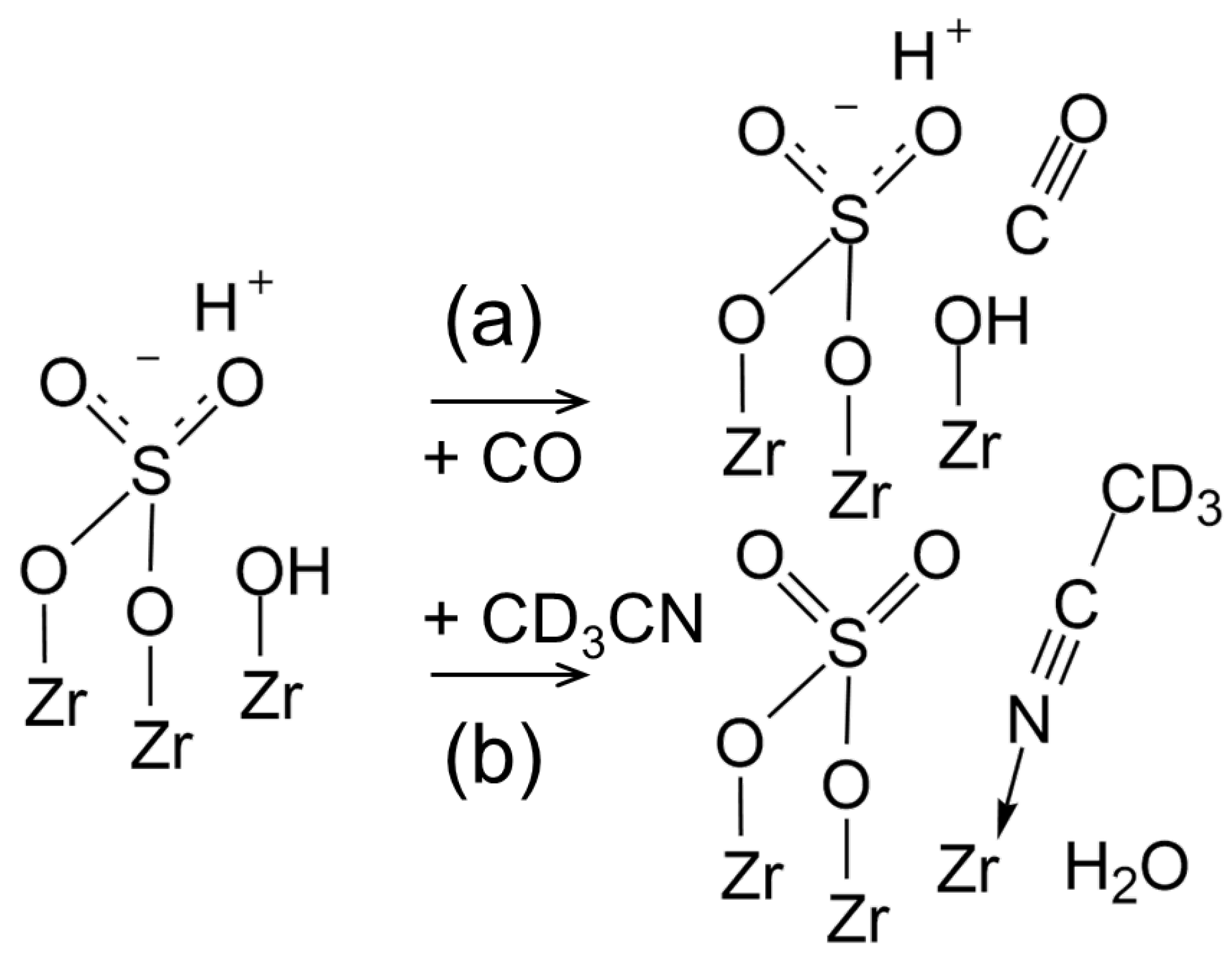
3.4. CO Adsorption
3.5. CD3CN Adsorption
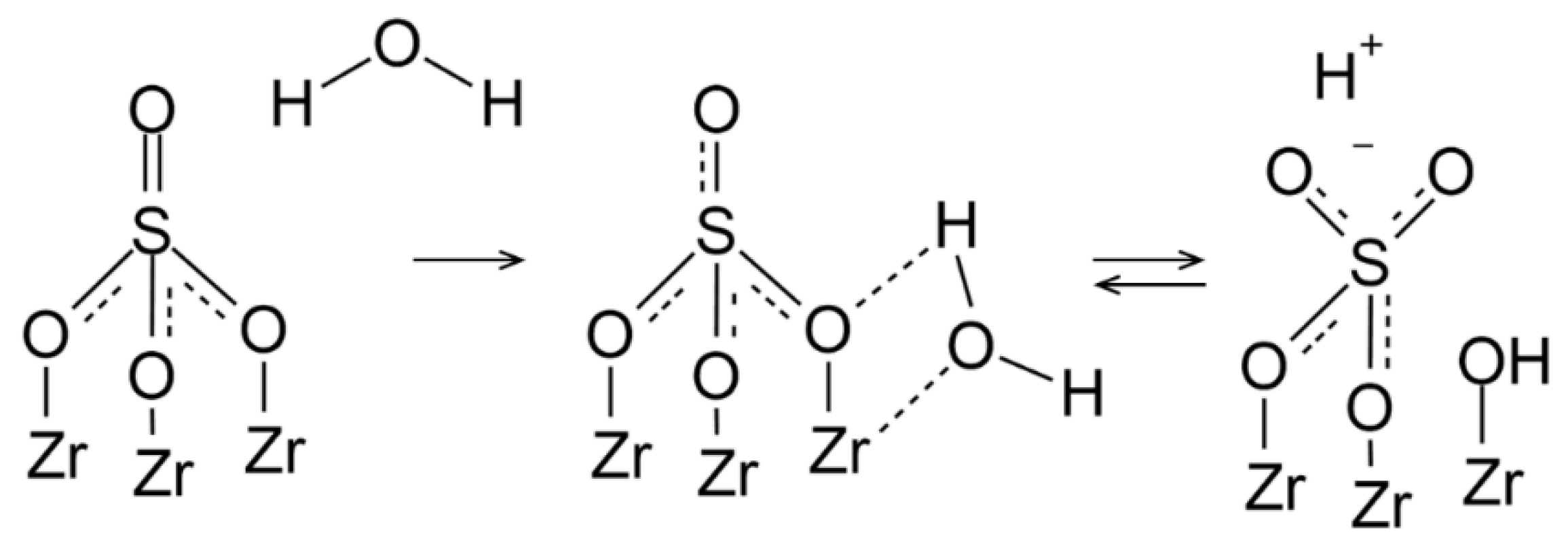
4. Discussion
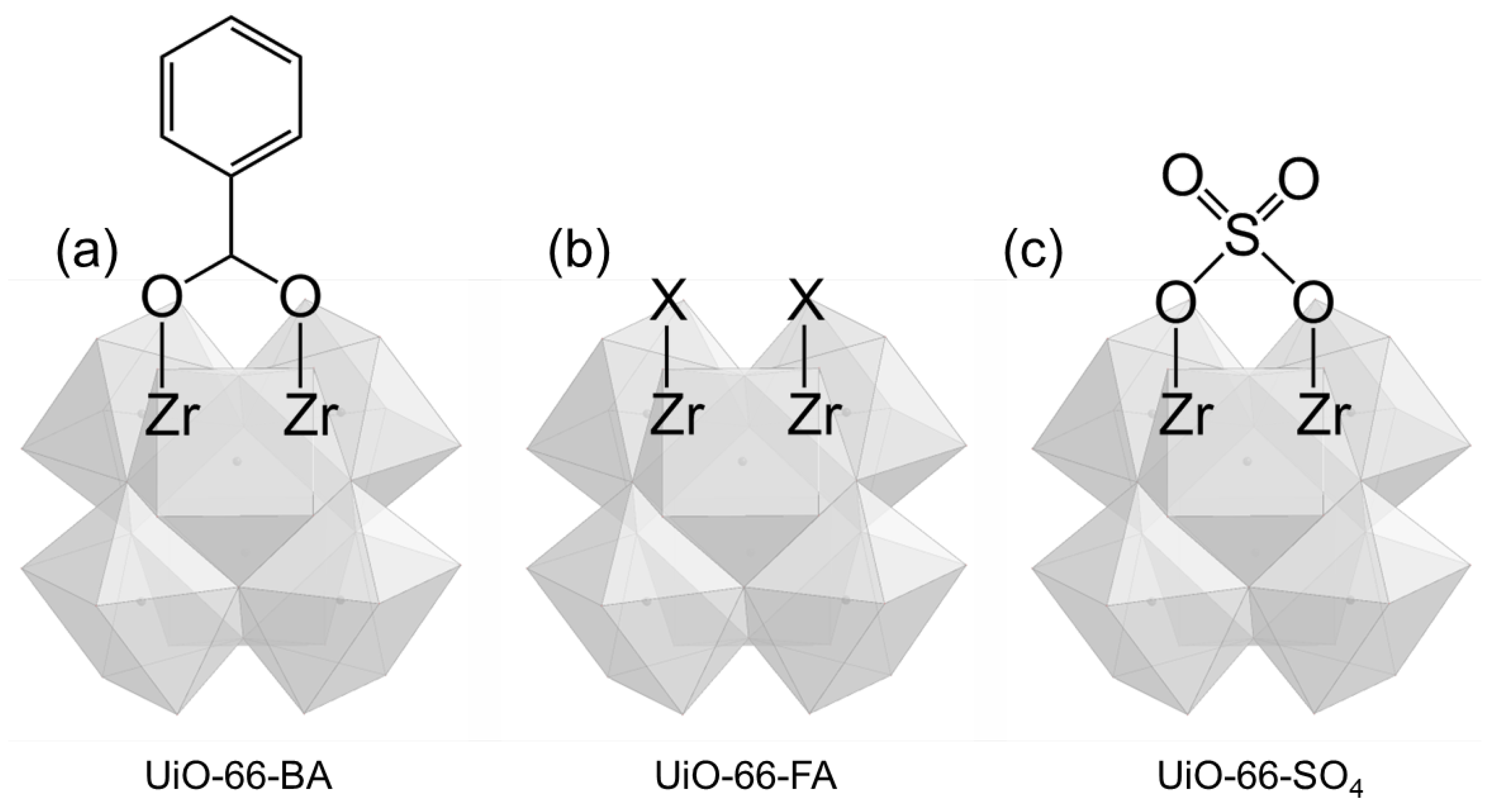
- UiO-66-BA—contains defects, but they are coordinated with benzoate ions, making the Zr sites inaccessible.
- UiO-66-FA—the benzoate ions have been removed, exposing Zr sites in defect pores.
- UiO-66-SO4—derived from UiO-66-FA by modifying it with sulfate (SO4) groups.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fatimah, I.; Yanti, I.; Suharto, T.E.; Sagadevan, S. ZrO2-based catalysts for biodiesel production: A review. Inorg. Chem. Commun. 2022, 143, 109808. [Google Scholar] [CrossRef]
- Mittal, A.; Kumar, G.; Saroha, B.; Peppel, T.; Kumar, V.; Kumar, S.; Kumar, N. ZrO2 based nanostructures: A sustainable, economical, green and efficient organocatalyst. J. Mol. Liq. 2024, 398, 124223. [Google Scholar] [CrossRef]
- Bugrova, T.A.; Dutov, V.V.; Svetlichnyi, V.A.; Cortés Corberán, V.; Mamontov, G.V. Oxidative dehydrogenation of ethane with CO2 over CrOx catalysts supported on Al2O3, ZrO2, CeO2 and CexZr1−xO2. Catal. Today 2019, 333, 71–80. [Google Scholar] [CrossRef]
- Kunamalla, A.; Maity, S.K. Production of green jet fuel from furanics via hydroxyalkylation-alkylation over mesoporous MoO3-ZrO2 and hydrodeoxygenation over Co/γ-Al2O3: Role of calcination temperature and MoO3 content in MoO3-ZrO2. Fuel 2023, 332, 125977. [Google Scholar] [CrossRef]
- Soultanidis, N.; Zhou, W.; Psarras, A.C.; Gonzalez, A.J.; Iliopoulou, E.F.; Kiely, C.J.; Wachs, I.E.; Wong, M.S. Relating n-pentane isomerization activity to the tungsten surface density of WOx/ZrO2. J. Am. Chem. Soc. 2010, 132, 13462–13471. [Google Scholar] [CrossRef]
- Yu, S.; Wu, S.; Li, L.; Ge, X. Upgrading bio-oil from waste cooking oil by esterification using SO42−/ZrO2 as catalyst. Fuel 2020, 276, 118019. [Google Scholar] [CrossRef]
- Brunet, S.; Lebeau, B.; Naboulsi, I.; Michelin, L.; Comparot, J.D.; Marichal, C.; Rigolet, S.; Bonne, M.; Blin, J.L. Effect of mesostructured zirconia support on the activity and selectivity of 4,6-dimethydibenzothiophene hydrodesulfurization. Catalysts 2020, 10, 1162. [Google Scholar] [CrossRef]
