_Hui.png)
Covalent Triazine Framework C6N6 as an Electrochemical Sensor for Hydrogen-Containing Industrial Pollutants. A DFT Study



Abstract
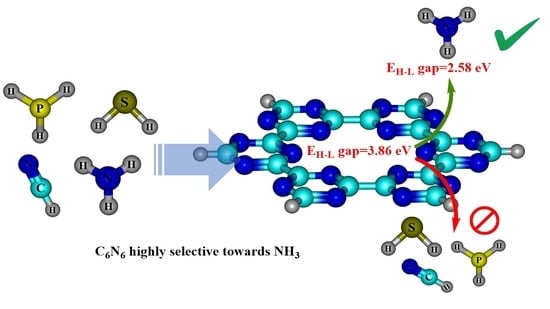
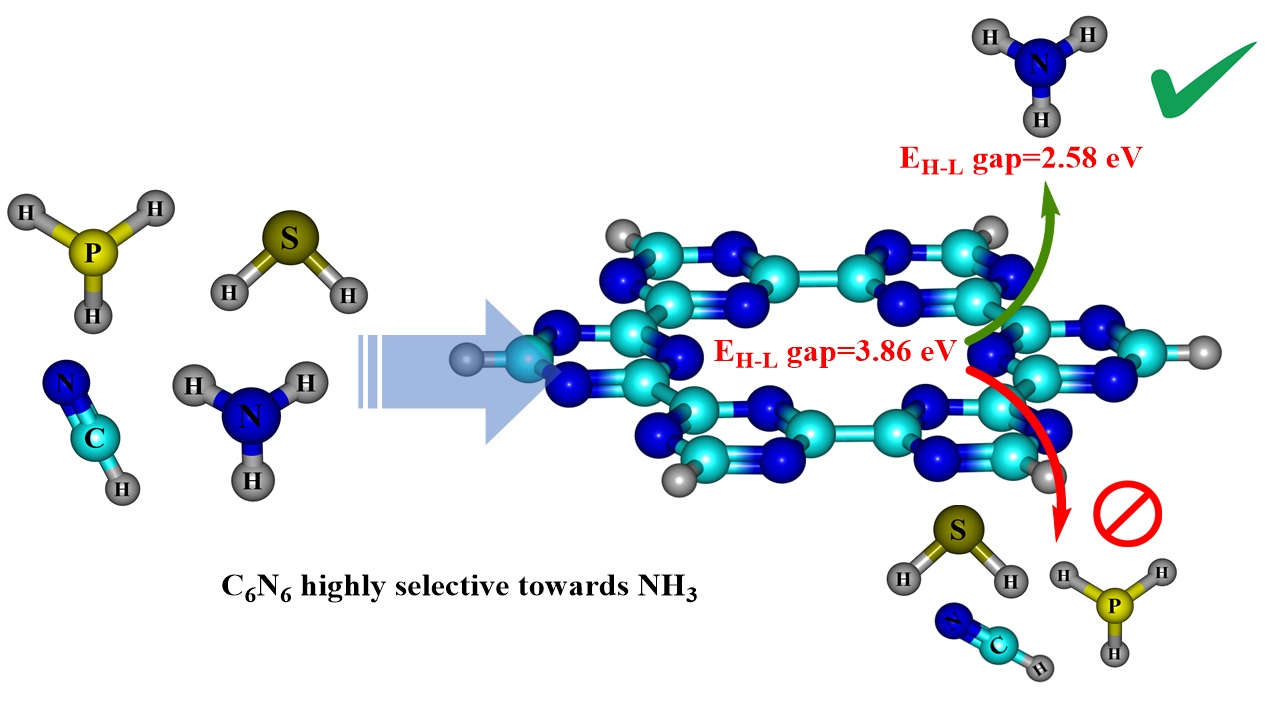
1. Introduction
2. Computational Methodology
3. Results and Discussion
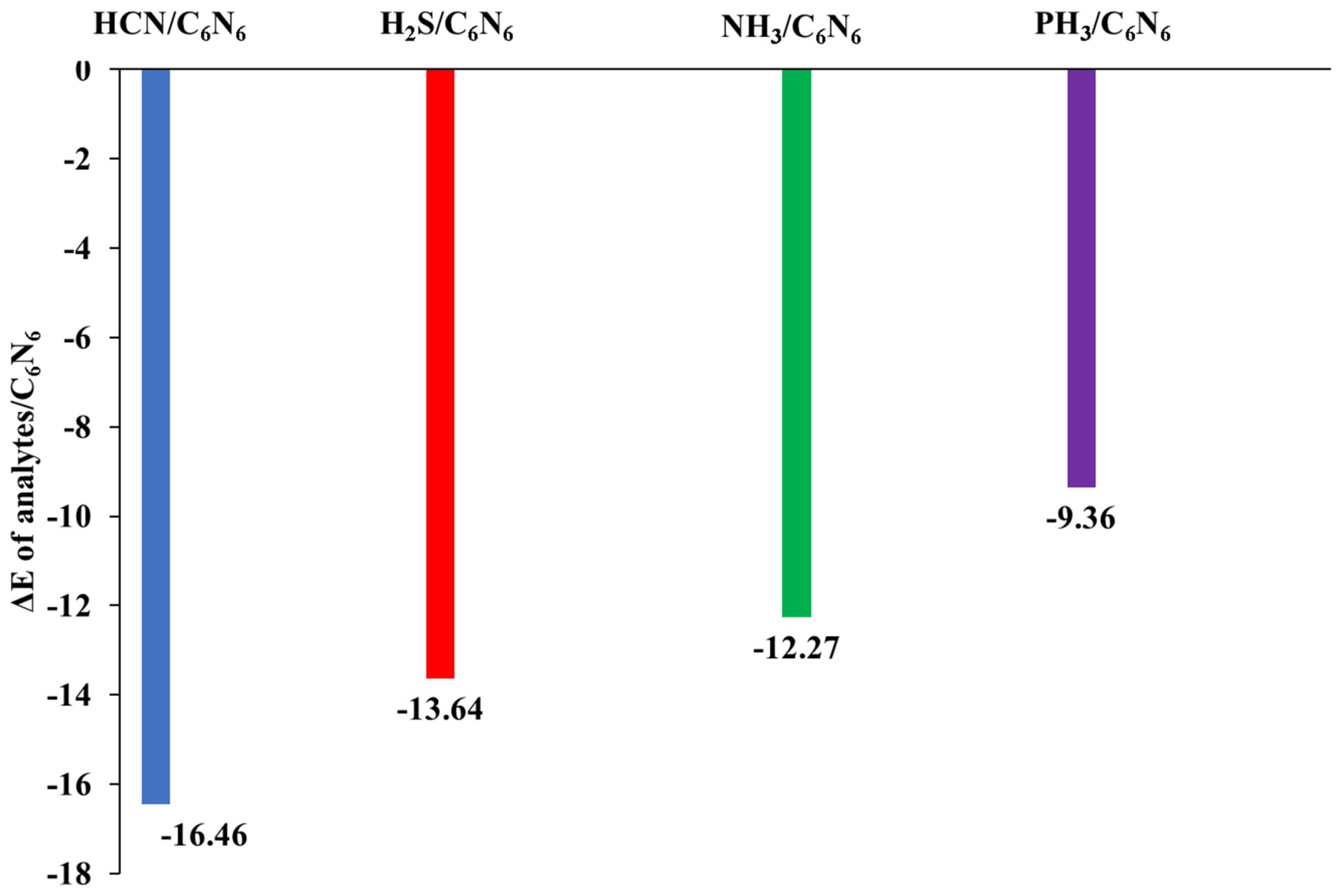
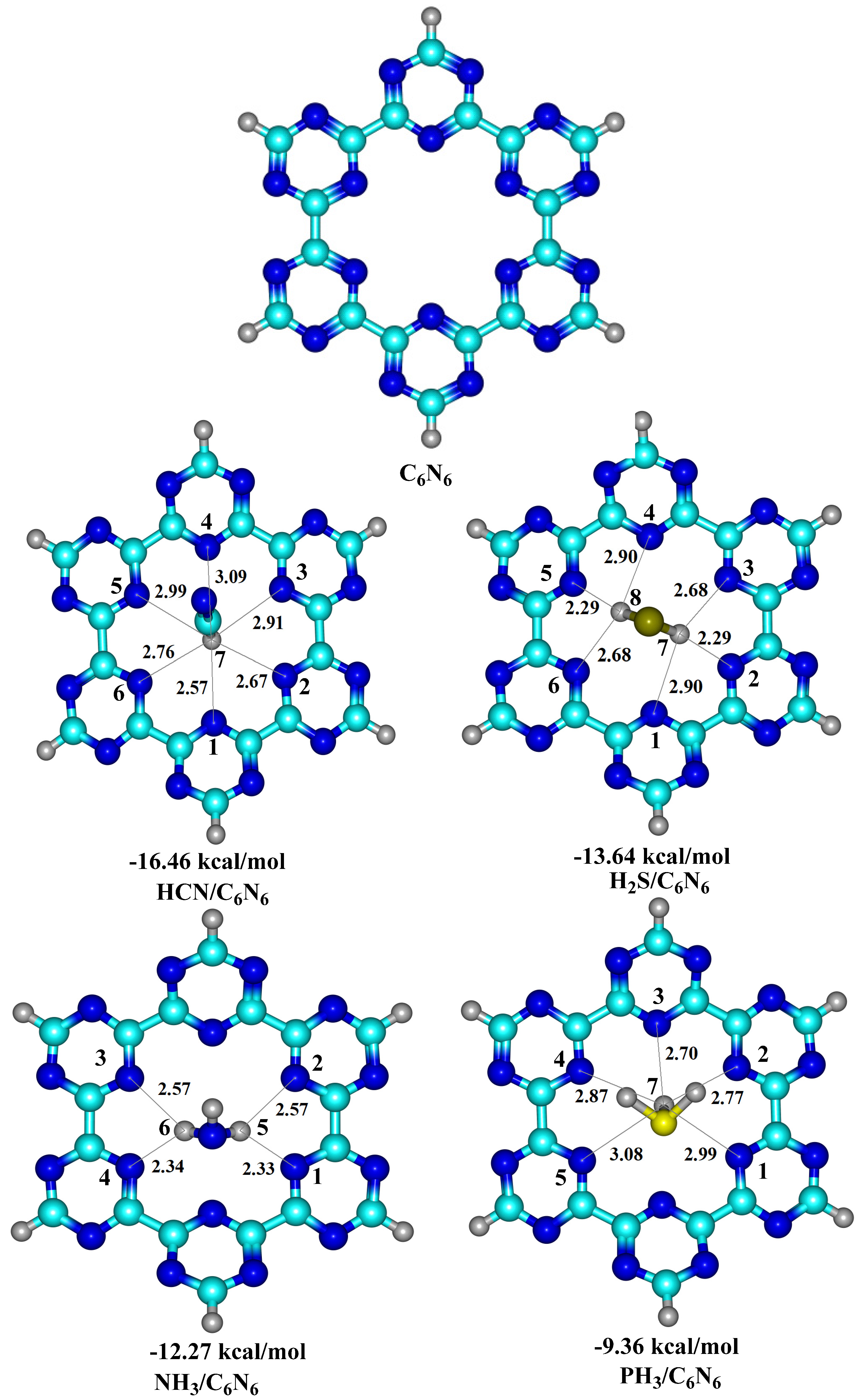
3.1. Geometry Optimization and Interaction Energy
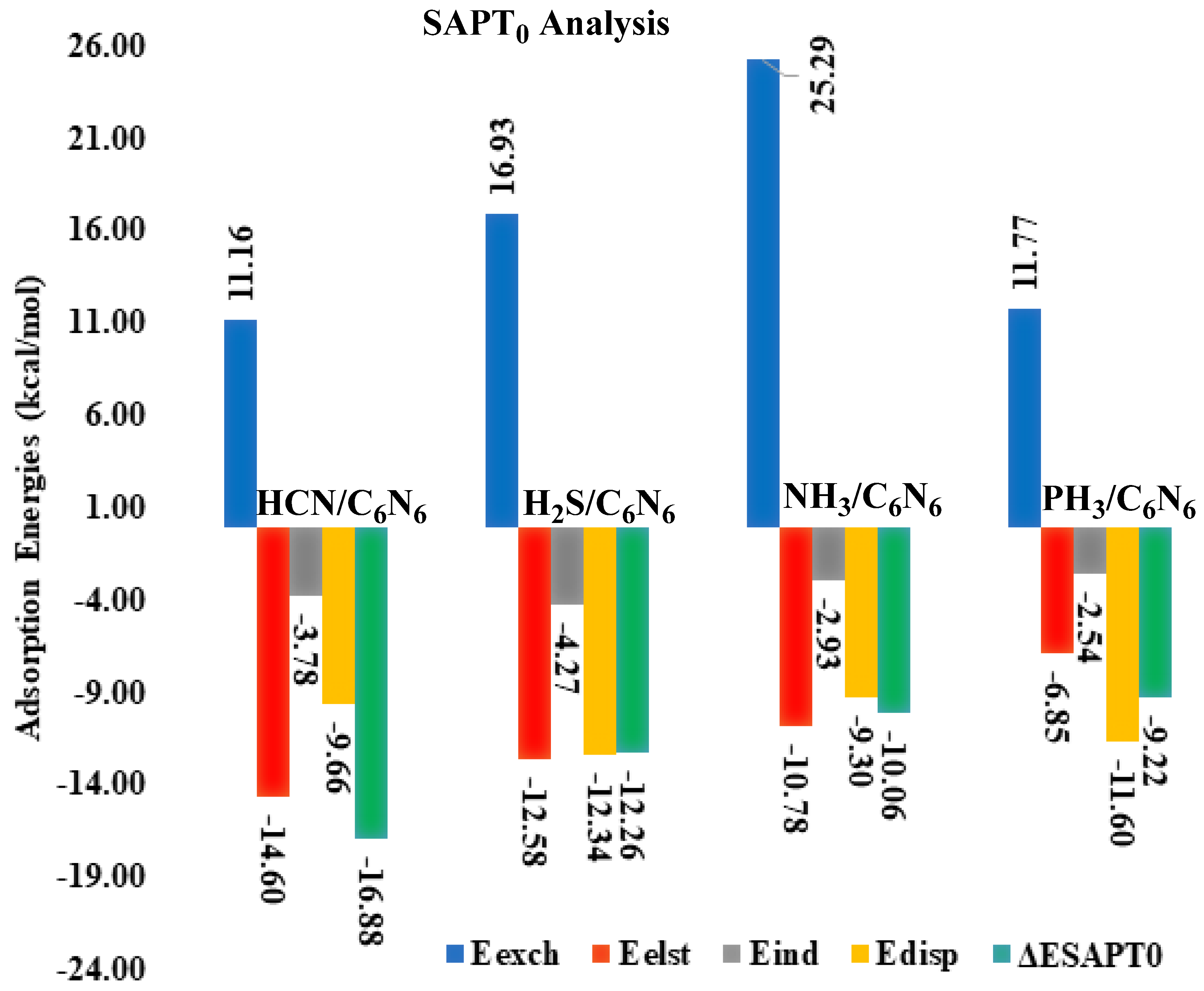
3.2. Symmetry Adapted Perturbation Theory (SAPT0) Analysis
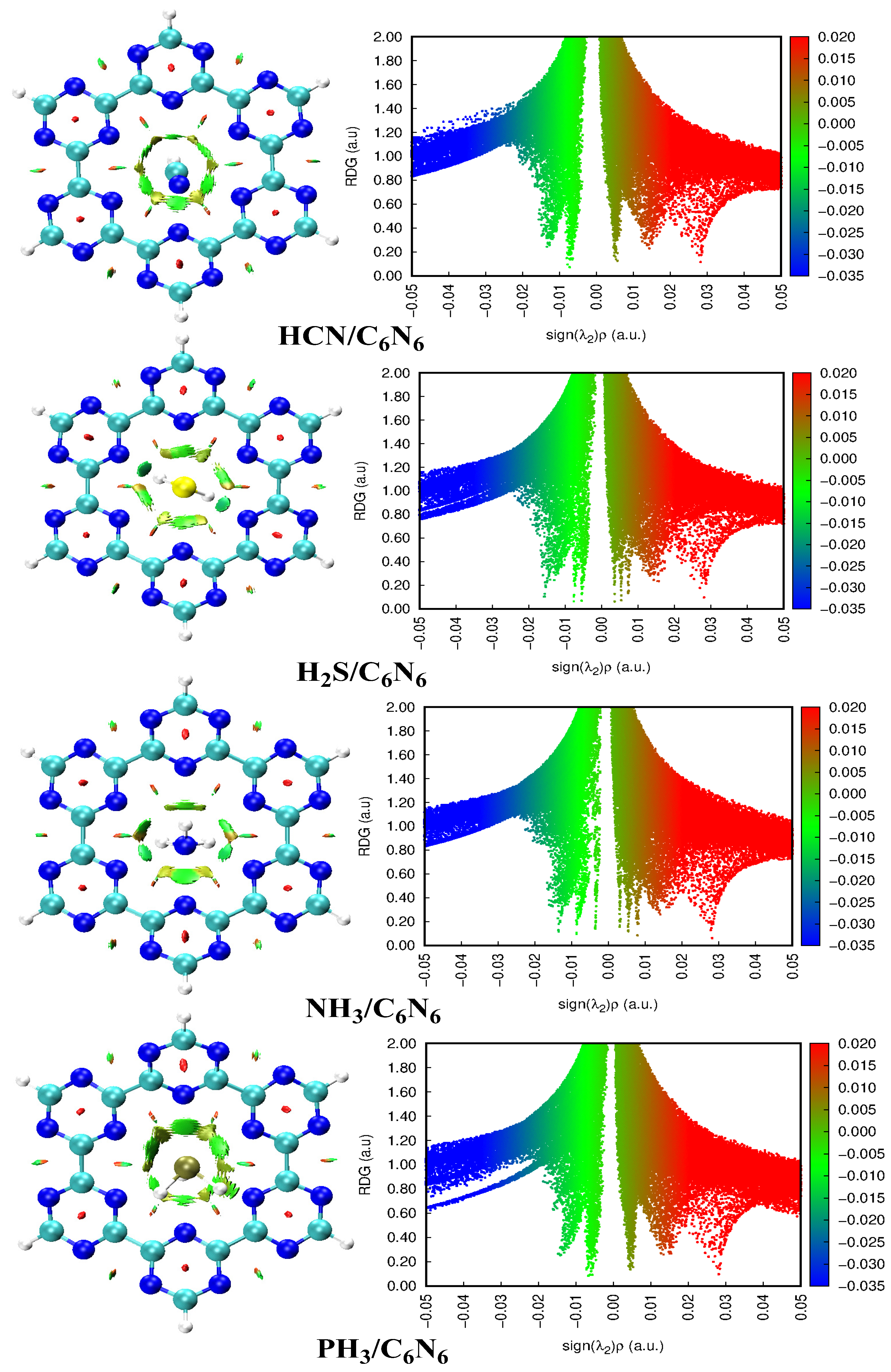
3.3. Non-Covalent Interaction (NCI) Analysis
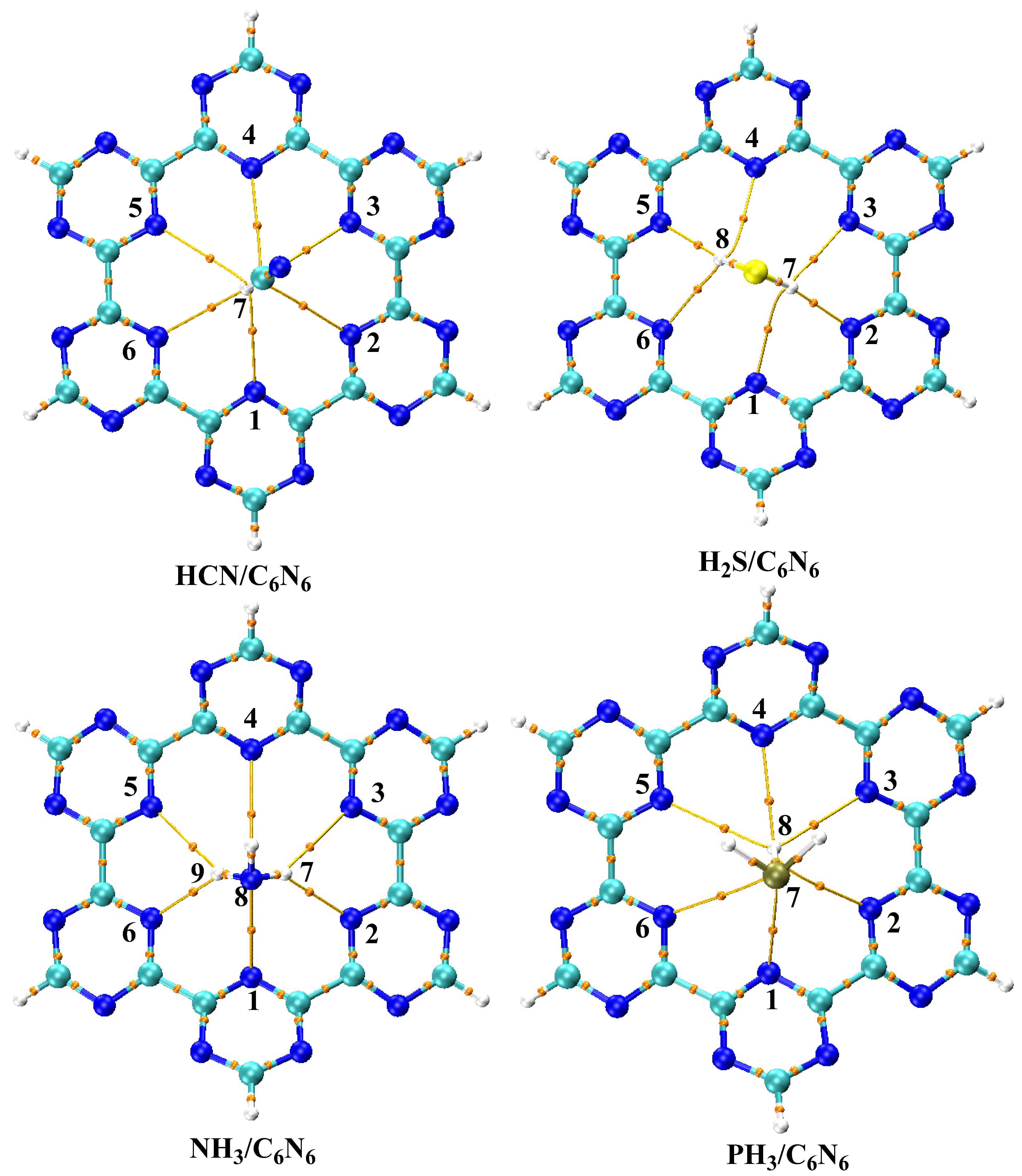
3.4. Quantum Theory of Atoms in Molecule (QTAIM) Analysis
4. Electronic Properties
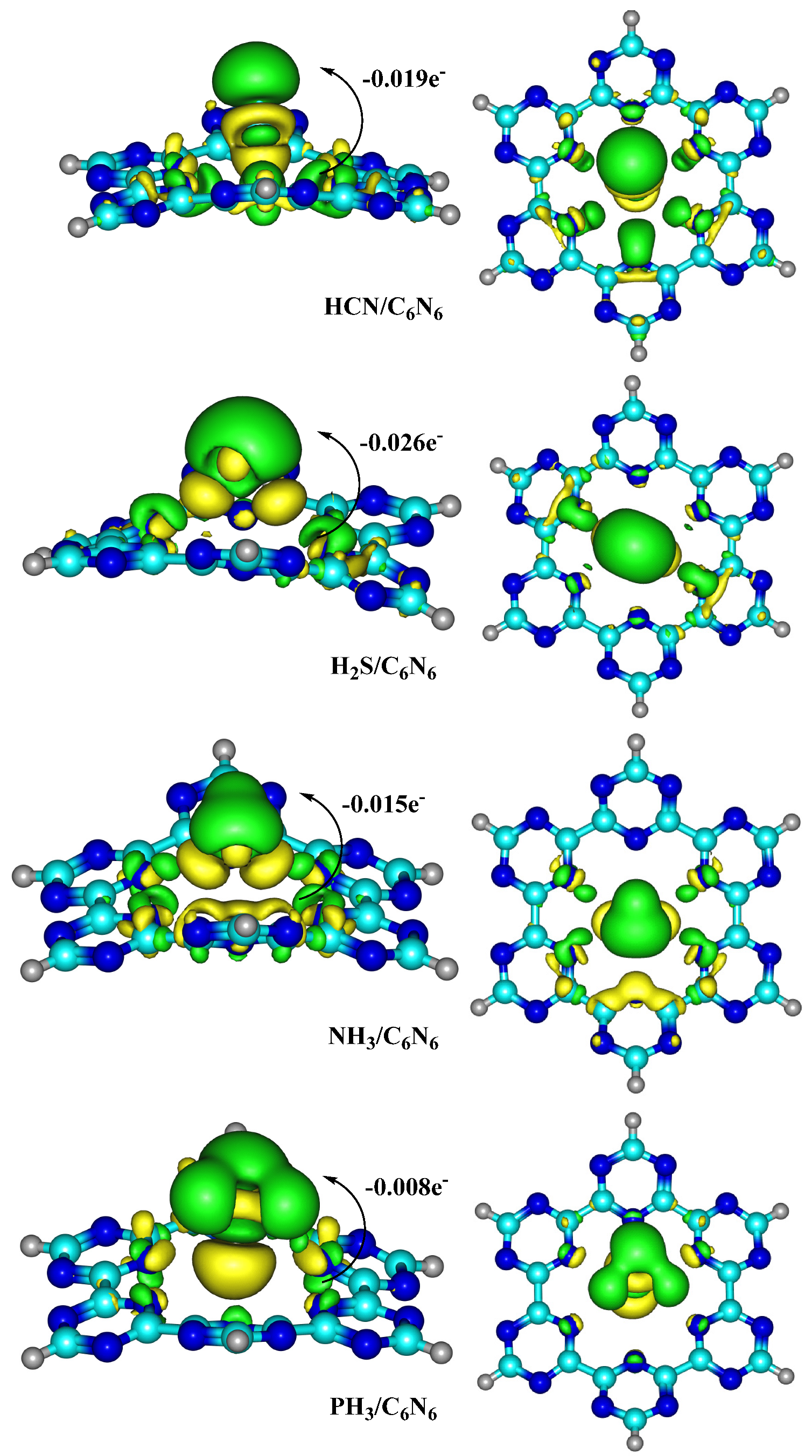
4.1. Natural Bond Orbital (NBO) and Electron Density Differences (EDD) Analyses
4.2. Frontier Molecular Orbital (FMO) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laplaza, R.; Peccati, F.; Boto, R.A.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-García, J. NCIPLOT and the Analysis of Noncovalent Interactions Using the Reduced Density Gradient. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1497. [Google Scholar] [CrossRef]
- Wang, Y.; Ning, P.; Zhao, R.; Li, K.; Wang, C.; Sun, X.; Song, X.; Lin, Q. A Cu-Modified Active Carbon Fiber Significantly Promoted H2S and PH3 Simultaneous Removal at a Low Reaction Temperature. Front. Environ. Sci. Eng. 2021, 15, 132. [Google Scholar] [CrossRef]
- Haridas, V.; Sukhananazerin, A.; Sneha, J.M.; Pullithadathil, B.; Narayanan, B. α-Fe2O3 Loaded Less-Defective Graphene Sheets as Chemiresistive Gas Sensor for Selective Sensing of NH3. Appl. Surf. Sci. 2020, 517, 146158. [Google Scholar] [CrossRef]
- Farooqi, B.A.; Yar, M.; Ashraf, A.; Farooq, U.; Ayub, K. Polyaniline emeraldine salt as selective electrochemical sensor for HBr over HCl: A systematic density functional theory study through oligomer approach. J. Mol. Model. 2020, 26, 332. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Yar, M.; Kosar, N.; Ayub, K.; Arshad, M.; Zahid, N.; Mahmood, T. First-principles study for exploring the adsorption behavior of G-series nerve agents on graphdyine surface. Comput. Theor. Chem. 2020, 1191, 113043. [Google Scholar] [CrossRef]
- Matthews, C.N. Dark Matter in the Solar System: Hydrogen Cyanide Polymers. Orig. Life Evol. Biosph. 1991, 21, 421–434. [Google Scholar] [CrossRef]
- Ding, Y.; Li, X.; Chen, C.; Ling, J.; Li, W.; Guo, Y.; Yan, J.; Zha, L.; Cai, J. A Rapid Evaluation of Acute Hydrogen Sulfide Poisoning in Blood Based on DNA-Cu/Ag Nanocluster Fluorescence Probe. Sci. Rep. 2017, 7, 9638. [Google Scholar] [CrossRef]
- Lemus, J.; Bedia, J.; Moya, C.; Alonso-Morales, N.; Gilarranz, M.A.; Palomar, J.; Rodriguez, J.J. Ammonia Capture from the Gas Phase by Encapsulated Ionic Liquids (ENILs). RSC Adv. 2016, 6, 61650–61660. [Google Scholar] [CrossRef]
