

Harshly Oxidized Activated Charcoal Enhances Protein Persulfidation with Implications for Neurodegeneration as Exemplified by Friedreich’s Ataxia

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Lines
2.2. PEG-OAC Synthesis
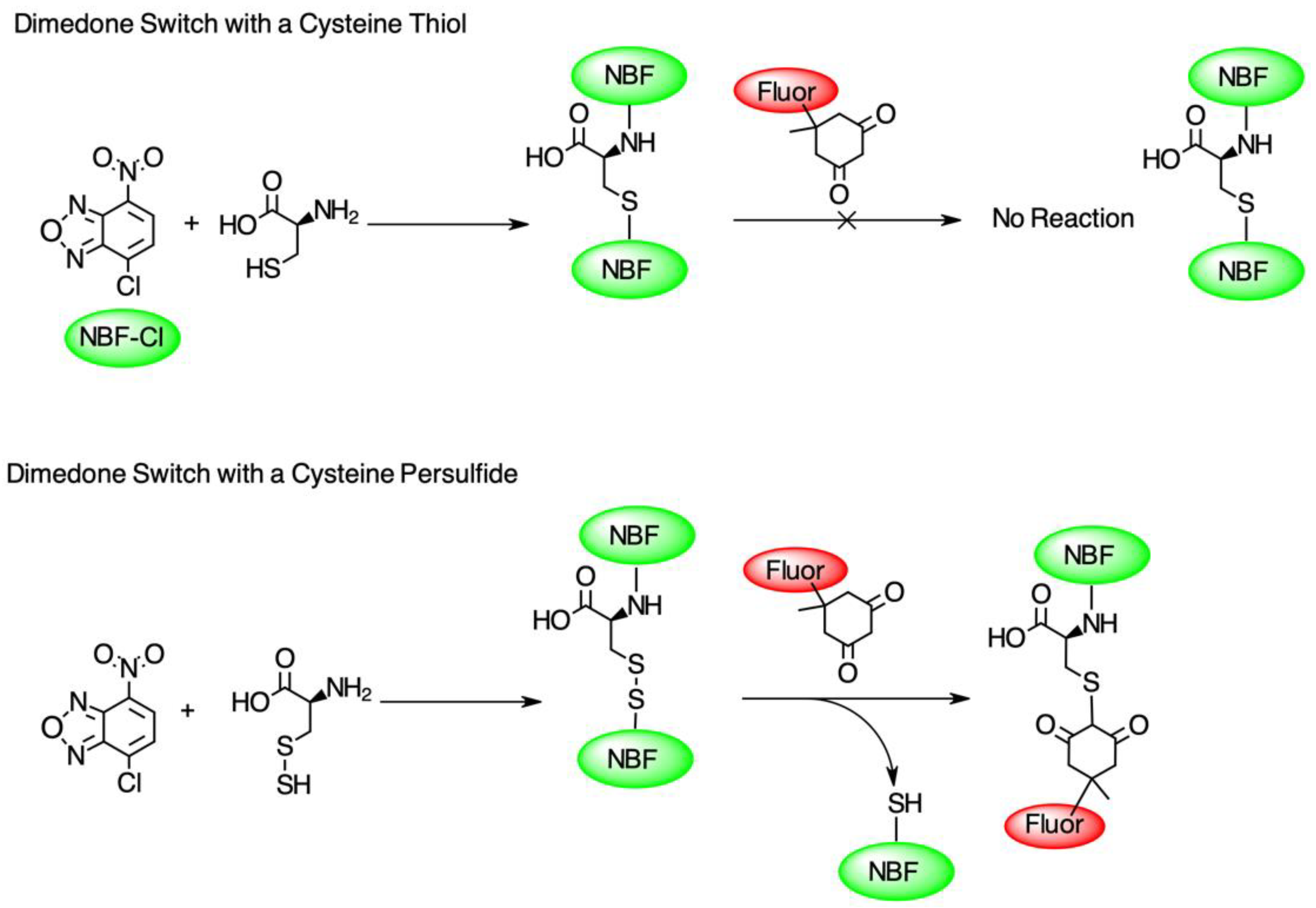
2.3. Protein Persulfidation Labeling Using the Dimedone Switch Method
2.4. Protein Quantification
2.5. SDS-PAGE
2.6. Gel Imaging
2.7. Western Blot
2.8. Redox Western Blot
2.9. Protein Aggregation
2.10. Quantification and Statistical Analysis
3. Results
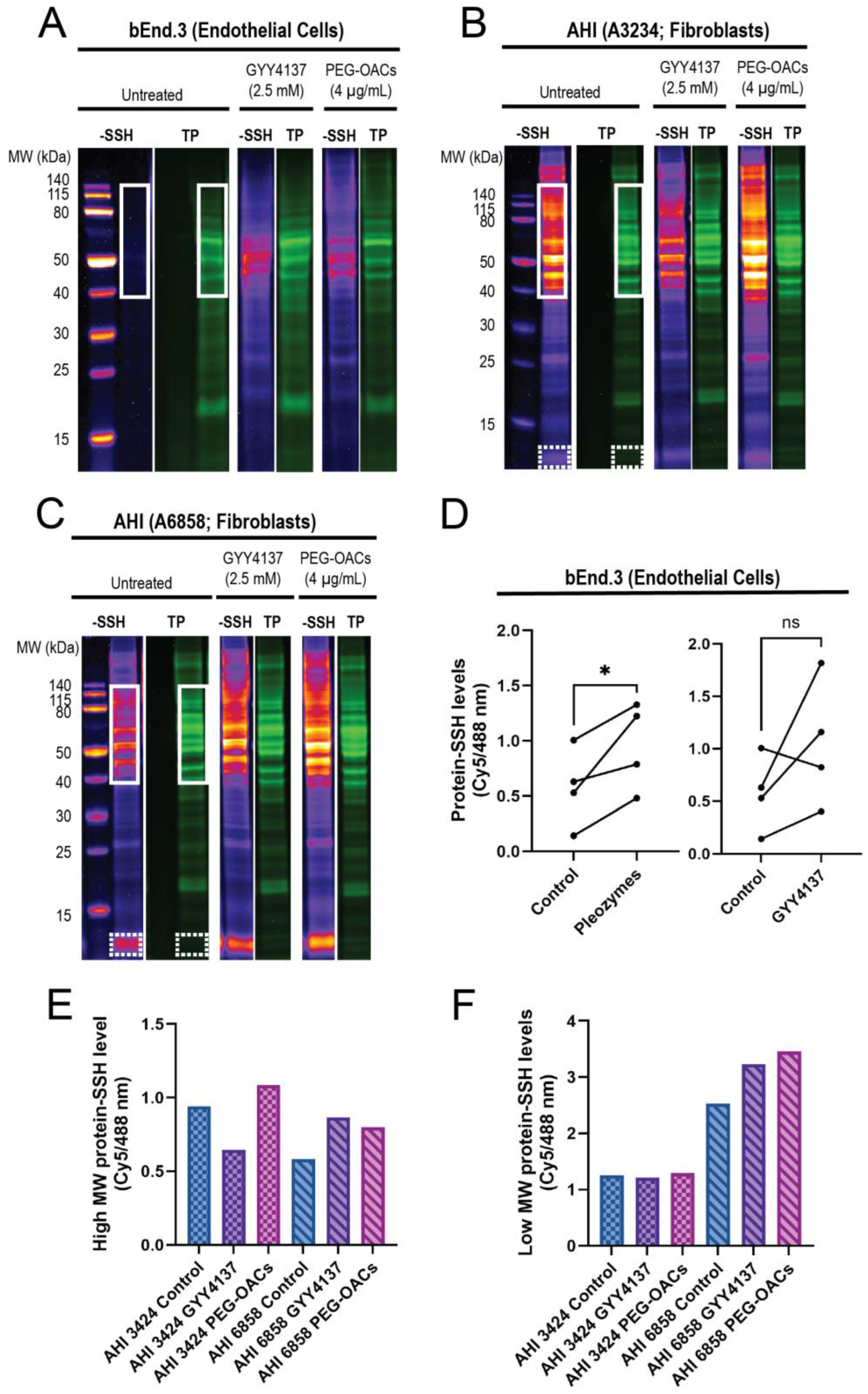
3.1. PEG-OACs Induced Protein Persulfidation
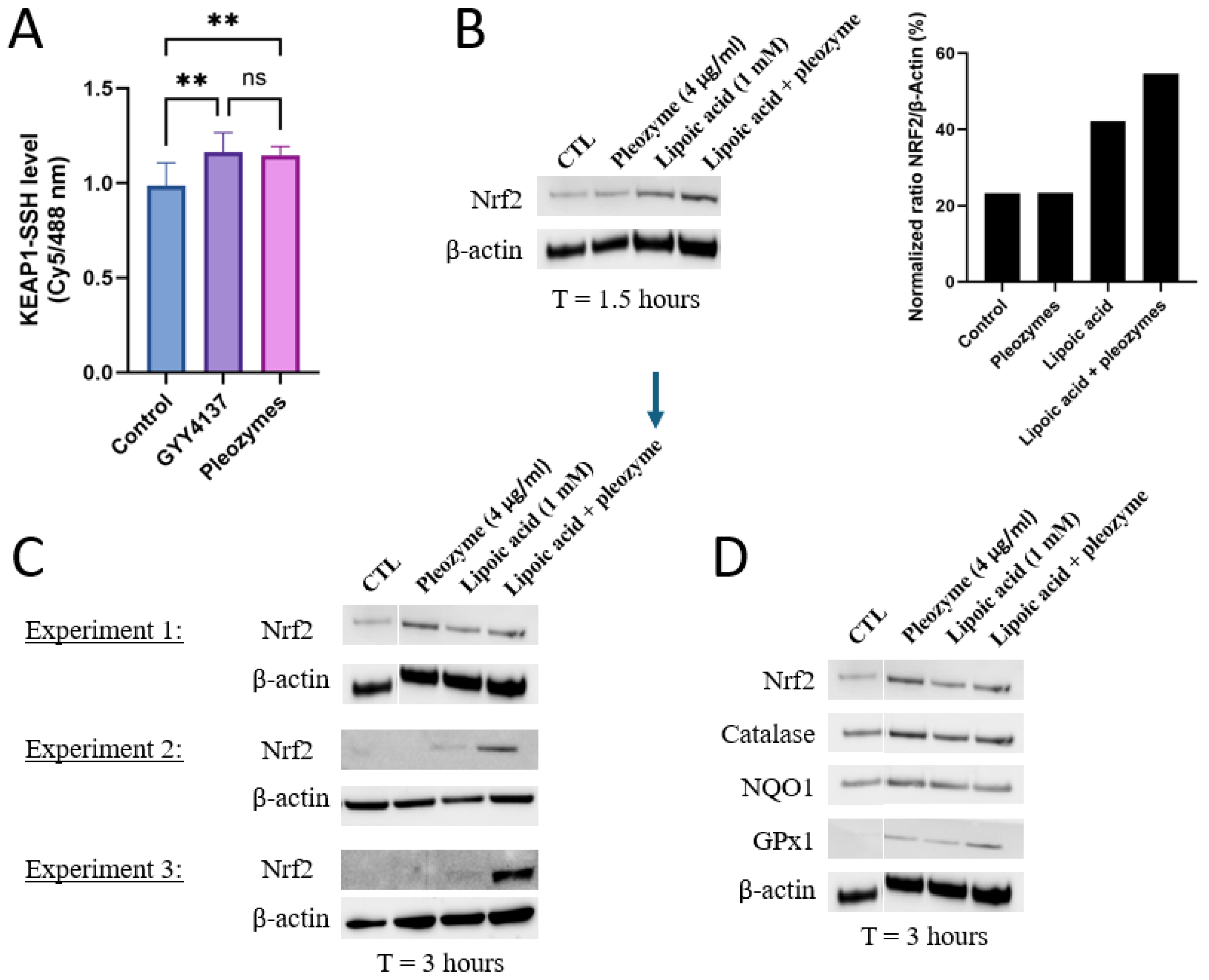
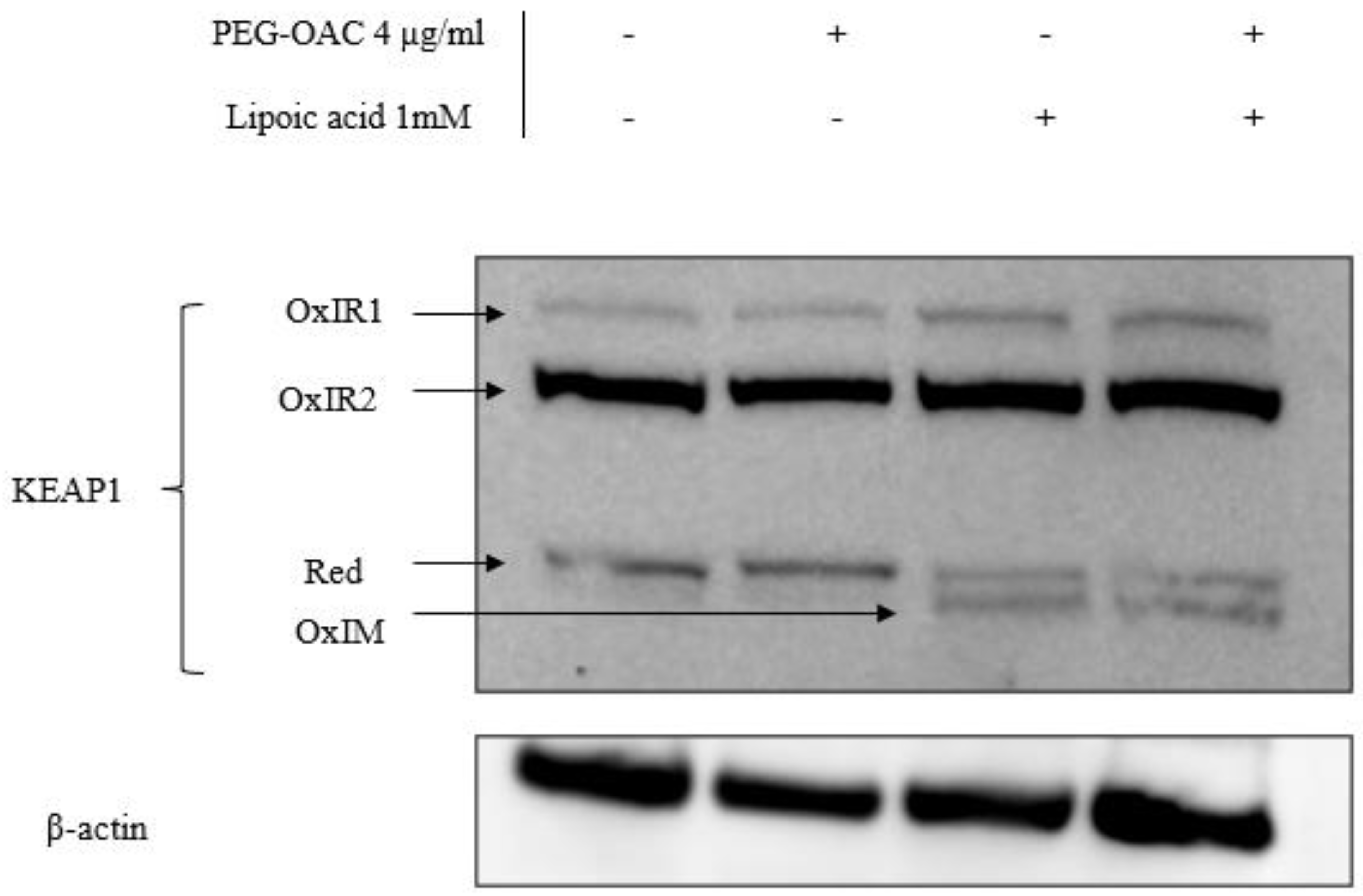
3.2. Pleozymes Induced the Expressions of Nrf2 When Intracellular H2S Pool Increased
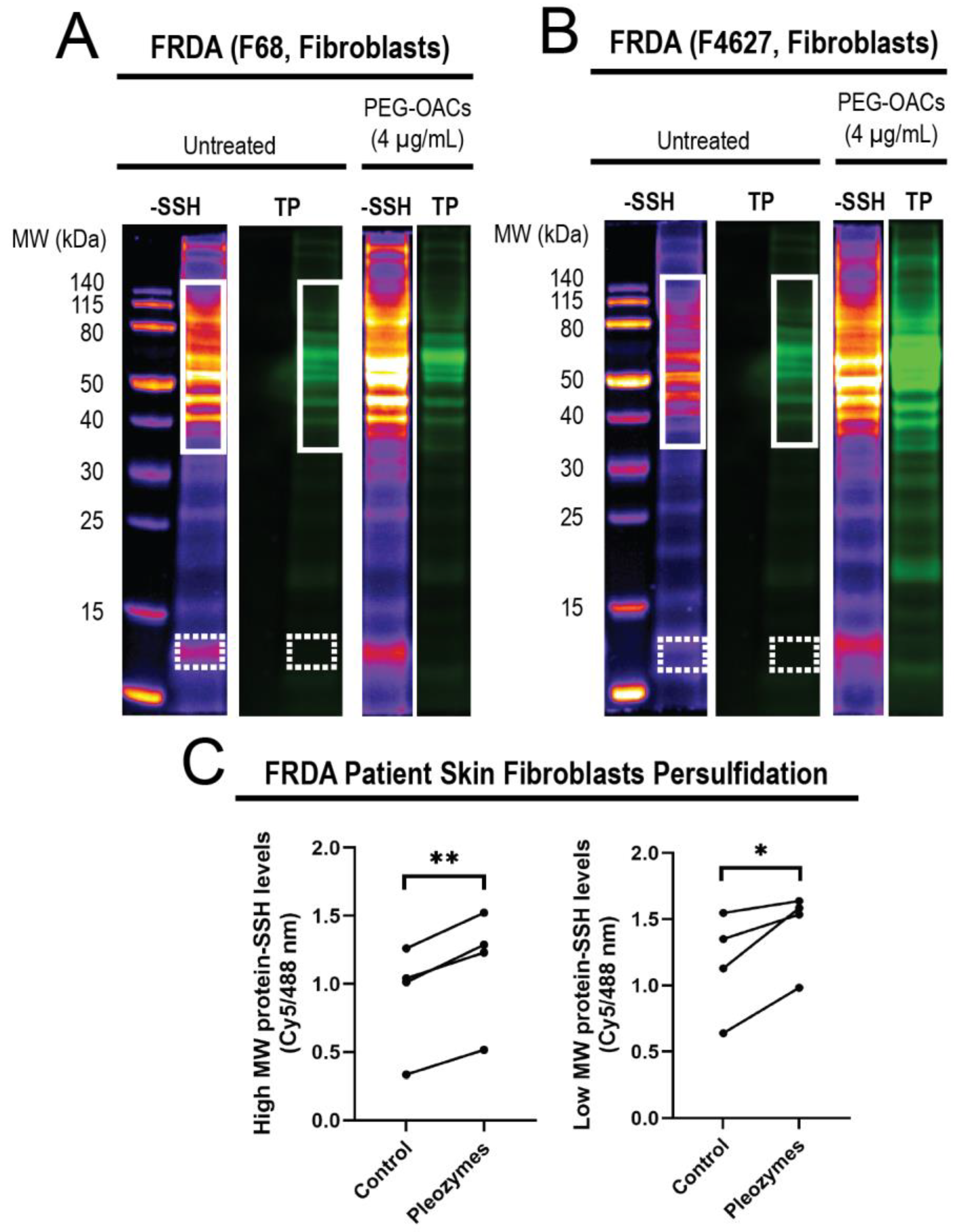
3.3. Pleozymes Increased Protein Persulfidation in Patient-Derived FRDA Skin Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, H.; Zhang, X.; Chen, Z.; Zhao, R.; Hou, N.; Liu, J.; Xun, L.; Liu, H. Developing Polysulfide-Sensitive GFPs for Real-Time Analysis of Polysulfides in Live Cells and Subcellular Organelles. Anal. Chem. 2019, 91, 3893–3901. [Google Scholar] [CrossRef]
- He, B.; Zhang, Z.; Huang, Z.; Duan, X.; Wang, Y.; Cao, J.; Li, L.; He, K.; Nice, E.C.; He, W.; et al. Protein persulfidation: Rewiring the hydrogen sulfide signaling in cell stress response. Biochem. Pharmacol. 2023, 209, 115444. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The Role of Protein Persulfidation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef]
- Luo, S.; Kong, C.; Ye, D.; Liu, X.; Wang, Y.; Meng, G.; Han, Y.; Xie, L.; Ji, Y. Protein Persulfidation: Recent Progress and Future Directions. Antioxid. Redox Signal. 2023, 39, 829–852. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef] [PubMed]
- Bithi, N.; Link, C.; Henderson, Y.O.; Kim, S.; Yang, J.; Li, L.; Wang, R.; Willard, B.; Hine, C. Dietary restriction transforms the mammalian protein persulfidome in a tissue-specific and cystathionine gamma-lyase-dependent manner. Nat. Commun. 2021, 12, 1745. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Biagiotti, S.; Bianchi, M.; Rossi, L.; Chessa, L.; Magnani, M. Activation of NRF2 by dexamethasone in ataxia telangiectasia cells involves KEAP1 inhibition but not the inhibition of p38. PLoS ONE 2019, 14, e0216668. [Google Scholar] [CrossRef] [PubMed]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Lamriben, L.; van Zadelhoff, G.; Hebert, D.N. Analysis of Disulfide Bond Formation. Curr. Protoc. Protein Sci. 2017, 90, 14.1.1–14.1.21. [Google Scholar] [CrossRef] [PubMed]
- Drekolia, M.K.; Karantanou, C.; Wittig, I.; Li, Y.; Fuhrmann, D.C.; Brune, B.; Katsouda, A.; Hu, J.; Papapetropoulos, A.; Bibli, S.I. Loss of cardiac mitochondrial complex I persulfidation impairs NAD(+) homeostasis in aging. Redox Biol. 2024, 69, 103014. [Google Scholar] [CrossRef]
- Vignane, T.; Filipovic, M.R. Emerging Chemical Biology of Protein Persulfidation. Antioxid. Redox Signal. 2023, 39, 19–39. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3beta and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef]
- Panagaki, T.; Janickova, L.; Petrovic, D.; Zuhra, K.; Ditroi, T.; Juranyi, E.P.; Bremer, O.; Ascencao, K.; Philipp, T.M.; Nagy, P.; et al. Neurobehavioral dysfunction in a mouse model of Down syndrome: Upregulation of cystathionine beta-synthase, H2S overproduction, altered protein persulfidation, synaptic dysfunction, endoplasmic reticulum stress, and autophagy. Geroscience 2024, 46, 4275–4314. [Google Scholar] [CrossRef] [PubMed]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first Friedreich’s ataxia drug. Nat. Rev. Drug Discov. 2023, 22, 258. [Google Scholar] [CrossRef]
- Scott, V.; Delatycki, M.B.; Tai, G.; Corben, L.A. New and Emerging Drug and Gene Therapies for Friedreich Ataxia. CNS Drugs 2024, 38, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2021, 89, 212–225. [Google Scholar] [CrossRef]
- Eisel, M.L.S.; Burns, M.; Ashizawa, T.; Byrne, B.; Corti, M.; Subramony, S.H. Emerging therapies in hereditary ataxias. Trends Mol. Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- McHugh, E.A.; Liopo, A.V.; Mendoza, K.; Robertson, C.S.; Wu, G.; Wang, Z.; Chen, W.; Beckham, J.L.; Derry, P.J.; Kent, T.A.; et al. Oxidized Activated Charcoal Nanozymes: Synthesis, and Optimization for In Vitro and In Vivo Bioactivity for Traumatic Brain Injury. Adv. Mater. 