

Towards Unbiased Fluorophore Counting in Superresolution Fluorescence Microscopy
 and
and
Abstract
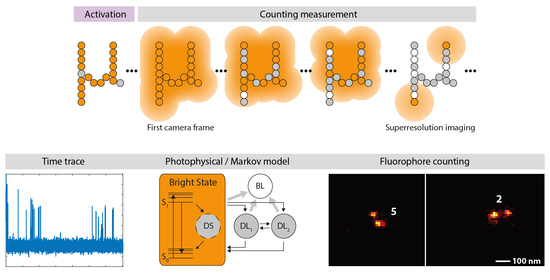
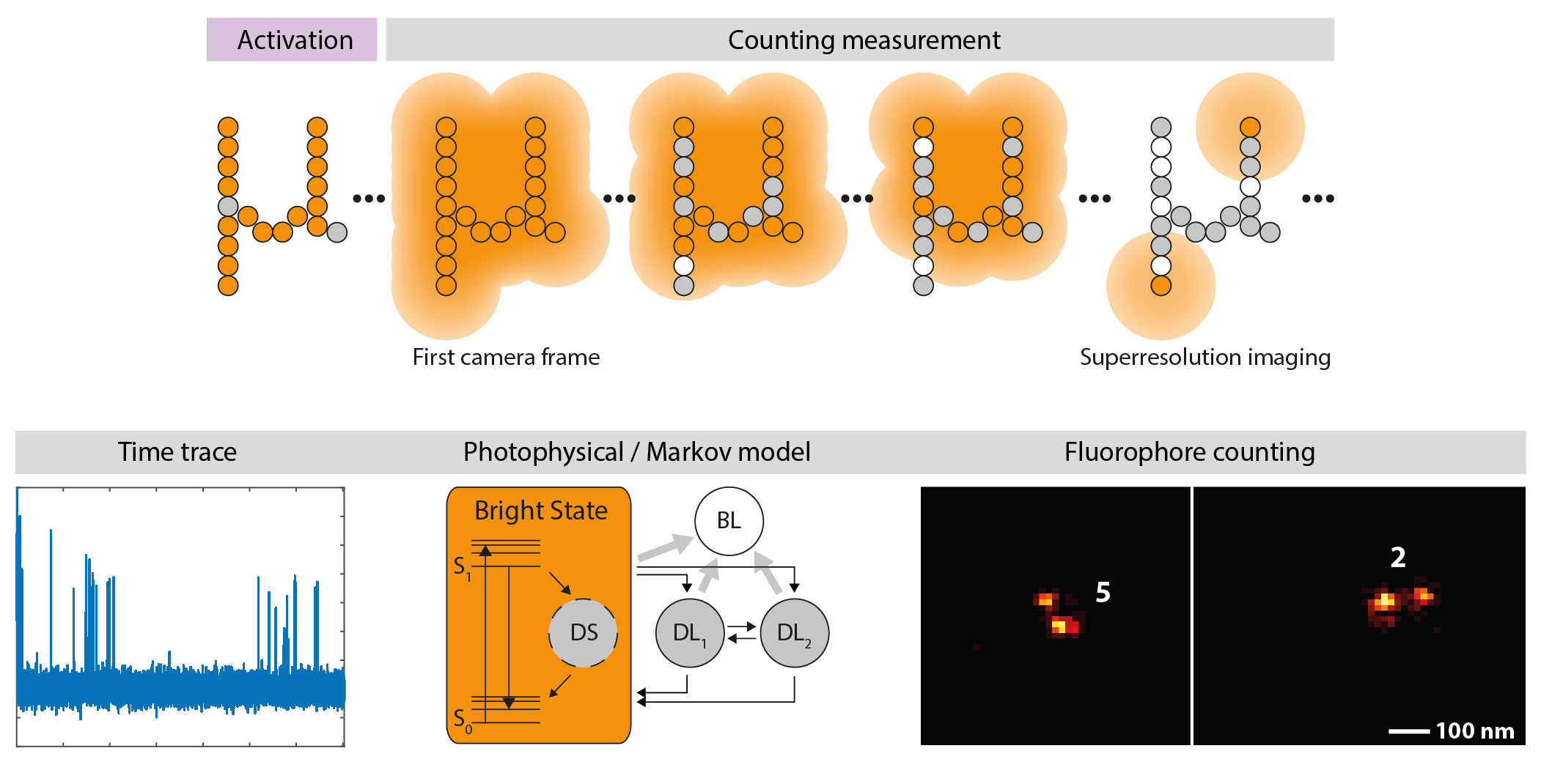{kind=link}
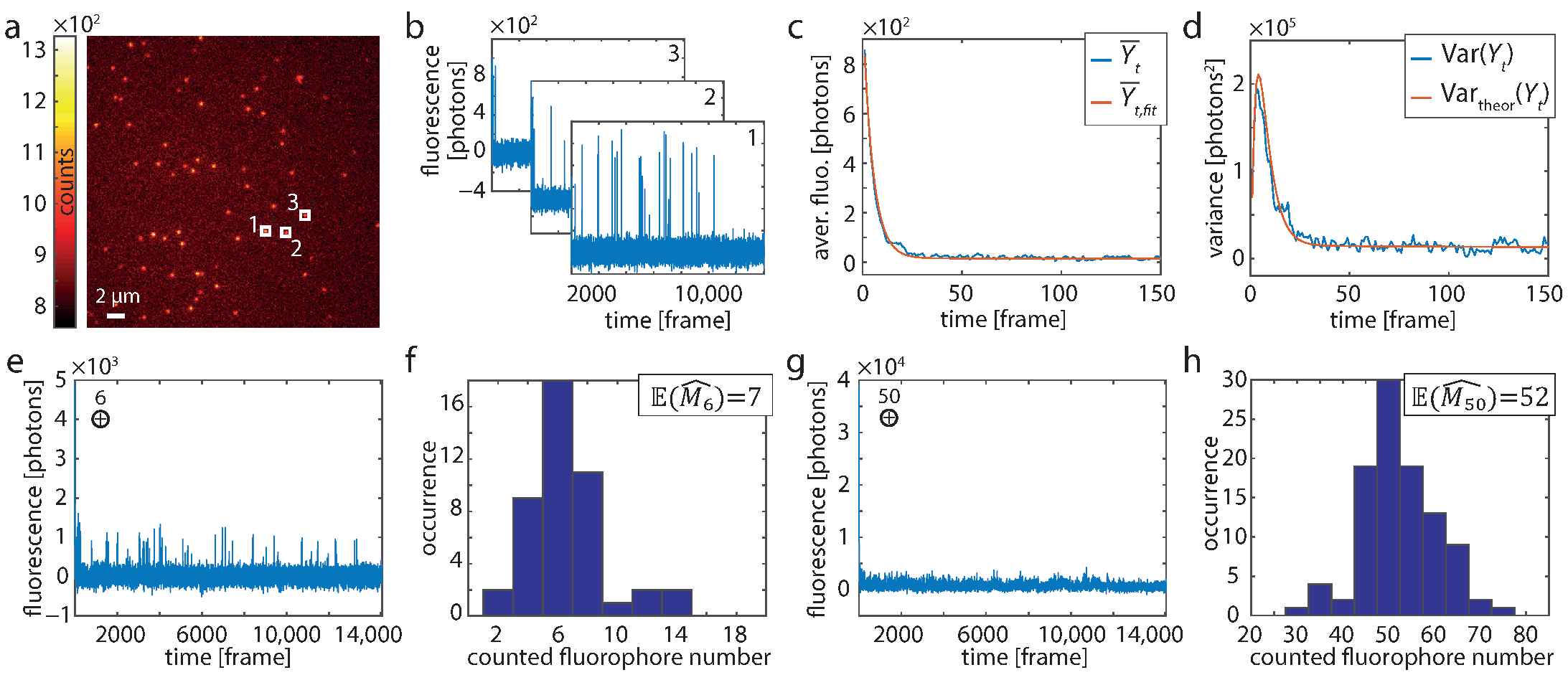{kind=link}
{kind=link}
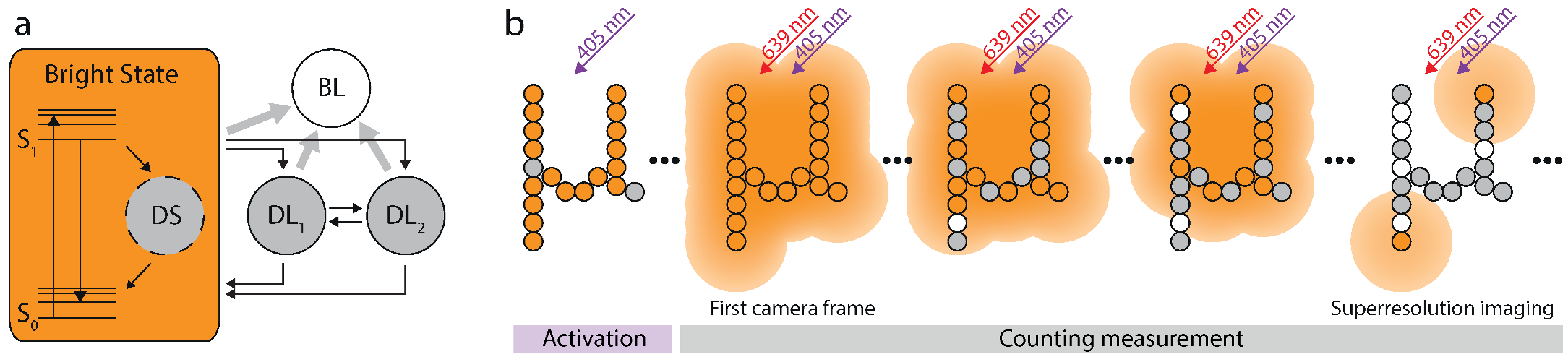{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Fluorescence Microscope
2.2. Sample Preparation
2.3. Imaging Buffer
2.4. Measurement Protocol for Fluorophore Counting
2.5. Data Correction and Analysis
2.6. Number of Dark States Verification
2.7. Excess Relative Variance
2.8. Fitting Averaged Fluorescence Traces
2.9. Pseudo Log-Likelihood Estimator
2.10. Simplified Estimator
3. Results
3.1. Hidden Two-Timescales Markov Model
3.2. Model for Alexa 647 and Single-Molecule Experiments
3.3. Counting under Real Conditions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giepmans, B.N.G.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. The Fluorescent Toolbox for Assessing Protein Location and Function. Science 2006, 312, 217–224. [Google Scholar] [CrossRef]
- Hess, S.T.; Huang, S.; Heikal, A.A.; Webb, W.W. Biological and Chemical Applications of Fluorescence Correlation Spectroscopy: A Review. Biochemistry 2002, 41, 697–705. [Google Scholar] [CrossRef]
- Manzo, C.; Garcia-Parajo, M.F. A review of progress in single particle tracking: From methods to biophysical insights. Rep. Prog. Phys. 2015, 78, 124601. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar] [CrossRef]
- Dong, C.Y.; French, T.; So, P.T.C.; Buehler, C.; Berland, K.M.; Gratton, E. Fluorescence-Lifetime Imaging Techniques for Microscopy. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 431–464. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [Google Scholar] [CrossRef]
- Esposito, A.; Schlachter, S.; Schierle, G.S.K.; Elder, A.D.; Diaspro, A.; Wouters, F.S.; Kaminski, C.F.; Iliev, A.I. Quantitative Fluorescence Microscopy Techniques. In Cytoskeleton Methods and Protocols; Gavin, R.H., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 117–142. [Google Scholar] [CrossRef]