- Bai, Z.; Dong, Y.; Wang, Z.; Zhu, T. Emission of Ammonia from Indoor Concrete Wall and Assessment of Human Exposure. Environ. Int. 2006, 32, 303–311. [Google Scholar] [CrossRef]
- Nath, N.S.; Bhattacharya, I.; Tuck, A.G.; Schlipalius, D.I.; Ebert, P.R. Mechanisms of Phosphine Toxicity. J. Toxicol. 2011, 2011, 494168. [Google Scholar] [CrossRef]
- Zhou, X.; Su, Z.; Chen, H.; Xiao, X.; Qin, Y.; Yang, L.; Yan, Z.; Sun, W. Capture of Pure Toxic Gases through Porous Materials from Molecular Simulations. Mol. Phys. 2018, 116, 2095–2107. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective Gas Adsorption and Separation in Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1477. [Google Scholar] [CrossRef] [PubMed]
- Yar, M.; Ahsan, F.; Gulzar, A.; Ayub, K. Adsorption and Sensor Applications of C2N Surface for G-Series and Mustard Series Chemical Warfare Agents. Microporous Mesoporous Mater. 2021, 317, 110984. [Google Scholar] [CrossRef]
- He, Y.; Li, D.; Gao, W.; Yin, H.; Chen, F.; Sun, Y. High-Performance NO2 Sensors based on Spontaneously Functionalized Hexagonal Boron Nitride Nanosheets via Chemical Exfoliation. Nanoscale 2019, 11, 21909–21916. [Google Scholar] [CrossRef]
- Wang, C.; Yin, L.; Zhang, L.; Xiang, D.; Gao, R. Metal Oxide Gas Sensors: Sensitivity and Influencing Factors. Sensors 2010, 10, 2088–2106. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Hupp, J.T. Metal−Organic Frameworks as Sensors: A ZIF-8 Based Fabry−Pérot Device as a Selective Sensor for Chemical Vapors and Gases. J. Am. Chem. Soc. 2010, 132, 7832–7833. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Effects of Functionalization, Catenation, and Variation of the Metal Oxide and Organic Linking Units on the Low-Pressure Hydrogen Adsorption Properties of Metal−Organic Frameworks. J. Am. Chem. Soc. 2006, 128, 1304–1315. [Google Scholar] [CrossRef]
- Rostami, Z.; Soleymanabadi, H. Investigation of Phosgene Adsorption Behavior on Aluminum Nitride Nanocones: Density Functional Study. J. Mol. Liq. 2017, 248, 473–478. [Google Scholar] [CrossRef]
- Deng, Z.-Y.; Zhang, J.-M.; Xu, K.-W. Adsorption of SO2 Molecule on Doped (8, 0) Boron Nitride Nanotube: A First-Principles Study. Phys. E Low Dimens. Syst. Nanostruct. 2016, 76, 47–51. [Google Scholar] [CrossRef]
- Broitman, E.; Gueorguiev, G.K.; Furlan, A.; Son, N.T.; Gellman, A.J.; Stafström, S.; Hultman, L. Water Adsorption on Fullerene-like Carbon Nitride Overcoats. Thin Solid Films 2008, 517, 1106–1110. [Google Scholar] [CrossRef]
- Yuan, J.; Li, G.; Yang, B.; Zhang, J.; Li, Z.; Chen, H. Selective Adsorption of Ethylene on Bimetallic CuVn+/0 (n = 1–5) Clusters: A Theoretical Study. Comput. Mater. Sci. 2016, 111, 489–496. [Google Scholar] [CrossRef]