2023, 36, e2211239. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, P.; Talakatta, G.; Mitra, J.; Wang, H.; Derry, P.J.; Nilewski, L.G.; McHugh, E.A.; Fabian, R.H.; Mendoza, K.; Vasquez, V.; et al. Pervasive Genomic Damage in Experimental Intracerebral Hemorrhage: Therapeutic Potential of a Mechanistic-Based Carbon Nanoparticle. ACS Nano 2020, 14, 2827–2846. [Google Scholar] [CrossRef] [PubMed]
- Derry, P.J.; Liopo, A.V.; Mouli, K.; McHugh, E.A.; Vo, A.T.T.; McKelvey, A.; Suva, L.J.; Wu, G.; Gao, Y.; Olson, K.R.; et al. Oxidation of Hydrogen Sulfide to Polysulfide and Thiosulfate by a Carbon Nanozyme: Therapeutic Implications with an Emphasis on Down Syndrome. Adv. Mater. 2023, 36, e2211241. [Google Scholar] [CrossRef]
- Liang, M.; Yan, X. Nanozymes: From New Concepts, Mechanisms, and Standards to Applications. Acc. Chem. Res. 2019, 52, 2190–2200. [Google Scholar] [CrossRef]
- Huq, R.; Samuel, E.L.; Sikkema, W.K.; Nilewski, L.G.; Lee, T.; Tanner, M.R.; Khan, F.S.; Porter, P.C.; Tajhya, R.B.; Patel, R.S.; et al. Preferential uptake of antioxidant carbon nanoparticles by T lymphocytes for immunomodulation. Sci. Rep. 2016, 6, 33808. [Google Scholar] [CrossRef] [PubMed]
- Fabian, R.H.; Derry, P.J.; Rea, H.C.; Dalmeida, W.V.; Nilewski, L.G.; Sikkema, W.K.A.; Mandava, P.; Tsai, A.L.; Mendoza, K.; Berka, V.; et al. Efficacy of Novel Carbon Nanoparticle Antioxidant Therapy in a Severe Model of Reversible Middle Cerebral Artery Stroke in Acutely Hyperglycemic Rats. Front. Neurol. 2018, 9, 199. [Google Scholar] [CrossRef]
- Bitner, B.R.; Marcano, D.C.; Berlin, J.M.; Fabian, R.H.; Cherian, L.; Culver, J.C.; Dickinson, M.E.; Robertson, C.S.; Pautler, R.G.; Kent, T.A.; et al. Antioxidant carbon particles improve cerebrovascular dysfunction following traumatic brain injury. ACS Nano 2012, 6, 8007–8014. [Google Scholar] [CrossRef] [PubMed]
