
Theoretical Investigation of Delafossite-Cu2ZnSnO4 as a Promising Photovoltaic Absorber
Abstract
:1. Introduction
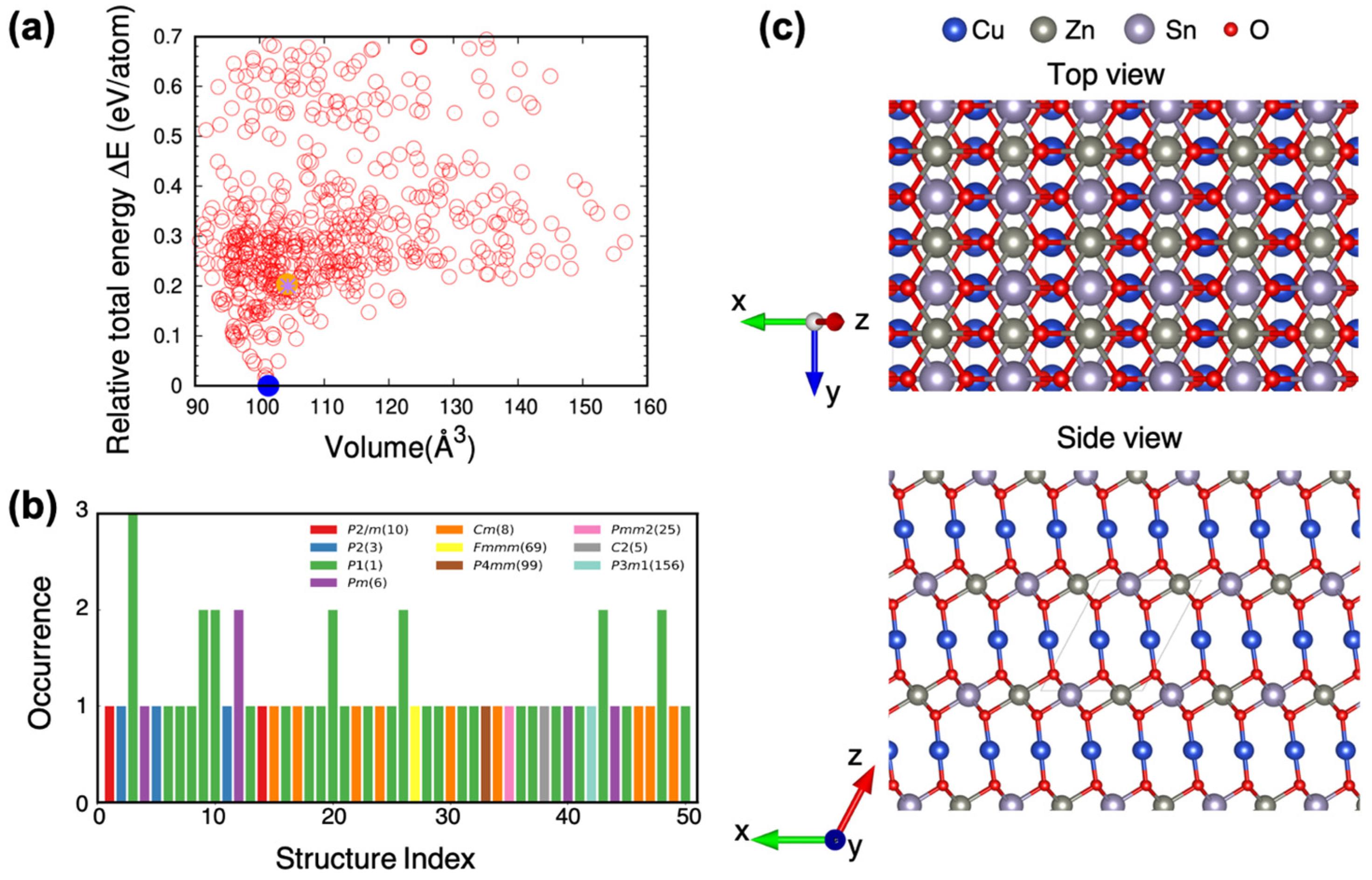
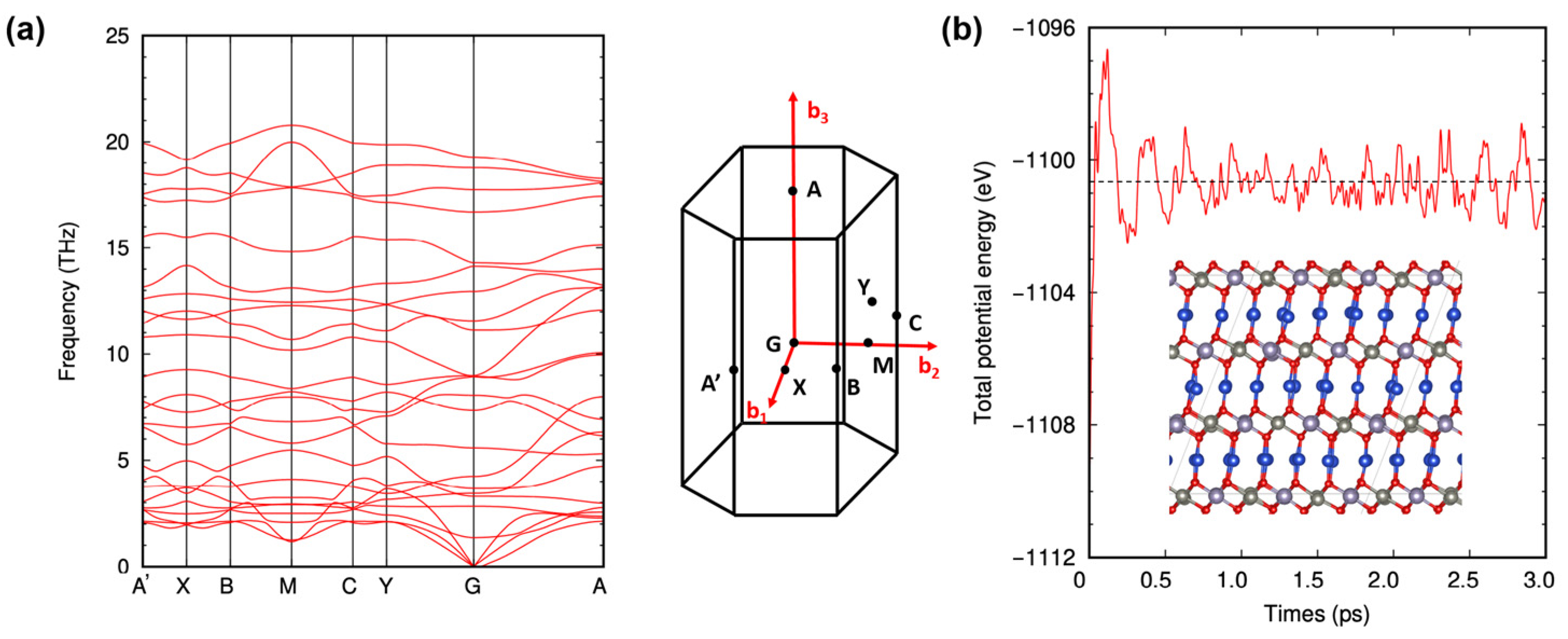
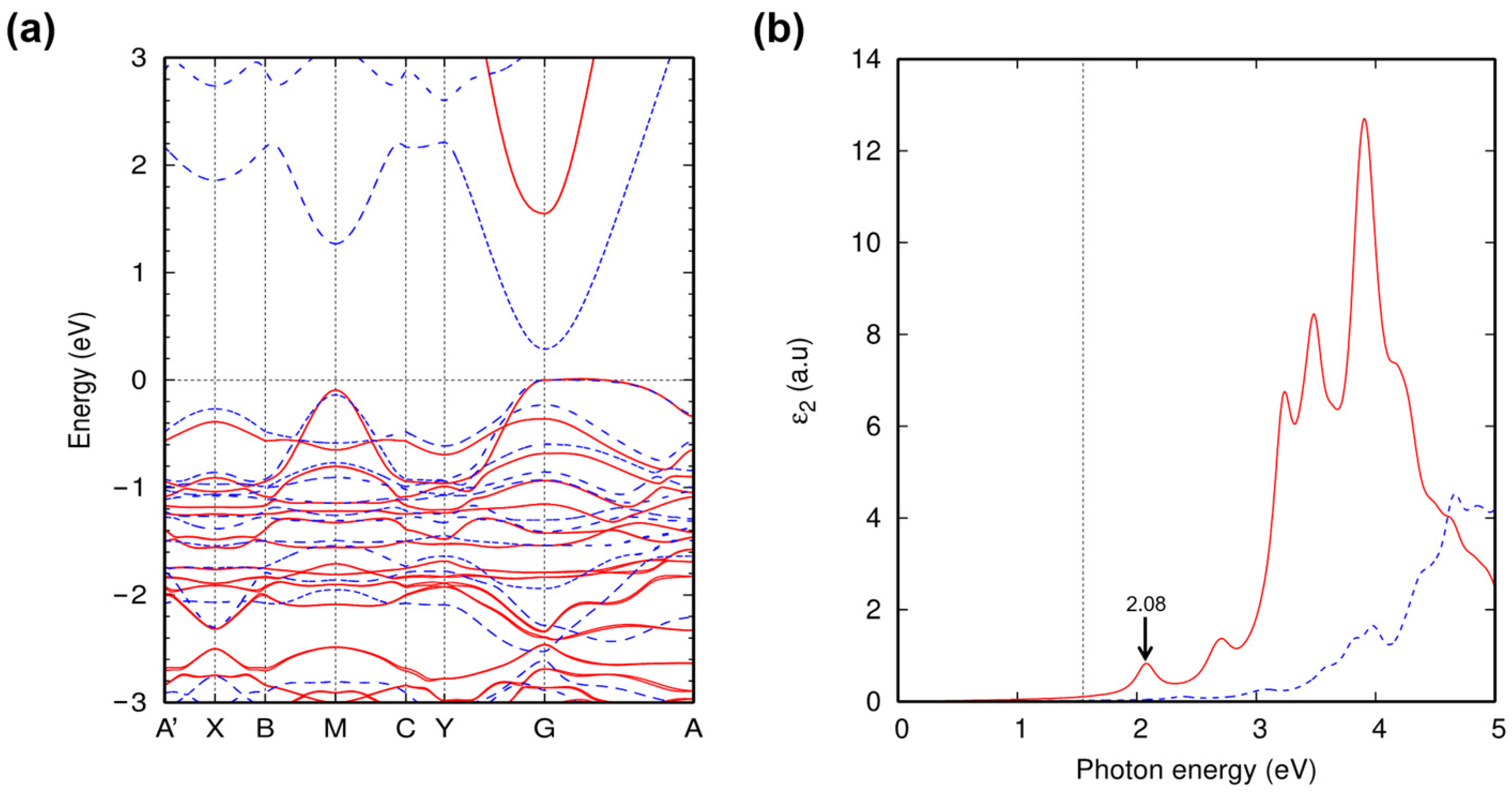
2. Results
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Crystal Prediction Calculations
Appendix A.2. Ab Initio Density Functional Theory (DFT) Calculations
Appendix A.3. Synthesis and Characterization
References
- Polman, A.; Knight, M.; Garnett, E.C.; Ehrler, B.; Sinke, W.C. Photovoltaic materials: Present efficiencies and future challenges. Science 2016, 352, aad4424. [Google Scholar] [CrossRef]
- Kayes, B.M.; Nie, H.; Twist, R.; Spruytte, S.G.; Reinhardt, F.; Kizilyalli, I.C.; Higashi, G.S. 27.6% conversion efficiency, a new record for single-junction solar cells under 1 sun illumination. In Proceedings of the 2011 37th IEEE Photovoltaic Specialists Conference, Seattle, WA, USA, 19–24 June 2011. (Unpublished). [Google Scholar]
- Jackson, P.; Hariskos, D.; Wuerz, R.; Kiowski, O.; Bauer, A.; Friedlmeier, T.M.; Powalla, M. Properties of Cu (In, Ga) Se2 solar cells with new record efficiencies up to 21.7%. Phys. Status Solidi (RRL)–Rapid Res. Lett. 2015, 9, 28–31. [Google Scholar] [CrossRef]
- Jeon, N.J.; Noh, J.H.; Yang, W.S.; Kim, Y.C.; Ryu, S.; Seo, J.; Seok, S.I. Compositional engineering of perovskite materials for high-performance solar cells. Nature 2015, 517, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, J.; Singh, U.P. Copper indium gallium selenide based solar cells—A review. Energy Environ. Sci. 2017, 10, 1306–1319. [Google Scholar] [CrossRef]
- Eyderman, S.; Deinega, A.; John, S. Near perfect solar absorption in ultra-thin-film GaAs photonic crystals. J. Mater. Chem. A 2014, 2, 761–769. [Google Scholar] [CrossRef]
