
Edge Magnetism in MoS2 Nanoribbons: Insights from a Simple One-Dimensional Model



Abstract
:1. Introduction
2. Methods
2.1. DFT Calculations
2.2. TB Formalism
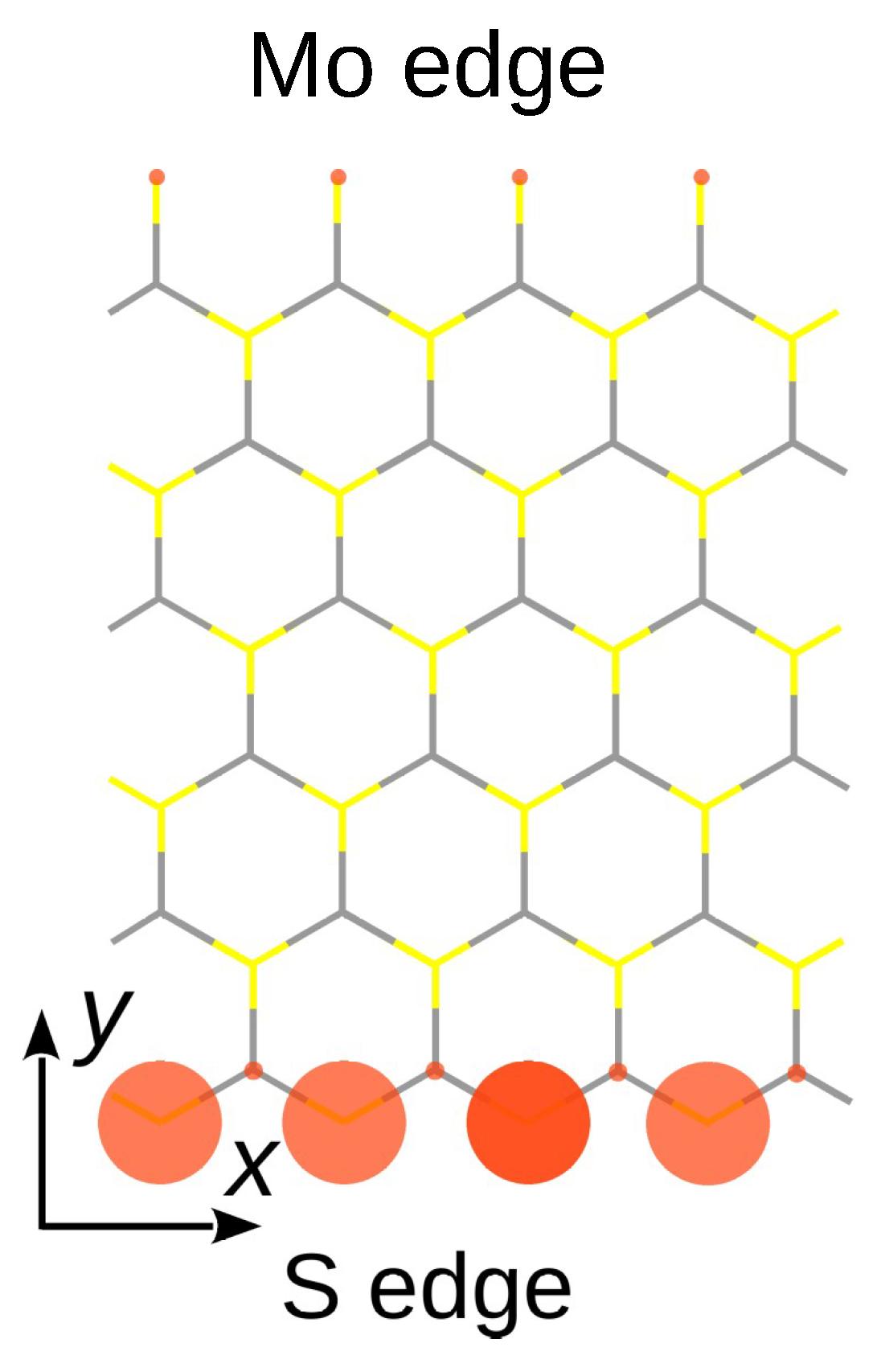
3. Atomic Chain Model
3.1. Motivation
3.2. Energetics
3.3. Perfect Magnetic Structures
3.4. Bloch Wall
3.5. Small Bloch Domains
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Spin Flip in a Ferromagnetic Linear Chain

References
- Kolobov, A.V.; Tominaga, J. Two-Dimensional Transition Metal Dichalcogenides; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–533. [Google Scholar]
- Manzeli, S.; Ovchinnikov, D.; Pasquier, D.; Yazyev, O.V.; Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2017, 2, 17033. [Google Scholar] [CrossRef]
- Yin, X.; Tang, C.; Zheng, Y.; Gao, J.; Wu, J.; Zhang, H.; Chhowalla, M.; Chen, W.; Wee, A.T.S. Recent developments in 2D transition metal dichalcogenides: Phase transition and applications of the (quasi-)metallic phases. Chem. Soc. Rev. 2021, 50, 10087–10115. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, H.; Zhang, Y.; Shi, J. Recent progress of two-dimensional metallic transition metal dichalcogenides: Syntheses, physical properties, and applications featured. J. Appl. Phys. 2022, 131, 060902. [Google Scholar] [CrossRef]
- Yagmurcukardes, M.; Peeters, F.M.; Senger, R.T.; Sahin, H. Nanoribbons: From fundamentals to state-of-the-art applications. Appl. Phys. Rev. 2016, 3, 041302. [Google Scholar] [CrossRef]
- Nakada, K.; Fujita, M.; Dresselhaus, G.; Dresselhaus, M.S. Edge state in graphene ribbons: Nanometer size effect and edge shape dependence. Phys. Rev. B 1996, 54, 17954. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Sasaki, K.I.; Nakanishi, T.; Enoki, T. Electronic states of graphene nanoribbons and analytical solutions. Sci. Technol. Adv. Mater. 2010, 11, 054504. [Google Scholar] [CrossRef] [PubMed]
- Lieb, E.H. Two theorems on the Hubbard model. Phys. Rev. Lett. 1989, 62, 1201–1204. [Google Scholar] [CrossRef]
- Davelou, D.; Kopidakis, G.; Kioseoglou, G.; Remediakis, I.N. MoS2 nanostructures: Semiconductors with metallic edges. Solid St. Comm. 2014, 192, 42–46. [Google Scholar] [CrossRef]