- Marcano, D.C.; Bitner, B.R.; Berlin, J.M.; Jarjour, J.; Lee, J.M.; Jacob, A.; Fabian, R.H.; Kent, T.A.; Tour, J.M. Design of poly(ethylene glycol)-functionalized hydrophilic carbon clusters for targeted therapy of cerebrovascular dysfunction in mild traumatic brain injury. J. Neurotrauma 2013, 30, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Mouli, K.; Liopo, A.V.; McHugh, E.A.; Underwood, E.; Zhao, J.; Dash, P.K.; Vo, A.T.T.; Malojirao, V.; Hegde, M.; Tour, J.M.; et al. Oxidized Carbon Nanoparticles Enhance Cellular Energetics with Application to Injured Brain. Adv. Healthc. Mater. 2024, e2401629. [Google Scholar] [CrossRef]
- Derry, P.J.; Nilewski, L.G.; Sikkema, W.K.A.; Mendoza, K.; Jalilov, A.; Berka, V.; McHugh, E.A.; Tsai, A.L.; Tour, J.M.; Kent, T.A. Catalytic oxidation and reduction reactions of hydrophilic carbon clusters with NADH and cytochrome C: Features of an electron transport nanozyme. Nanoscale 2019, 11, 10791–10807. [Google Scholar] [CrossRef]
- Wu, G.; Berka, V.; Derry, P.J.; Mendoza, K.; Kakadiaris, E.; Roy, T.; Kent, T.A.; Tour, J.M.; Tsai, A.L. Critical Comparison of the Superoxide Dismutase-like Activity of Carbon Antioxidant Nanozymes by Direct Superoxide Consumption Kinetic Measurements. ACS Nano 2019, 13, 11203–11213. [Google Scholar] [CrossRef]
- Samuel, E.L.; Marcano, D.C.; Berka, V.; Bitner, B.R.; Wu, G.; Potter, A.; Fabian, R.H.; Pautler, R.G.; Kent, T.A.; Tsai, A.L.; et al. Highly efficient conversion of superoxide to oxygen using hydrophilic carbon clusters. Proc. Natl. Acad. Sci. USA 2015, 112, 2343–2348. [Google Scholar] [CrossRef]
- Jalilov, A.S.; Zhang, C.; Samuel, E.L.; Sikkema, W.K.; Wu, G.; Berka, V.; Kent, T.A.; Tsai, A.L.; Tour, J.M. Mechanistic Study of the Conversion of Superoxide to Oxygen and Hydrogen Peroxide in Carbon Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 15086–15092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Polak, U.; Clark, A.D.; Bhalla, A.D.; Chen, Y.Y.; Li, J.; Farmer, J.; Seyer, L.; Lynch, D.; Butler, J.S.; et al. Establishment and Maintenance of Primary Fibroblast Repositories for Rare Diseases-Friedreich’s Ataxia Example. Biopreserv. Biobank. 2016, 14, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Napierala, J.S.; Li, Y.; Lu, Y.; Lin, K.; Hauser, L.A.; Lynch, D.R.; Napierala, M. Comprehensive analysis of gene expression patterns in Friedreich’s ataxia fibroblasts by RNA sequencing reveals altered levels of protein synthesis factors and solute carriers. Dis. Model. Mech. 2017, 10, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ho, E.S.; Cotticelli, M.G.; Xu, P.; Napierala, J.S.; Hauser, L.A.; Napierala, M.; Himes, B.E.; Wilson, R.B.; Lynch, D.R.; et al. Skin fibroblast metabolomic profiling reveals that lipid dysfunction predicts the severity of Friedreich’s ataxia. J. Lipid Res. 2022, 63, 100255. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Corrales, C.; Linsenmeier, M.; Alalami, H.; Sepanj, N.; Bitan, G. Examination of SOD1 aggregation modulators and their effect on SOD1 enzymatic activity as a proxy for potential toxicity. FASEB J. 2020, 34, 11957–11969. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef]
- Kimura, Y.; Koike, S.; Shibuya, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci. Rep. 2017, 7, 10459. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Talwar, D.; Barayeu, U.; Schilling, D.; Luzarowski, M.; Sokolowski, M.; Glatt, S.; Dick, T.P. 3-Mercaptopyruvate sulfur transferase is a protein persulfidase. Nat. Chem. Biol. 2023, 19, 507–517. [Google Scholar] [CrossRef]
- Crinelli, R.; Zara, C.; Galluzzi, L.; Buffi, G.; Ceccarini, C.; Smietana, M.; Mari, M.; Magnani, M.; Fraternale, A. Activation of NRF2 and ATF4 Signaling by the Pro-Glutathione Molecule I-152, a Co-Drug of N-Acetyl-Cysteine and Cysteamine. Antioxidants 2021, 10, 175. [Google Scholar] [CrossRef]
- Karunatilleke, N.C.; Fast, C.S.; Ngo, V.; Brickenden, A.; Duennwald, M.L.; Konermann, L.; Choy, W.Y. Nrf2, the Major Regulator of the Cellular Oxidative Stress Response, is Partially Disordered. Int. J. Mol. Sci. 2021, 22, 7434. [Google Scholar] [CrossRef]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K(110) and K(533) Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [CrossRef]
- Amin, K.N.; Rajaguru, P.; Suzuki, T.; Sarkar, K.; Ganesan, K.; Ramkumar, K.M. Quantitative proteomic analyses uncover regulatory roles of Nrf2 in human endothelial cells. Cell Stress Chaperones 2023, 28, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, X.; Wu, W.; Wang, J.; Xie, H.; Wu, Z. Regeneration of glutathione by alpha-lipoic acid via Nrf2/ARE signaling pathway alleviates cadmium-induced HepG2 cell toxicity. Environ. Toxicol. Pharmacol. 2017, 51, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.; Lim, J.W.; Kim, H. alpha-Lipoic Acid Inhibits IL-8 Expression by Activating Nrf2 Signaling in Helicobacter pylori-infected Gastric Epithelial Cells. Nutrients 2019, 11, 2524. [Google Scholar] [CrossRef] [PubMed]