- Leijtens, T.; Eperon, G.E.; Noel, N.K.; Habisreutinger, S.N.; Petrozza, A.; Snaith, H.J. Stability of metal halide perovskite solar cells. Adv. Energy Mater. 2015, 5, 1500963. [Google Scholar] [CrossRef]
- Park, N.-G.; Grätzel, M.; Miyasaka, T.; Zhu, K.; Emery, K. Towards stable and commercially available perovskite solar cells. Nat. Energy 2016, 1, 1–8. [Google Scholar] [CrossRef]
- Todorov, T.K.; Reuter, K.B.; Mitzi, D.B. High-efficiency solar cell with earth-abundant liquid-processed absorber. Adv. Mater. 2010, 22, E156–E159. [Google Scholar] [CrossRef]
- Zandi, S.; Seresht, M.J.; Khan, A.; Gorji, N.E. Simulation of heat loss in Cu2ZnSn4SxSe4−x thin film solar cells: A coupled optical-electrical-thermal modeling. Renew. Energy 2022, 181, 320–328. [Google Scholar] [CrossRef]
- Olekseyuk, I.; Gulay, L.; Dydchak, I.; Piskach, L.; Parasyuk, O.; Marchuk, O. Single crystal preparation and crystal structure of the Cu2Zn/Cd, Hg/SnSe4 compounds. J. Alloys Compd. 2002, 340, 141–145. [Google Scholar] [CrossRef]
- Ito, K. Copper Zinc tin Sulfide-Based Thin-Film Solar Cells; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Wang, W.; Winkler, M.T.; Gunawan, O.; Gokmen, T.; Todorov, T.K.; Zhu, Y.; Mitzi, D.B. Device characteristics of CZTSSe thin-film solar cells with 12.6% efficiency. Adv. Energy Mater. 2014, 4, 1301465. [Google Scholar] [CrossRef]
- Kumar, M.; Dubey, A.; Adhikari, N.; Venkatesan, S.; Qiao, Q. Strategic review of secondary phases, defects and defect-complexes in kesterite CZTS–Se solar cells. Energy Environ. Sci. 2015, 8, 3134–3159. [Google Scholar] [CrossRef]
- Ruhle, S.; Anderson, A.Y.; Barad, H.-N.; Kupfer, B.; Bouhadana, Y.; Rosh-Hodesh, E.; Zaban, A. All-oxide photovoltaics. J. Phys. Chem. Lett. 2012, 3, 3755–3764. [Google Scholar] [CrossRef] [PubMed]