- Hanafi, S.A.; El-Syed, H.A.; Elmelawy, M.S.; Sultan, E.S.A. Characterization and catalytic activities of ZrO2-TiO2 supported hydrodesulfurization catalysts. Petrol. Sci. Technol. 2009, 27, 83–95. [Google Scholar] [CrossRef]
- Hongloi, N.; Rahman, T.; Feyzbar-Khalkhali-Nejad, F.; Prapainainar, C.; Wongsurakul, P.; Aransiola, E.; Zhang, L.; Bargiela, P.; Baltrusaitis, J.; Prapainainar, P.; et al. Palm oil deoxygenation with glycerol as a hydrogen donor for renewable fuel production using nickel-molybdenum catalysts: The effect of support. Fuel Process. Technol. 2025, 270, 108196. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, M.; Zhang, W.; Yang, C.; Li, C. Isomerization of n-Butane over SO42-/Al2O3-ZrO2 in a Circulated Fluidized Bed Reactor: Prospects for Commercial Application. Ind. Eng. Chem. Res. 2017, 56, 8456–8464. [Google Scholar] [CrossRef]
- Bouvier, P.; Djurado, E.; Ritter, C.; Dianoux, A.J.; Lucazeau, G. Low temperature phase transformation of nanocrystalline tetragonal ZrO2 by neutron and Raman scattering studies. Int. J. Inorg. Mater. 2001, 3, 647–654. [Google Scholar] [CrossRef]
- Valmalette, J.C.; Isa, M. Size Effects on the Stabilization of Ultrafine Zirconia Nanoparticles. Chem. Mater. 2002, 14, 5098–5102. [Google Scholar] [CrossRef]
- Marin, R.; Sponchia, G.; Back, M.; Riello, P. Determining europium compositional fluctuations in partially stabilized zirconia nanopowders: A non-line-broadening-based method. Acta Crystallogr. B 2016, 72, 29–38. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Avreyska, V.; Klissurski, D.; Marinova, T. Surface species formed after NO adsorption and NO + O2 coadsorption on ZrO2 and sulfated ZrO2: An FTIR spectroscopic study. Langmuir 2002, 18, 1619–1625. [Google Scholar] [CrossRef]
- Bardin, B.B.; Davis, R.J. A comparison of cesium-containing heteropolyacid and sulfated zirconia catalysts for isomerization of light alkanes. Top. Catal. 1998, 6, 77–86. [Google Scholar] [CrossRef]