- Shay, K.P.; Michels, A.J.; Li, W.; Kong, A.N.; Hagen, T.M. Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta 2012, 1823, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Briggs, A.; Devireddy, M.; Xian, M.; Gao, Y. Are the beneficial effects of ‘antioxidant’ lipoic acid mediated through metabolism of reactive sulfur species? Free Radic. Biol. Med. 2020, 146, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich ataxia. Arch. Neurol. 2008, 65, 1296–1303. [Google Scholar] [CrossRef]
- Telot, L.; Rousseau, E.; Lesuisse, E.; Garcia, C.; Morlet, B.; Leger, T.; Camadro, J.M.; Serre, V. Quantitative proteomics in Friedreich’s ataxia B-lymphocytes: A valuable approach to decipher the biochemical events responsible for pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 997–1009. [Google Scholar] [CrossRef]
- Fan, F.; Jiang, H.; Hou, Y.; Zhang, Y.; Zhao, Q.; Zeng, Y.; Meng, X.; Wang, X. Barrier Functional Integrity Recording on bEnd.3 Vascular Endothelial Cells via Transendothelial Electrical Resistance Detection. J. Vis. Exp. 2023, e65938. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jin, H.; Zhao, Z.G. Epidermal growth factor treatment has protective effects on the integrity of the blood-brain barrier against cerebral ischemia injury in bEnd3 cells. Exp. Ther. Med. 2019, 17, 2397–2402. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, J.; Liu, Y.; Liu, W.; Yu, G.; Huang, Y.; Yang, Y.; Chen, X.; Chen, T. Brain-Targeted Biomimetic Nanodecoys with Neuroprotective Effects for Precise Therapy of Parkinson’s Disease. ACS Cent. Sci. 2022, 8, 1336–1349. [Google Scholar] [CrossRef]
- Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener. 2022, 11, 36. [Google Scholar] [CrossRef]
- Kazirod, K.; Myszka, M.; Dulak, J.; Loboda, A. Hydrogen sulfide as a therapeutic option for the treatment of Duchenne muscular dystrophy and other muscle-related diseases. Cell. Mol. Life Sci. 2022, 79, 608. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, M.R. Persulfidation (S-sulfhydration) and H2S. Handb. Exp. Pharmacol. 2015, 230, 29–59. [Google Scholar] [CrossRef]