- Erlemann, S.; Neuner, A.; Gombos, L.; Gibeaux, R.; Antony, C.; Schiebel, E. An extended γ-tubulin ring functions as a stable platform in microtubule nucleation. J. Cell Biol. 2012, 197, 59–74. [Google Scholar] [CrossRef]
- Reyes-Lamothe, R.; Sherratt, D.J.; Leake, M.C. Stoichiometry and Architecture of Active DNA Replication Machinery in Escherichia coli. Science 2010, 328, 498–501. [Google Scholar] [CrossRef]
- Yano, M.; Ono, K.; Ohkusa, T.; Suetsugu, M.; Kohno, M.; Hisaoka, T.; Kobayashi, S.; Hisamatsu, Y.; Yamamoto, T.; Kohno, M.; et al. Altered Stoichiometry of FKBP12.6 Versus Ryanodine Receptor as a Cause of Abnormal Ca2+ Leak Through Ryanodine Receptor in Heart Failure. Circulation 2000, 102, 2131–2136. [Google Scholar] [CrossRef]
- Morin, T.J.; Kobertz, W.R. Counting membrane-embedded KCNE β-subunits in functioning K+ channel complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 1478–1482. [Google Scholar] [CrossRef]
- Osteen, J.D.; Sampson, K.J.; Kass, R.S. The cardiac IKs channel, complex indeed. Proc. Natl. Acad. Sci. USA 2010, 107, 18751–18752. [Google Scholar] [CrossRef]
- Nakajo, K.; Ulbrich, M.H.; Kubo, Y.; Isacoff, E.Y. Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc. Natl. Acad. Sci. USA 2010, 107, 18862–18867. [Google Scholar] [CrossRef]
- Coffman, V.C.; Wu, J.Q. Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem. Sci. 2012, 37, 499–506. [Google Scholar] [CrossRef]
- Hell, S.W. Microscopy and its focal switch. Nat. Methods 2009, 6, 24–32. [Google Scholar] [CrossRef]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef]
- Aspelmeier, T.; Egner, A.; Munk, A. Modern Statistical Challenges in High-Resolution Fluorescence Microscopy. Annu. Rev. Stat. Its Appl. 2015, 2, 163–202. [Google Scholar] [CrossRef]