- Yan, G.X.; Wang, A.; Wachs, I.E.; Baltrusaitis, J. Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production. Appl. Catal. A 2019, 572, 210–225. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Butova, V.V.; Budnyk, A.P.; Charykov, K.M.; Vetlitsyna-Novikova, K.S.; Lamberti, C.; Soldatov, A.V. Water as a structure-driving agent between the UiO-66 and MIL-140A metal-organic frameworks. Chem. Commun. 2019, 55, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Atzori, C.; Shearer, G.C.; Maschio, L.; Civalleri, B.; Bonino, F.; Lamberti, C.; Svelle, S.; Lillerud, K.P.; Bordiga, S. Effect of benzoic acid as a modulator in the structure of UiO-66: An experimental and computational study. J. Phys. Chem. C 2017, 121, 9312–9324. [Google Scholar] [CrossRef]
- Butova, V.V.; Zdravkova, V.R.; Burachevskaia, O.A.; Gorban, I.E.; Soldatov, M.A.; Hadjiivanov, K.I. FTIR traced modification of defect-engineered UiO-66 for enhanced accessibility of zirconium sites. Microporous Mesoporous Mater. 2025, 382, 113372. [Google Scholar] [CrossRef]
- Butova, V.V.; Zdravkova, V.R.; Burachevskaia, O.A.; Tereshchenko, A.A.; Shestakova, P.S.; Hadjiivanov, K.I. In situ FTIR spectroscopy for scanning accessible active sites in defect-engineered UiO-66. Nanomaterials 2023, 13, 1675. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Morales, J.M.; Lozano, L.A.; Castillejos-López, E.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A.; Zamaro, J.M. Direct sulfation of a Zr-based metal-organic framework to attain strong acid catalysts. Microporous Mesoporous Mater. 2019, 290, 109686. [Google Scholar] [CrossRef]
- Jiang, J.; Gándara, F.; Zhang, Y.-B.; Na, K.; Yaghi, O.M.; Klemperer, W.G. Superacidity in sulfated metal–organic framework-808. J. Am. Chem. Soc. 2014, 136, 12844–12847. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, R.; Ye, B.; Hou, Z. Acetalization of glycerol over sulfated UiO-66 under mild condition. J. Ind. Eng. Chem. 2022, 110, 357–366. [Google Scholar] [CrossRef]
- Li, H.; Wang, T.; Chu, H.; Rokhum, S.L.; Zhang, Y.; Yu, H.; Xiao, Q.; Guo, M.; Ma, X.; Li, S.; et al. In–situ modification of UiO–66(Zr) organic ligand to synthesize highly recyclable solid acid for biodiesel production. Chem. Eng. Res. Des. 2024, 205, 713–721. [Google Scholar] [CrossRef]
- Nazari, M.; Amini, A.; Eden, N.T.; Duke, M.C.; Cheng, C.; Hill, M.R. Highly-Efficient Sulfonated UiO-66(Zr) Optical Fiber for Rapid Detection of Trace Levels of Pb2+. Int. J. Mol. Sci. 2021, 22, 6053. [Google Scholar] [CrossRef]