- Bollinger, M.V.; Lauritsen, J.V.; Jacobsen, K.W.; Nørskov, J.K.; Helveg, S.; Besenbacher, F. One-dimensional metallic edge states in MoS2. Phys. Rev. Lett. 2001, 87, 196803. [Google Scholar] [CrossRef]
- Cui, P.; Choi, J.H.; Chen, W.; Shih, J.; Zeng, C.K.; Li, Z.; Zhang, Z. Contrasting structural reconstructions, electronic properties, and magnetic orderings along different edges of zigzag transition metal dichalcogenide nanoribbons. Nano Lett. 2017, 17, 1097–1101. [Google Scholar] [CrossRef]
- Vojvodic, A.; Hinnemann, B.; Nørskov, J.K. Magnetic edge states in MoS2 characterized using density-functional theory. Phys. Rev. B 2009, 80, 125416. [Google Scholar] [CrossRef]
- Vancsó, P.; Hagymási, I.; Castenetto, P.; Lambin, P. Stability of edge magnetism against disorder in zigzag MoS2 nanoribbons. Phys. Rev. Mater. 2019, 3, 094003. [Google Scholar] [CrossRef]
- Deng, X.; Li, Z.; Yang, J. One-dimensional magnetic order stabilized in edge-reconstructed MoS2 nanoribbon via bias voltage. J. Phys. Chem. Lett. 2020, 11, 7531–7535. [Google Scholar] [CrossRef]
- Zhang, J.; Mei Soon, J.; Ping Loh, K.; Yin, J.; Ding, J.; Sullivian, M.B.; Wu, P. Magnetic molybdenum disulfide nanosheet films. Nano Lett. 2007, 7, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Si, M.; Li, J.; Zhang, J.; Zhang, Z.; Yang, Z.; Xue, D. Ferromagnetism in freestanding MoS2 nanosheets. Nanoscale Res. Lett. 2013, 8, 129. [Google Scholar] [CrossRef]
- Huo, N.; Li, Y.; Kang, J.; Li, R.; Xia, Q.; Li, J. Edge-states ferromagnetism of WS2 nanosheets. Appl. Phys. Lett. 2014, 104, 202406. [Google Scholar] [CrossRef]
- Qi, R.; Wang, S.; Wang, M.; Liu, W.; Yan, Z.; Bi, X.; Huang, Q. Towards well-defined MoS2 nanoribbons on a large scale. Chem. Comm. 2017, 53, 9757–9760. [Google Scholar] [CrossRef]
- Zhou, Q.; Su, S.; Cheng, P.; Hu, X.; Zeng, M.; Gao, X.; Zhang, Z.; Liu, J.M. Robust ferromagnetism in zigzag-edge rich MoS2 pyramids. Nanoscale 2018, 10, 11578–11584. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metal. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmentend-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Cappelluti, E.; Roldán, R.; Silva-Guillén, J.A.; Ordejón, P.; Guinea, F. Tight-binding model and direct-gap/indirect-gap transition in single-layer and multilayer MoS2. Phys. Rev. B 2013, 88, 075409. [Google Scholar] [CrossRef]