- Klar, T.A.; Engel, E.; Hell, S.W. Breaking Abbe’s diffraction resolution limit in fluorescence microscopy with stimulated emission depletion beams of various shapes. Phys. Rev. E 2001, 64. [Google Scholar] [CrossRef]
- Ta, H.; Keller, J.; Haltmeier, M.; Saka, S.K.; Schmied, J.; Opazo, F.; Tinnefeld, P.; Munk, A.; Hell, S.W. Mapping molecules in scanning far-field fluorescence nanoscopy. Nat. Commun. 2015, 6, 7977. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2013, 331, 1642–1645. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef]
- Egner, A.; Geisler, C.; von Middendorff, C.; Bock, H.; Wenzel, D.; Medda, R.; Andresen, M.; Stiel, A.C.; Jakobs, S.; Eggeling, C.; et al. Fluorescence Nanoscopy in Whole Cells by Asynchronous Localization of Photoswitching Emitters. Biophys. J. 2007, 93, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; Wurm, C.A.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 2008, 5, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, J.Y.; Lee, A.; Bustamante, C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM). Proc. Natl. Acad. Sci. USA 2012, 109, 17436–17441. [Google Scholar] [CrossRef]
- Annibale, P.; Vanni, S.; Scarselli, M.; Rothlisberger, U.; Radenovic, A. Quantitative Photo Activated Localization Microscopy: Unraveling the Effects of Photoblinking. PLoS ONE 2011, 6, 17436–17441. [Google Scholar] [CrossRef]
- Rollins, G.C.; Shin, J.Y.; Bustamante, C.; Pressé, S. Stochastic approach to the molecular counting problem in superresolution microscopy. Proc. Natl. Acad. Sci. USA 2014, 112, E110–E118. [Google Scholar] [CrossRef]
- Karathanasis, C.; Fricke, F.; Hummer, G.; Heilemann, M. Molecule Counts in Localization Microscopy with Organic Fluorophores. ChemPhysChem 2017, 18, 942–948. [Google Scholar] [CrossRef]
- Hummer, G.; Fricke, F.; Heilemann, M. Model-independent counting of molecules in single-molecule localization microscopy. Mol. Biol. Cell 2016, 27, 3637–3644. [Google Scholar] [CrossRef]