- Raveendran, S.R.D.; Teh, L.P.; Othaman, R.; Chia, C.H. Enhanced carbon dioxide adsorption performance of UiO-66-SO3H with a mixed ligand strategy. J. Environ. Chem. Eng. 2024, 12, 113404. [Google Scholar] [CrossRef]
- Syed, Z.H.; Mian, M.R.; Patel, R.; Xie, H.; Pengmei, Z.; Chen, Z.; Son, F.A.; Goetjen, T.A.; Chapovetsky, A.; Fahy, K.M.; et al. Sulfated Zirconium Metal–Organic Frameworks as Well-Defined Supports for Enhancing Organometallic Catalysis. J. Am. Chem. Soc. 2022, 144, 16883–16897. [Google Scholar] [CrossRef]
- Tahir, Z.; Aslam, M.; Gilani, M.A.; Bilad, M.R.; Anjum, M.W.; Zhu, L.-P.; Khan, A.L. SO3H functionalized UiO-66 nanocrystals in Polysulfone based mixed matrix membranes: Synthesis and application for efficient CO2 capture. Sep. Purif. Technol. 2019, 224, 524–533. [Google Scholar] [CrossRef]
- Shi, Q.; Wu, Q.; Li, H.; Shi, D.; Zhao, Y.; Jiao, Q. Enhanced catalytic performance of UiO-66 via a sulfuric acid post-synthetic modification strategy with partial etching. Appl. Catal. A 2020, 602, 117733. [Google Scholar] [CrossRef]
- Chadwick, A.V.; Mountjoy, G.; Nield, V.M.; Poplett, I.J.F.; Smith, M.E.; Strange, J.H.; Tucker, M.G. Solid-state NMR and X-ray studies of the structural evolution of nanocrystalline zirconia. Chem. Mater. 2001, 13, 1219–1229. [Google Scholar] [CrossRef]
- Jentoft, F.C.; Hahn, A.; Kröhnert, J.; Lorenz, G.; Jentoft, R.E.; Ressler, T.; Wild, U.; Schlögl, R.; Häßner, C.; Köhler, K. Incorporation of manganese and iron into the zirconia lattice in promoted sulfated zirconia catalysts. J. Catal. 2004, 224, 124–137. [Google Scholar] [CrossRef]
- Jentoft, R.E.; Hahn, A.H.P.; Jentoft, F.C.; Ressler, T. In situ XANES study of Mn in promoted sulfated zirconia catalysts. Phys. Chem. Chem. Phys. 2005, 7, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Hadjiivanov, K. Use of overtones and combination modes for the identification of surface NOx anionic species by IR spectroscopy. Catal. Lett. 2000, 68, 157–161. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Lavalley, J.C. FT-IR spectroscopic study of CO adsorption on monoclinic zirconia of different hydroxylation degrees. Catal. Commun. 