- Broitman, E.; Furlan, A.; Gueorguiev, G.K.; Czigány, Z.; Tarditi, A.M.; Gellman, A.J.; Stafström, S.; Hultman, L. Water Adsorption on Phosphorous-Carbide Thin Films. Surf. Coat. Technol. 2009, 204, 1035–1039. [Google Scholar] [CrossRef]
- Korotcenkov, G. Metal Oxides for Solid-State Gas Sensors: What Determines Our Choice? Mater. Sci. Eng. B 2007, 139, 1–23. [Google Scholar] [CrossRef]
- Sahoo, H.R.; Kralj, J.G.; Jensen, K.F. Multistep Continuous-Flow Microchemical Synthesis Involving Multiple Reactions and Separations. Angew. Chemie Int. Ed. 2007, 46, 5704–5708. [Google Scholar] [CrossRef] [PubMed]
- Sangiovanni, D.G.; Gueorguiev, G.K.; Kakanakova-Georgieva, A. Ab Initio Molecular Dynamics of Atomic-Scale Surface Reactions: Insights into Metal Organic Chemical Vapor Deposition of AlN on Graphene. Phys. Chem. Chem. Phys. 2018, 20, 17751–17761. [Google Scholar] [CrossRef]
- Anota, E.C.; Juárez, A.R.; Castro, M.; Cocoletzi, H.H. A Density Functional Theory Analysis for the Adsorption of the Amine Group on Graphene and Boron Nitride Nanosheets. J. Mol. Model. 2013, 19, 321–328. [Google Scholar] [CrossRef]
- Limon, P.; Miralrio, A.; Castro, M. Adsorption and Dissociation of Carbon Monoxide on Iron and Iron-Carbon Clusters: Fen + 2CO and FenC + 2CO, N = 4 and 7. A Theoretical Study. Comput. Theor. Chem. 2018, 1129, 37–47. [Google Scholar] [CrossRef]
- Online, V.A. RSC Advances Long Range Corrected-WPBE Based Analysis of the H2O Adsorption on Magnetic BC3 Nanosheets. RSC Adv. 2016, 6, 20409–20421. [Google Scholar] [CrossRef]
- Baughman, R.H. Carbon Nanotubes—The Route Toward Applications. Science 2002, 297, 787–792. [Google Scholar] [CrossRef]
- Rafiee, M.A.; Rafiee, J.; Wang, Z.; Song, H.; Yu, Z.-Z.; Koratkar, N. Enhanced Mechanical Properties of Nanocomposites at Low Graphene Content. ACS Nano 2009, 3, 3884–3890. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, C.; Wang, Y.; Qiu, L.; Li, D. Liquid-Mediated Dense Integration of Graphene Materials for Compact Capacitive Energy Storage. Science 2013, 341, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Liu, Q.; Tang, K.; Zheng, J.; Zhou, J.; Qin, R.; Gao, Z.; Yu, D.; Lu, J. Tunable Bandgap in Silicene and Germanene. Nano Lett. 2012, 12, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liang, T.; Shi, M.; Chen, H. Graphene-Like Two-Dimensional Materials. Chem. Rev. 2013, 113, 3766–3798. [Google Scholar] [CrossRef]
- Vogt, P.; De Padova, P.; Quaresima, C.; Avila, J.; Frantzeskakis, E.; Asensio, M.C.; Resta, A.; Ealet, B.; Le Lay, G. Silicene: Compelling Experimental Evidence for Graphenelike Two-Dimensional Silicon. Phys. Rev. Lett. 2012, 108, 155501. [Google Scholar] [CrossRef] [PubMed]
- Jose, D.; Datta, A. Structures and Chemical Properties of Silicene: Unlike Graphene. Acc. Chem. Res. 2014, 47, 593–602. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef]