- Whelan, E.; Steuber, F.W.; Gunnlaugsson, T.; Schmitt, W. Tuning photoactive metal–organic frameworks for luminescence and photocatalytic applications. Coord. Chem. Rev. 2021, 437, 213757. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B 2000, 62, 4927. [Google Scholar] [CrossRef]
- Rühle, S. Tabulated values of the Shockley–Queisser limit for single junction solar cells. Sol. Energy 2016, 130, 139–147. [Google Scholar] [CrossRef]
- Yu, L.; Zunger, A. Identification of potential photovoltaic absorbers based on first-principles spectroscopic screening of materials. Phys. Rev. Lett. 2012, 108, 068701. [Google Scholar] [CrossRef]
- Peng, H.; Bikowski, A.; Zakutayev, A.; Lany, S. Pathway to oxide photovoltaics via band-structure engineering of SnO. APL Mater. 2016, 4, 106103. [Google Scholar] [CrossRef]
- Živković, A.; Roldan, A.; De Leeuw, N.H. Density functional theory study explaining the underperformance of copper oxides as photovoltaic absorbers. Phys. Rev. B 2019, 99, 035154. [Google Scholar] [CrossRef]
- Thind, A.S.; Kavadiya, S.; Kouhnavard, M.; Wheelus, R.; Cho, S.B.; Lin, L.-Y.; Kacica, C.; Mulmudi, H.K.; Unocic, K.A.; Borisevich, A.Y. KBaTeBiO6: A lead-free, inorganic double-perovskite semiconductor for photovoltaic applications. Chem. Mater. 2019, 31, 4769–4778. [Google Scholar] [CrossRef]
- Kangsabanik, J.; Alam, A. Ab initio discovery of stable double perovskite oxides Na2BIO6 (B = Bi, In) with promising optoelectronic properties. J. Phys. Chem. Lett. 2020, 11, 5148–5155. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Paudel, T.R.; Dong, S.; Tsymbal, E.Y. Hexagonal rare-earth manganites as promising photovoltaics and light polarizers. Phys. Rev. B 2015, 92, 125201. [Google Scholar] [CrossRef]