2001, 2, 129–133. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Panayotov, D.A.; Mihaylov, M.Y.; Ivanova, E.Z.; Chakarova, K.K.; Andonova, S.M.; Drenchev, N.L. Power of infrared and raman spectroscopies to characterize metal-organic frameworks and investigate their interaction with guest molecules. Chem. Rev. 2021, 121, 1286–1424. [Google Scholar] [CrossRef]
- Butova, V.V.; Burachevskaya, O.A.; Ozhogin, I.V.; Borodkin, G.S.; Starikov, A.G.; Bordiga, S.; Damin, A.; Lillerud, K.P.; Soldatov, A.V. UiO-66 type MOFs with mixed-linkers—1,4-benzenedicarboxylate and 1,4-naphthalenedicarboxylate: Effect of the modulator and post-synthetic exchange. Microporous Mesoporous Mater. 2020, 305, 110324. [Google Scholar] [CrossRef]
- Shearer, G.C.; Vitillo, J.G.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Functionalizing the defects: Postsynthetic ligand exchange in the metal organic framework UiO-66. Chem. Mater. 2016, 28, 7190–7193. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic computing system JANA2006: General features. Z. Krist. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Bhagwat, M.; Ramaswamy, V. Synthesis of nanocrystalline zirconia by amorphous citrate route: Structural and thermal (HTXRD) studies. Mater. Res. Bull. 2004, 39, 1627–1640. [Google Scholar] [CrossRef]
- Igawa, N.; Ishii, Y. Crystal Structure of Metastable Tetragonal Zirconia up to 1473 K. J. Am. Chem. Soc. 2001, 84, 1169–1171. [Google Scholar] [CrossRef]
- Lutterotti, L.; Scardi, P. Simultaneous structure and size-strain refinement by the rietveld method. J. Appl. Crystallog. 1990, 23, 246–252. [Google Scholar] [CrossRef]
- Butova, V.V.; Aboraia, A.M.; Solayman, M.; Yahia, I.S.; Zahran, H.Y.; Abd El-Rehim, A.F.; Algarni, H.; Khabiri, G.; Soldatov, A.V. The joint effect of naphthalene-system and defects on dye removal by UiO-66 derivatives. Microporous Mesoporous Mater. 2021, 325, 111314. [Google Scholar] [CrossRef]