- Meric, I.; Han, M.Y.; Young, A.F.; Ozyilmaz, B.; Kim, P.; Shepard, K.L. Current Saturation in Zero-Bandgap, Top-Gated Graphene Field-Effect Transistors. Nat. Nanotechnol. 2008, 3, 654–659. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Chen, Y.-B.; Zhou, K.-G.; Liu, C.-H.; Zeng, J.; Zhang, H.-L.; Peng, Y. Improving Gas Sensing Properties of Graphene by Introducing Dopants and Defects: A First-Principles Study. Nanotechnology 2009, 20, 185504. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Routh, P.; Kim, D.-H.; Huang, W.; Chen, P. Heteroatom-Doped Graphene Materials: Syntheses, Properties and Applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, C.-T.; Liu, K.-K.; Tite, T.; Su, C.-Y.; Chang, C.-H.; Lee, Y.-H.; Chu, C.-W.; Wei, K.-H.; Kuo, J.-L.; et al. Opening an Electrical Band Gap of Bilayer Graphene with Molecular Doping. ACS Nano 2011, 5, 7517–7524. [Google Scholar] [CrossRef]
- Cote, A.P. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef]
- Ma, H.; Liu, B.; Li, B.; Zhang, L.; Li, Y.-G.; Tan, H.-Q.; Zang, H.-Y.; Zhu, G. Cationic Covalent Organic Frameworks: A Simple Platform of Anionic Exchange for Porosity Tuning and Proton Conduction. J. Am. Chem. Soc. 2016, 138, 5897–5903. [Google Scholar] [CrossRef]
- Ning, G.-H.; Liu, C.; Loh, K.P.; Leng, K.; Tang, W.; Gao, Q.; Xu, H.-S.; Tian, B.; Li, X. Highly Photoluminescent Two-Dimensional Imine-Based Covalent Organic Frameworks for Chemical Sensing. Chem. Commun. 2018, 54, 2349–2352. [Google Scholar] [CrossRef]
- Zhu, M.W.; Xu, S.Q.; Wang, X.Z.; Chen, Y.; Dai, L.; Zhao, X. The Construction of Fluorescent Heteropore Covalent Organic Frameworks and Their Applications in Spectroscopic and Visual Detection of Trinitrophenol with High Selectivity and Sensitivity. Chem. Commun. 2018, 54, 2308–2311. [Google Scholar] [CrossRef]
- Cui, F.; Liang, R.; Qi, Q.; Jiang, G.; Zhao, X. Efficient Removal of Cr(VI) from Aqueous Solutions by a Dual-Pore Covalent Organic Framework. Adv. Sustain. Syst. 2019, 3, 1800150. [Google Scholar] [CrossRef]
- Jansone-Popova, S.; Moinel, A.; Schott, J.A.; Mahurin, S.M.; Popovs, I.; Veith, G.M.; Moyer, B.A. Guanidinium-Based Ionic Covalent Organic Framework for Rapid and Selective Removal of Toxic Cr(VI) Oxoanions from Water. Environ. Sci. Technol. 2019, 53, 878–883. [Google Scholar] [CrossRef]
- Jiang, Q.; Huang, H.; Tang, Y.; Zhang, Y.; Zhong, C. Highly Porous Covalent Triazine Frameworks for Reversible Iodine Capture and Efficient Removal of Dye. Ind. Eng. Chem. Res. 2018, 57, 15114–15121. [Google Scholar] [CrossRef]
- Fayyaz, F.; Yar, M.; Gulzar, A.; Ayub, K. First principles calculations of the adsorption of fluorouracil and nitrosourea on CTF-0; organic frameworks as drug delivery systems for cancer treatment. J. Mol. Liq. 2022, 356, 118941. [Google Scholar] [CrossRef]