- Monfort, B.; Want, K.; Gervason, S.; D’Autreaux, B. Recent Advances in the Elucidation of Frataxin Biochemical Function Open Novel Perspectives for the Treatment of Friedreich’s Ataxia. Front. Neurosci. 2022, 16, 838335. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.T.; Devarie-Baez, N.O.; Hamsath, A.; Fu, X.D.; Xian, M. S-Persulfidation: Chemistry, Chemical Biology, and Significance in Health and Disease. Antioxid. Redox Signal. 2020, 33, 1092–1114. [Google Scholar] [CrossRef]
- Yu, B.; Kang, T.; Xu, Y.; Liu, Y.; Ma, Y.; Ke, B. Prodrugs of Persulfide and Sulfide: Is There a Pharmacological Difference between the Two in the Context of Rapid Exchanges among Various Sulfur Species In Vivo? Angew. Chem. Int. Ed. Engl. 2022, 61, e202201668. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Matsubara, H.; Hara, H. Potential therapeutic effects of Nrf2 activators on intracranial hemorrhage. J. Cereb. Blood Flow. Metab. 2021, 41, 1483–1500. [Google Scholar] [CrossRef]
- Loan, J.J.M.; Al-Shahi Salman, R.; McColl, B.W.; Hardingham, G.E. Activation of Nrf2 to Optimise Immune Responses to Intracerebral Haemorrhage. Biomolecules 2022, 12, 1438. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Locascio, L.M.; Dore, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11, 2377. [Google Scholar] [CrossRef] [PubMed]
- Okuda, D.T.; Kantarci, O.; Lebrun-Frenay, C.; Sormani, M.P.; Azevedo, C.J.; Bovis, F.; Hua, L.H.; Amezcua, L.; Mowry, E.M.; Hotermans, C.; et al. Dimethyl Fumarate Delays Multiple Sclerosis in Radiologically Isolated Syndrome. Ann. Neurol. 2023, 93, 604–614. [Google Scholar] [CrossRef]
- Profeta, V.; McIntyre, K.; Wells, M.; Park, C.; Lynch, D.R. Omaveloxolone: An activator of Nrf2 for the treatment of Friedreich ataxia. Expert. Opin. Investig. Drugs 2023, 32, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ishima, Y.; Chuang, V.T.G.; Sakai, M.; Osafune, H.; Ando, H.; Shimizu, T.; Okuhira, K.; Watanabe, H.; Maruyama, T.; et al. Distribution of Polysulfide in Human Biological Fluids and Their Association with Amylase and Sperm Activities. Molecules 2019, 24, 1689. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ishima, Y.; Shibata, A.; Chuang, V.T.G.; Sawa, T.; Ihara, H.; Watanabe, H.; Xian, M.; Ouchi, Y.; Shimizu, T.; et al. Quantitative determination of polysulfide in albumins, plasma proteins and biological fluid samples using a novel combined assays approach. Anal. Chim. Acta 2017, 969, 18–25. [Google Scholar] [CrossRef]
- Lindahl, S.; Xian, M. Recent development of polysulfides: Chemistry and biological applications. Curr. Opin. Chem. Biol. 2023, 75, 102325. [Google Scholar] [CrossRef]
- Xu, W.C.; Liang, J.Z.; Li, C.; He, Z.X.; Yuan, H.Y.; Huang, B.Y.; Liu, X.L.; Tang, B.; Pang, D.W.; Du, H.N.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, A.T.T.; Khan, U.; Liopo, A.V.; Mouli, K.; Olson, K.R.; McHugh, E.A.; Tour, J.M.; Pooparayil Manoj, M.; Derry, P.J.; Kent, T.A. Harshly Oxidized Activated Charcoal Enhances Protein Persulfidation with Implications for Neurodegeneration as Exemplified by Friedreich’s Ataxia. Nanomaterials 2024, 14, 2007. https://doi.org/10.3390/nano14242007
Vo ATT, Khan U, Liopo AV, Mouli K, Olson KR, McHugh EA, Tour JM, Pooparayil Manoj M, Derry PJ, Kent TA. Harshly Oxidized Activated Charcoal Enhances Protein Persulfidation with Implications for Neurodegeneration as Exemplified by Friedreich’s Ataxia. Nanomaterials. 2024; 14(24):2007. https://doi.org/10.3390/nano14242007
Chicago/Turabian StyleVo, Anh T. T., Uffaf Khan, Anton V. Liopo, Karthik Mouli, Kenneth R. Olson, Emily A. McHugh, James M. Tour, Madhavan Pooparayil Manoj, Paul J. Derry, and Thomas A. Kent. 2024. "Harshly Oxidized Activated Charcoal Enhances Protein Persulfidation with Implications for Neurodegeneration as Exemplified by Friedreich’s Ataxia" Nanomaterials 14, no. 24: 2007. https://doi.org/10.3390/nano14242007
APA StyleVo, A. T. T., Khan, U., Liopo, A. V., Mouli, K., Olson, K. R., McHugh, E. A., Tour, J. M., Pooparayil Manoj, M., Derry, P. J., & Kent, T. A. (2024). Harshly Oxidized Activated Charcoal Enhances Protein Persulfidation with Implications for Neurodegeneration as Exemplified by Friedreich’s Ataxia. Nanomaterials, 14(24), 2007. https://doi.org/10.3390/nano14242007