- Butova, V.V.; Burachevskaya, O.A.; Muratidi, M.A.; Surzhikova, I.I.; Zolotukhin, P.V.; Medvedev, P.V.; Gorban, I.E.; Kuzharov, A.A.; Soldatov, M.A. Loading of the Model Amino Acid Leucine in UiO-66 and UiO-66-NH2: Optimization of Metal-Organic Framework Carriers and Evaluation of Host-Guest Interactions. Inorg. Chem. 2021, 60, 5694–5703. [Google Scholar] [CrossRef]
- Øien, S.; Wragg, D.; Reinsch, H.; Svelle, S.; Bordiga, S.; Lamberti, C.; Lillerud, K.P. Detailed Structure Analysis of Atomic Positions and Defects in Zirconium Metal–Organic Frameworks. Cryst. Growth Des. 2014, 14, 5370–5372. [Google Scholar] [CrossRef]
- Noda, L.K.; Gonçalves, N.S.; de Borba, S.M.; Silveira, J.A. Raman spectroscopy and thermal analysis of sulfated ZrO2 prepared by two synthesis routes. Vib. Spectrosc. 2007, 44, 101–107. [Google Scholar] [CrossRef]
- Butova, V.V.; Budnyk, A.P.; Charykov, K.M.; Vetlitsyna-Novikova, K.S.; Bugaev, A.L.; Guda, A.A.; Damin, A.; Chavan, S.M.; Øien-ØDegaard, S.; Lillerud, K.P.; et al. Partial and complete substitution of the 1,4-benzenedicarboxylate linker in UiO-66 with 1,4-naphthalenedicarboxylate: Synthesis, characterization, and H2—Adsorption properties. Inorg. Chem. 2019, 58, 1607–1620. [Google Scholar] [CrossRef]
- Marlowe, J.; Acharya, S.; Zuber, A.; Tsilomelekis, G. Characterization of sulfated SnO2-ZrO2 catalysts and their catalytic performance on the tert-butylation of phenol. Catalysts 2020, 10, 726. [Google Scholar] [CrossRef]
- Adeeva, V.; Dehaan, J.W.; Janchen, J.; Lei, G.D.; Schunemann, V.; Vandeven, L.J.M.; Sachtler, W.M.H.; Vansanten, R.A. Acid sites in sulfated and metal-promoted zirconium dioxide catalysts. J. Catal. 1995, 151, 364–372. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic properties of sulfated zirconia: An infrared spectroscopic study. J. Catal. 1995, 152, 341–349. [Google Scholar] [CrossRef]
- Morterra, C.; Peñarroya Mentruit, M.; Cerrato, G. Acetonitrile adsorption as an IR spectroscopic probe for surface acidity/basicity of pure and modified zirconias. Phys. Chem. Chem. Phys. 2002, 4, 676–687. [Google Scholar] [CrossRef]
- Kustov, L.M.; Kazansky, V.B.; Figueras, F.; Tichit, D. Investigation of the acidic properties of ZrO2 modified by SO42− anions. J. Catal. 1994, 150, 143–149. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.C.; Morrow, B.A. An infrared study of sulfated zirconia. Mater. Chem. Phys. 1988, 19, 147–156. [Google Scholar] [CrossRef]