- Liao, L.; Li, M.; Yin, Y.; Chen, J.; Zhong, Q.; Du, R.; Liu, S.; He, Y.; Fu, W.; Zeng, F. Advances in the Synthesis of Covalent Triazine Frameworks. ACS Omega 2023, 8, 4527–4542. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Yar, M.; Ans, M.; Gilani, M.A.; Ludwig, R.; Hashmi, M.A.; Hussain, M.; Muhammad, S.; Ayub, K. Computational Investigation of a Covalent Triazine Framework (CTF-0) as an Efficient Electrochemical Sensor. RSC Adv. 2022, 12, 3909–3923. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yuan, W.; Hua, Z.; Tang, Y.; Yin, F.; Xia, D. Triazine-Based Two-Dimensional Organic Polymer for Selective NO2 Sensing with Excellent Performance. ACS Appl. Mater. Interfaces 2020, 12, 3919–3927. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, L.; Jin, S.; Tan, B. Covalent Triazine Frameworks: Synthesis and Applications. J. Mater. Chem. A 2019, 7, 5153–5172. [Google Scholar] [CrossRef]
- Liang, D.; Jing, T.; Ma, Y.; Hao, J.; Sun, G.; Deng, M. Photocatalytic Properties of G-C6N6/g-C3N4 Heterostructure: A Theoretical Study. J. Phys. Chem. C 2016, 120, 24023–24029. [Google Scholar] [CrossRef]
- Zhang, W.X.; Yan, H.M.; He, C. G-C6N6 Monolayer: A Highly Sensitive Molecule Sensor for Biomarker Volatiles of Liver Cirrhosis. Appl. Surf. Sci. 2021, 566, 150716. [Google Scholar] [CrossRef]
- Alkhalifah, M.A.; Yar, M.; Bayach, I.; Sheikh, N.S.; Ayub, K. Covalent Organic Framework (C6N6) as a Drug Delivery Platform for Fluorouracil to Treat Cancerous Cells: A DFT Study. Materials 2022, 15, 7425. [Google Scholar] [CrossRef]
- Bafekry, A.; Stampfl, C.; Akgenc, B.; Mortazavi, B.; Ghergherehchi, M.; Nguyen, C.V. Embedding of Atoms into the Nanopore Sites of the C6N6 and C6N8 Porous Carbon Nitride Monolayers with Tunable Electronic Properties. Phys. Chem. Chem. Phys. 2020, 22, 6418–6433. [Google Scholar] [CrossRef]
- Pagliai, M.; Caporali, S.; Muniz-Miranda, M.; Pratesi, G.; Schettino, V. SERS, XPS, and DFT Study of Adenine Adsorption on Silver and Gold Surfaces. J. Phys. Chem. Lett. 2012, 3, 242–245. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Qi, S.-D.; Niu, X.-Y.; Hu, J.; Ren, C.-L.; Chen, H.-L.; Chen, X.-G. Metallic nanoparticles immobilized in magnetic metal-organic frameworks: Preparation and application as highly active, magnetically isolable and reusable catalysts. Catal. Sci. Technol. 2014, 4, 3013–3024. [Google Scholar] [CrossRef]
- Yar, M.; Ayub, K. Expanding the Horizons of Covalent Organic Frameworks to Electrochemical Sensors; A Case Study of CTF-FUM. Microporous Mesoporous Mater. 2020, 300, 110146. [Google Scholar] [CrossRef]
- Farooqi, B.A.; Yar, M.; Ashraf, A.; Farooq, U.; Ayub, K. Graphene-Polyaniline Composite as Superior Electrochemical Sensor for Detection of Cyano Explosives. Eur. Polym. J. 2020, 138, 109981. [Google Scholar] [CrossRef]
- Yar, M.; Hashmi, M.A.; Ayub, K. Nitrogenated Holey Graphene (C2N) Surface as Highly Selective Electrochemical Sensor for Ammonia. J. Mol. Liq. 2019, 296, 111929. [Google Scholar] [CrossRef]