- Wanzhong, L.; Jian, S.; Chong, D. Layer-dependent electronic and optical properties of tin monoxide: A potential candidate in photovoltaic applications. Phys. Chem. Chem. Phys. 2022, 24, 7611–7616. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kong, M.; Kang, M.; Kim, M.; Kim, S.; Kim, Y.; Yoon, S.; Ok, J.M. Growth of delafossite CuAlO2 single crystals in a reactive crucible. J. Phys. Condens. Matter 2022, 51, 024002. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Huhn, W.P.; Lange, B.; Yu, V.W.-Z.; Yoon, M.; Blum, V. GPU acceleration of all-electron electronic structure theory using localized numeric atom-centered basis functions. Comput. Phys. Commun. 2020, 254, 107314. [Google Scholar] [CrossRef]
- Yu, V.W.-Z.; Moussa, J.; Kůs, P.; Marek, A.; Messmer, P.; Yoon, M.; Lederer, H.; Blum, V. GPU-acceleration of the ELPA2 distributed eigensolver for dense symmetric and hermitian eigenproblems. Comput. Phys. Commun. 2021, 262, 107808. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Wright, J.N.A.S. Numerical Optimization; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Hedin, L. New method for calculating the one-particle Green’s function with application to the electron-gas problem. Phys. Rev. 1965, 139, A796. [Google Scholar] [CrossRef]
- Deslippe, J.; Samsonidze, G.; Jain, M.; Cohen, M.L.; Louie, S.G. Coulomb-hole summations and energies for G W calculations with limited number of empty orbitals: A modified static remainder approach. Phys. Rev. B 2013, 87, 165124. [Google Scholar] [CrossRef]
- Salpeter, E.E.; Bethe, H.A. A relativistic equation for bound-state problems. Phys. Rev. 1951, 84, 1232. [Google Scholar] [CrossRef]
- Albrecht, S.; Reining, L.; Del Sole, R.; Onida, G. Ab initio calculation of excitonic effects in the optical spectra of semiconductors. Phys. Rev. Lett. 1998, 80, 4510. [Google Scholar] [CrossRef]