- Chakarova, K.; Strauss, I.; Mihaylov, M.; Drenchev, N.; Hadjiivanov, K. Evolution of acid and basic sites in UiO-66 and UiO-66-NH2 metal-organic frameworks: FTIR study by probe molecules. Microporous Mesoporous Mater. 2019, 281, 110–122. [Google Scholar] [CrossRef]
- Escalona Platero, E.; Peñarroya Mentruit, M.; Otero Areán, C.; Zecchina, A. FTIR studies on the acidity of sulfated zirconia prepared by thermolysis of zirconium sulfate. J. Catal. 1996, 162, 268–276. [Google Scholar] [CrossRef]
- Lange, F.; Hadjiivanov, K.; Schmelz, H.; Knözinger, H. Low temperature infrared study of carbon monoxide adsorption on sulfated titania. Catal. Lett. 1992, 16, 97–107. [Google Scholar] [CrossRef]
- Torbina, V.V.; Salaev, M.A.; Paukshtis, E.A.; Liotta, L.F.; Vodyankina, O.V. Effect of linker substituent nature on performance of active sites in UiO-66: Combined FT-IR and DFT study. Int. J. Mol. Sci. 2023, 24, 14893. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; Taylor & Francis: Abingdon, UK, 1998. [Google Scholar]
- Nakamoto, K. Applications in Inorganic Chemistry. In Infrared and Raman Spectra of Inorganic and Coordination Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 149–354. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2004; p. 368. [Google Scholar]
- Degenhardt, J.; McQuillan, A.J. In Situ ATR-FTIR spectroscopic study of adsorption of perchlorate, sulfate, and thiosulfate ions onto chromium(III) oxide hydroxide thin films. Langmuir 1999, 15, 4595–4602. [Google Scholar] [CrossRef]
- Schnaars, D.D.; Wilson, R.E. Uranium(IV) sulfates: Investigating structural periodicity in the tetravalent actinides. Inorg. Chem. 2012, 51, 9481–9490. [Google Scholar] [CrossRef]
- Fry, A.M.; Sweeney, O.T.; Adam Phelan, W.; Drichko, N.; Siegler, M.A.; McQueen, T.M. Unique edge-sharing sulfate-transition metal coordination in Na2M(SO4)2 (M=Ni and Co). J. Solid State Chem. 2015, 222, 129–135. [Google Scholar] [CrossRef]