- Yar, M.; Hashmi, M.A.; Ayub, K. The C2N Surface as a Highly Selective Sensor for the Detection of Nitrogen Iodide from a Mixture of NX3 (X = Cl, Br, I) Explosives. RSC Adv. 2020, 10, 31997–32010. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Tomilin, F.N.; Pozdnyakova, A.A.; Vasilyeva, N.Y.; Malyar, Y.N.; Kuznetsova, S.A.; Avramov, P.V. Theoretical DFT Interpretation of Infrared Spectra of Biologically Active Arabinogalactan Sulphated Derivatives. Chem. Pap. 2020, 74, 4103–4113. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van Der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Pan, S.; Saha, R.; Mandal, S.; Mondal, S.; Gupta, A.; Fernández-Herrera, M.A.; Merino, G.; Chattaraj, P.K. Selectivity in Gas Adsorption by Molecular Cucurbit[6]Uril. J. Phys. Chem. C 2016, 120, 13911–13921. [Google Scholar] [CrossRef]
- Keinan, S.; Contreras-García, J.; Johnson, E.R.; Yang, W.; Mori-Sánchez, P.; Cohen, A.J. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of Hydrogen-Bond Interaction Potentials from the Electron Density: Integration of Noncovalent Interaction Regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A.; Kawazoe, Y. Unravelling the Nature of Binding of Cubane and Substituted Cubanes within Cucurbiturils: A DFT and NCI Study. J. Mol. Liq. 2018, 260, 18–29. [Google Scholar] [CrossRef]
- Mohammadi, M.D.; Abdullah, H.Y. The Adsorption of Chlorofluoromethane on Pristine, Al-, Ga-, P-, and As-Doped Boron Nitride Nanotubes: A PBC-DFT, NBO, and QTAIM Study. ChemistrySelect 2020, 5, 12115–12124. [Google Scholar] [CrossRef]
- Chu, Z.Q.; Stampfl, C.; Duan, X.M. Boron-Doped g-C6N6 Layer as a Metal-Free Photoelectrocatalyst for N2 Reduction Reaction. J. Phys. Chem. C 2019, 123, 28739–28743. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.-L.; Wang, M.-S. Effects of Atypical Hydrogen Bonds and π–π Interactions on Nonlinear Optical Properties: Dimers of Triangular Structures Based on Perylene, Naphthalene, and Pyromellitic Diimides. Langmuir 2022, 39, 357–366. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.; Ahsan, F.; Yar, M.; Ayub, K. Theoretical Investigation of Double-Cubed Polycationic Cluster (Sb7Se8Cl2)3+ for the Storage of Helium and Neon. Mater. Sci. Semicond. Process. 2022, 148, 106756. [Google Scholar] [CrossRef]
- Srinivasu, K.; Modak, B.; Ghosh, S.K. Porous Graphitic Carbon Nitride: A Possible Metal-Free Photocatalyst for Water Splitting. J. Phys. Chem. C 2014, 118, 26479–26484. [Google Scholar] [CrossRef]
- Li, T.; Zhang, W.-D.; Liu, Y.; Li, Y.; Cheng, C.; Zhu, H.; Yan, X.; Li, Z.; Gu, Z.-G. A Two-Dimensional Semiconducting Covalent Organic Framework with Nickel(ii) Coordination for High Capacitive Performance. J. Mater. Chem. A 2019, 7, 19676–19681. [Google Scholar] [CrossRef]