- Benedict, L.X.; Shirley, E.L.; Bohn, R.B. Optical absorption of insulators and the electron-hole interaction: An ab initio calculation. Phys. Rev. Lett. 1998, 80, 4514. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Louie, S.G. Electron correlation in semiconductors and insulators: Band gaps and quasiparticle energies. Phys. Rev. B 1986, 34, 5390. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Electron-hole excitations in semiconductors and insulators. Phys. Rev. Lett. 1998, 81, 2312. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLME (%) | SLME (%) | ||
|---|---|---|---|
| Cu2ZnSnO4 | 28.2 | Cu2O | 0.5 |
| CuGaO2 | 32.6 | Cu2O:Zn | 8.0 |
| CuInO2 | 31.9 | Ba2SnNbO6 | 26.5 |
| SnO | 5.3 | Na2Tl0.25Bi0.75O6 | 15.5 |
| 2D-SnO | 13.4 | SrBaVBiO6 | 16.8 |
| Sn0.75Zn0.25O | 22.2 | CdTe (ref) | 30.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.-H.; Kang, M.; Hwang, S.W.; Yeom, S.; Yoon, M.; Ok, J.M.; Yoon, S. Theoretical Investigation of Delafossite-Cu2ZnSnO4 as a Promising Photovoltaic Absorber. Nanomaterials 2023, 13, 3111. https://doi.org/10.3390/nano13243111
Kang S-H, Kang M, Hwang SW, Yeom S, Yoon M, Ok JM, Yoon S. Theoretical Investigation of Delafossite-Cu2ZnSnO4 as a Promising Photovoltaic Absorber. Nanomaterials. 2023; 13(24):3111. https://doi.org/10.3390/nano13243111
Chicago/Turabian StyleKang, Seoung-Hun, Myeongjun Kang, Sang Woon Hwang, Sinchul Yeom, Mina Yoon, Jong Mok Ok, and Sangmoon Yoon. 2023. "Theoretical Investigation of Delafossite-Cu2ZnSnO4 as a Promising Photovoltaic Absorber" Nanomaterials 13, no. 24: 3111. https://doi.org/10.3390/nano13243111
APA StyleKang, S.-H., Kang, M., Hwang, S. W., Yeom, S., Yoon, M., Ok, J. M., & Yoon, S. (2023). Theoretical Investigation of Delafossite-Cu2ZnSnO4 as a Promising Photovoltaic Absorber. Nanomaterials, 13(24), 3111. https://doi.org/10.3390/nano13243111