- Majzlan, J.; Alpers, C.; Bender Koch, C.; McCleskey, R.; Myneni, S.; Neil, J. Vibrational, X-ray absorption, and Mössbauer spectra of sulfate minerals from the weathered massive sulfide deposit at Iron Mountain, California. Chem. Geol. 2011, 284, 296–305. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Coordination | νs, cm−1 | νas, cm−1 | Ref. | |||
|---|---|---|---|---|---|---|---|
| Free SO42− | no | inactive | 1104 | [58] | |||
| [Co(NH3)6]SO4Br | monodentate | 970 | 1044–1032 | 1143–1117 | [59] | ||
| [Cr(H2O)5SO4Cl]·0.5H2O | 978 | 1040 | 1130 | [61] | |||
| [U(SO4)2(H2O)7]·2H2O | 996 | 1042 | 1144 | [62] | |||
| [Co2(NH3)8(NH2)(SO4)](NO3)3 | bidentate | 995 | 1170 | 1060–1050 | 1105 | [59] | |
| Na2Co(SO4)2 | 970 | 1220 | 1080 | 1160 | [63] | ||
| Fe3+0.67Fe3+4(SO4)6(OH)2·20H2O (Ferricopiapite) | 993 | 1213 | 1019 | 1125 | [64] | ||
| Organic sulfates | C-O-(SO2)-O-C | 1450–1350 | 1230–1150 | [60] | |||
| Sample | S-ZrO2 | UiO-66-BA | UiO-66-FA | UiO-66-SO4 | ||||
|---|---|---|---|---|---|---|---|---|
| Activation temperature | 25 °C | 450 °C | 25 °C | 250 °C | 25 °C | 250 °C | 25 °C | 250 °C |
| CO | ND | 2195 | ND | ND | ND | 2173 | ND | ND |
| CD3CN | 2300 | 2305 | ND | 2300 | 2303 | 2303 | 2303 | 2300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butova, V.V.; Burachevskaia, O.A.; Drenchev, N.L.; Tereshchenko, A.A.; Hadjiivanov, K.I. Probing Acidic and Defective Sites in Sulfated UiO-66 and ZrO2 via Adsorptive FTIR Spectroscopy. Nanomaterials 2025, 15, 779. https://doi.org/10.3390/nano15110779
Butova VV, Burachevskaia OA, Drenchev NL, Tereshchenko AA, Hadjiivanov KI. Probing Acidic and Defective Sites in Sulfated UiO-66 and ZrO2 via Adsorptive FTIR Spectroscopy. Nanomaterials. 2025; 15(11):779. https://doi.org/10.3390/nano15110779
Chicago/Turabian StyleButova, Vera V., Olga A. Burachevskaia, Nikola L. Drenchev, Andrei A. Tereshchenko, and Konstantin I. Hadjiivanov. 2025. "Probing Acidic and Defective Sites in Sulfated UiO-66 and ZrO2 via Adsorptive FTIR Spectroscopy" Nanomaterials 15, no. 11: 779. https://doi.org/10.3390/nano15110779
APA StyleButova, V. V., Burachevskaia, O. A., Drenchev, N. L., Tereshchenko, A. A., & Hadjiivanov, K. I. (2025). Probing Acidic and Defective Sites in Sulfated UiO-66 and ZrO2 via Adsorptive FTIR Spectroscopy. Nanomaterials, 15(11), 779. https://doi.org/10.3390/nano15110779