- Trendafilova, N.; Bauer, G.; Mihaylov, T. DFT and AIM Studies of Intramolecular Hydrogen Bonds in Dicoumarols. Chem. Phys. 2004, 302, 95–104. [Google Scholar] [CrossRef]
- Ghogomu, J.N.; Nkungli, N.K. DFT Studies and Topological Analyses of Electron Density on Acetophenone and Propiophenone Thiosemicarbazone Derivatives as Covalent Inhibitors of Falcipain-2, a Major Plasmodium Falciparum Cysteine Protease. Phys. Chem. Res 2017, 5, 795–817. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Solimannejad, M.; Grabowski, S.J. Dihydrogen Bonding vs Metal−σ Interaction in Complexes between H2 and Metal Hydride. J. Phys. Chem. A 2011, 115, 201–210. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chemie Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Theoretical Determination of Molecular Structure and Conformation. 15. Three-Membered Rings: Bent Bonds, Ring Strain, and Surface Delocalization. J. Am. Chem. Soc. 1985, 107, 3800–3810. [Google Scholar] [CrossRef]
- Du, J.; Sun, X.; Jiang, G. Exploring the Interaction Natures in Plutonyl (VI) Complexes with Topological Analyses of Electron Density. Int. J. Mol. Sci. 2016, 17, 414. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Yar, M.; Ayub, K. Covalent Triazine Framework (CTF-0) Surface as a Smart Sensing Material for the Detection of CWAs and Industrial Pollutants. Mater. Sci. Semicond. Process. 2022, 139, 106334. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
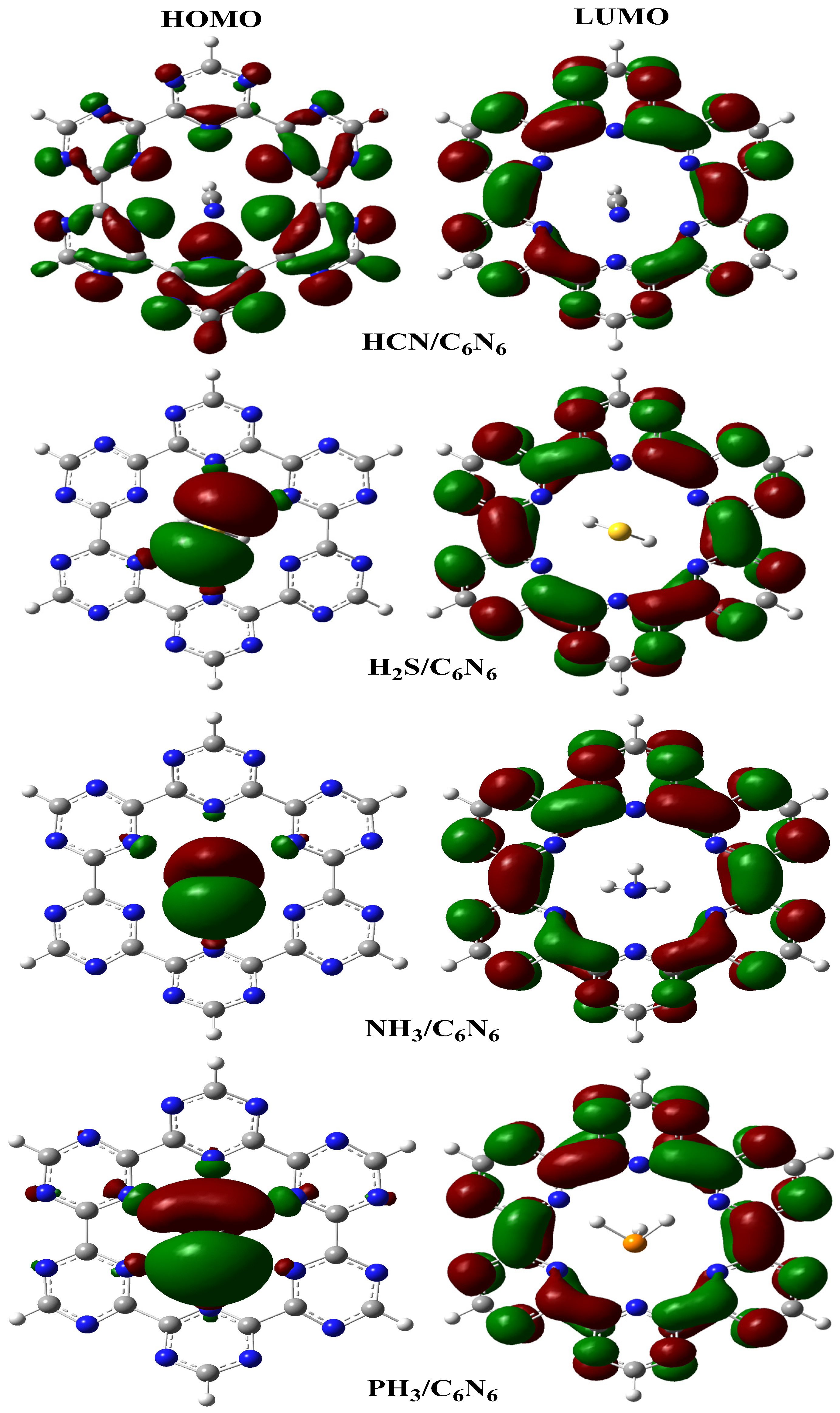{kind=link}
| Analytes@C6N6 | Eexch | Eelst | (%) | Eind | (%) | Edisp | (%) | ESAPT0 |
|---|---|---|---|---|---|---|---|---|
| HCN@C6N6 | 11.16 | −14.60 | (52) | −3.78 | (13) | −9.66 | (34) | −16.88 |
| H2S@C6N6 | 16.93 | −12.58 | (43) | −4.27 | (15) | −12.34 | (42) | −12.26 |
| NH3@C6N6 | 25.29 | −10.78 | (47) | −2.93 | (13) | −9.30 | (40) | −10.06 |
| PH3@C6N6 | 11.77 | −6.85 | (33) | −2.54 | (12) | −11.60 | (55) | −9.22 |
| Analyte@C6N6 | Analyte-C6N6 | ρ | ∇2ρ | G (r) | V (r) | H (r) | V(r)/G(r) | Eint (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| HCN@C6N6 | H7-N1 | 0.00724 | 0.02687 | 0.00540 | −0.00408 | 0.00132 | −0.76 | −1.28 |
| H7-N2 | 0.00706 | 0.02429 | 0.00494 | −0.00381 | 0.00113 | −0.77 | −1.19 | |
| H7-N3 | 0.00780 | 0.02622 | 0.00547 | −0.00439 | 0.00108 | −0.80 | −1.38 | |
| H7-N4 | 0.00735 | 0.02451 | 0.00508 | −0.00404 | 0.00104 | −0.79 | −1.27 | |
| H7-N5 | 0.00659 | 0.02509 | 0.00489 | −0.00352 | 0.00138 | −0.72 | −1.10 | |
| H7-N6 | 0.00814 | 0.02847 | 0.00597 | −0.00483 | 0.00115 | −0.81 | −1.51 | |
| H2S@ C6N6 | H7-N1 | 0.00549 | 0.01895 | 0.00380 | −0.00285 | 0.00000 | −0.75 | −0.90 |
| H7-N2 | 0.01586 | 0.03965 | 0.00992 | −0.00992 | 0.00000 | −1.00 | −3.11 | |
| H8-N3 | 0.00763 | 0.02552 | 0.00536 | −0.00434 | 0.00102 | −0.81 | −1.36 | |
| H8-N4 | 0.00549 | 0.01895 | 0.00379 | −0.00285 | 0.00094 | −0.75 | −0.89 | |
| H8-N5 | 0.01585 | 0.03963 | 0.00991 | −0.00991 | 0.00000 | −1.00 | −3.11 | |