- Staudt, T.; Aspelmeier, T.; Laitenberger, O.; Geisler, C.; Egner, A.; Munk, A. Statistical Molecule Counting in Super-Resolution Fluorescence Microscopy: Towards Quantitative Nanoscopy. Stat. Sci. 2020, 35, 92–111. [Google Scholar] [CrossRef]
- Schmied, J.J.; Raab, M.; Forthmann, C.; Pibiri, E.; Wünsch, B.; Dammeyer, T.; Tinnefeld, P. DNA origami–based standards for quantitative fluorescence microscopy. Nat. Protoc. 2014, 9, 1367–1391. [Google Scholar] [CrossRef]
- Dempsey, G.T.; Vaughan, J.C.; Chen, K.H.; Bates, M.; Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 2011, 8, 1027–1036. [Google Scholar] [CrossRef]
- Hirsch, M.; Wareham, R.J.; Martin-Fernandez, M.L.; Hobson, M.P.; Rolfe, D.J. A Stochastic Model for Electron Multiplication Charge-Coupled Devices—From Theory to Practice. PLoS ONE 2013, 8, e53671. [Google Scholar] [CrossRef]
- Dempsey, G.T.; Bates, M.; Kowtoniuk, W.E.; Liu, D.R.; Tsien, R.Y.; Zhuang, X. Photoswitching Mechanism of Cyanine Dyes. J. Am. Chem. Soc. 2009, 51, 18192–18193. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laitenberger, O.; Aspelmeier, T.; Staudt, T.; Geisler, C.; Munk, A.; Egner, A. Towards Unbiased Fluorophore Counting in Superresolution Fluorescence Microscopy. Nanomaterials 2023, 13, 459. https://doi.org/10.3390/nano13030459
Laitenberger O, Aspelmeier T, Staudt T, Geisler C, Munk A, Egner A. Towards Unbiased Fluorophore Counting in Superresolution Fluorescence Microscopy. Nanomaterials. 2023; 13(3):459. https://doi.org/10.3390/nano13030459
Chicago/Turabian StyleLaitenberger, Oskar, Timo Aspelmeier, Thomas Staudt, Claudia Geisler, Axel Munk, and Alexander Egner. 2023. "Towards Unbiased Fluorophore Counting in Superresolution Fluorescence Microscopy" Nanomaterials 13, no. 3: 459. https://doi.org/10.3390/nano13030459
APA StyleLaitenberger, O., Aspelmeier, T., Staudt, T., Geisler, C., Munk, A., & Egner, A. (2023). Towards Unbiased Fluorophore Counting in Superresolution Fluorescence Microscopy. Nanomaterials, 13(3), 459. https://doi.org/10.3390/nano13030459