| H7-N6 | 0.00763 | 0.02552 | 0.00536 | −0.00434 | 0.00102 | −0.81 | −1.36 | |
| NH3@C6N6 | N8-N1 | 0.00773 | 0.02558 | 0.00573 | −0.00506 | 0.00067 | −0.88 | −1.59 |
| H7-N2 | 0.01365 | 0.04227 | 0.00991 | −0.00925 | 0.00066 | −0.93 | −2.90 | |
| H7-N3 | 0.00867 | 0.02856 | 0.00627 | −0.00540 | 0.00087 | −0.86 | −1.69 | |
| N8-N4 | 0.00357 | 0.01222 | 0.00261 | −0.00217 | 0.00044 | −0.83 | −0.68 | |
| H9-N5 | 0.00874 | 0.02871 | 0.00631 | −0.00545 | 0.00087 | −0.86 | −1.71 | |
| H9-N6 | 0.01360 | 0.04224 | 0.00989 | −0.00922 | 0.00067 | −0.93 | −2.89 | |
| PH3@C6N6 | P7-N1 | 0.00687 | 0.02064 | 0.00445 | −0.00374 | 0.00071 | −0.84 | −1.17 |
| H8-N2 | 0.00562 | 0.01924 | 0.00390 | −0.00300 | 0.00091 | −0.77 | −0.94 | |
| H8-N3 | 0.00673 | 0.02215 | 0.00456 | −0.00358 | 0.00098 | −0.79 | −1.12 | |
| H8-N4 | 0.00730 | 0.02305 | 0.00482 | −0.00388 | 0.00094 | −0.80 | −1.22 | |
| H8-N5 | 0.00601 | 0.02070 | 0.00416 | −0.00315 | 0.00101 | −0.76 | −0.99 | |
| H7-N6 | 0.00625 | 0.02024 | 0.00435 | −0.00365 | 0.00071 | −0.84 | −1.14 |
| Analyte@C6N6 | HOMO (eV) | LUMO (eV) | Gap |
|---|---|---|---|
| C6N6 | −7.16 | −3.30 | 3.86 |
| HCN@C6N6 | −7.43 | −3.41 | 4.02 |
| H2S@C6N6 | −6.07 | −3.31 | 2.76 |
| NH3@C6N6 | −5.85 | −3.27 | 2.58 |
| PH3@C6N6 | −6.62 | −3.27 | 3.35 |
| S.No | Complexes | Eads (kcal/mol) | Ref | S.No | Complexes | Eads (kcal/mol) | Ref |
|---|---|---|---|---|---|---|---|
| 1 | NH3@CTF-FUM | −5.27 | [5] | 8 | HCN@CTF-0 | −5.99 | [86] |
| 2 | H2S@CTF-FUM | −3.79 | 9 | H2S@CTF-0 | −5.46 | ||
| 3 | PH3@CTF-FUM | −2.43 | 10 | NH3@CTF-0 | −6.65 | ||
| 4 | HCN@C2N | −15.24 | HCN@C6N6 | −16.46 | current work | ||
| 5 | NH3@C2N | −10.68 | PH3@C6N6 | −13.64 | |||
| 6 | H2S@C2N | −8.54 | NH3@C6N6 | −12.27 | |||
| 7 | PH3@C2N | −6.91 | PH3@C6N6 | −9.36 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammud, H.H.; Yar, M.; Bayach, I.; Ayub, K. Covalent Triazine Framework C6N6 as an Electrochemical Sensor for Hydrogen-Containing Industrial Pollutants. A DFT Study. Nanomaterials 2023, 13, 1121. https://doi.org/10.3390/nano13061121
Hammud HH, Yar M, Bayach I, Ayub K. Covalent Triazine Framework C6N6 as an Electrochemical Sensor for Hydrogen-Containing Industrial Pollutants. A DFT Study. Nanomaterials. 2023; 13(6):1121. https://doi.org/10.3390/nano13061121
Chicago/Turabian StyleHammud, Hassan H., Muhammad Yar, Imene Bayach, and Khurshid Ayub. 2023. "Covalent Triazine Framework C6N6 as an Electrochemical Sensor for Hydrogen-Containing Industrial Pollutants. A DFT Study" Nanomaterials 13, no. 6: 1121. https://doi.org/10.3390/nano13061121
APA StyleHammud, H. H., Yar, M., Bayach, I., & Ayub, K. (2023). Covalent Triazine Framework C6N6 as an Electrochemical Sensor for Hydrogen-Containing Industrial Pollutants. A DFT Study. Nanomaterials, 13(6), 1121. https://doi.org/10.3390/nano13061121