- Roldán, R.; López-Sancho, M.P.; Guinea, F.; Cappelluti, E.; Silva-Guillén, J.A.; Ordejón, P. Momentum dependence of spin-orbit interaction effects in single-layer and multi-layer transition metal dichalcogenides. 2D Mater. 2014, 1, 034003. [Google Scholar] [CrossRef]
- Bollinger, M.V.; Jacobsen, K.W.; Nørskov, J.K. Atomic and electronic structure of MoS2 nanoparticles. Phys. Rev. B 2003, 67, 085410. [Google Scholar] [CrossRef]
- Brito, F.M.O.; Li, L.; Lopes, J.M.V.P.; Castro, E.V. Edge magnetism in transition metal dichalcogenide nanoribbons: Mean field theory and determinant quantum Monte Carlo. Phys. Rev. B 2022, 105, 195130. [Google Scholar] [CrossRef]
- Szczȩśniak, D.; Kais, S. Gap states and valley-spin filtering in transition metal dichalcogenide monolayers. Phys. Rev. B 2020, 101, 115423. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Cheng, Y.C.; Schwingenschlögl, U. Giant spin-orbit-induced spin splitting in two-dimensional transition-metal dichalcogenide semiconductors. Phys. Rev. B 2011, 84, 153402. [Google Scholar] [CrossRef]
- Ridolfi, E.; Le, D.; Rahman, T.S.; Mucciolo, E.R.; Lewenkopf, C.H. A tight-binding model for MoS2 monolayers. J. Phys. Condens. Matter 2015, 27, 365501. [Google Scholar] [CrossRef]
- Stoner, E.C. Ferromagnetism. Rep. Prog. Phys. 1948, 11, 43. [Google Scholar]
- Kaplan, T.A. Single-band Hubbard model with spin-orbit coupling. Zeitschr. Phys. B Cond. Matter 1983, 49, 313–317. [Google Scholar] [CrossRef]
- Lieb, E.H.; Wu, F.Y. The one-dimensional Hubbard model: A reminiscence. Physica A 2003, 321, 1–27. [Google Scholar] [CrossRef]
- The Hubbard model at half a century. Nat. Phys. 2013, 9, 523. [CrossRef]
- Honet, A.; Henrard, L.; Meunier, V. Exact and many-body perturbation solutions of the Hubbard model applied to linear chains. AIP Advances 2022, 12, 035238. [Google Scholar] [CrossRef]
- França, V.V.; Vieira, D.; Capelle, K. Simple parameterization for the ground-state energy of the infinite Hubbard chain incorporating Mott physics, spin-dependent phenomena and spatial inhomogeneity. New J. Phys. 2012, 14, 073021. [Google Scholar] [CrossRef]





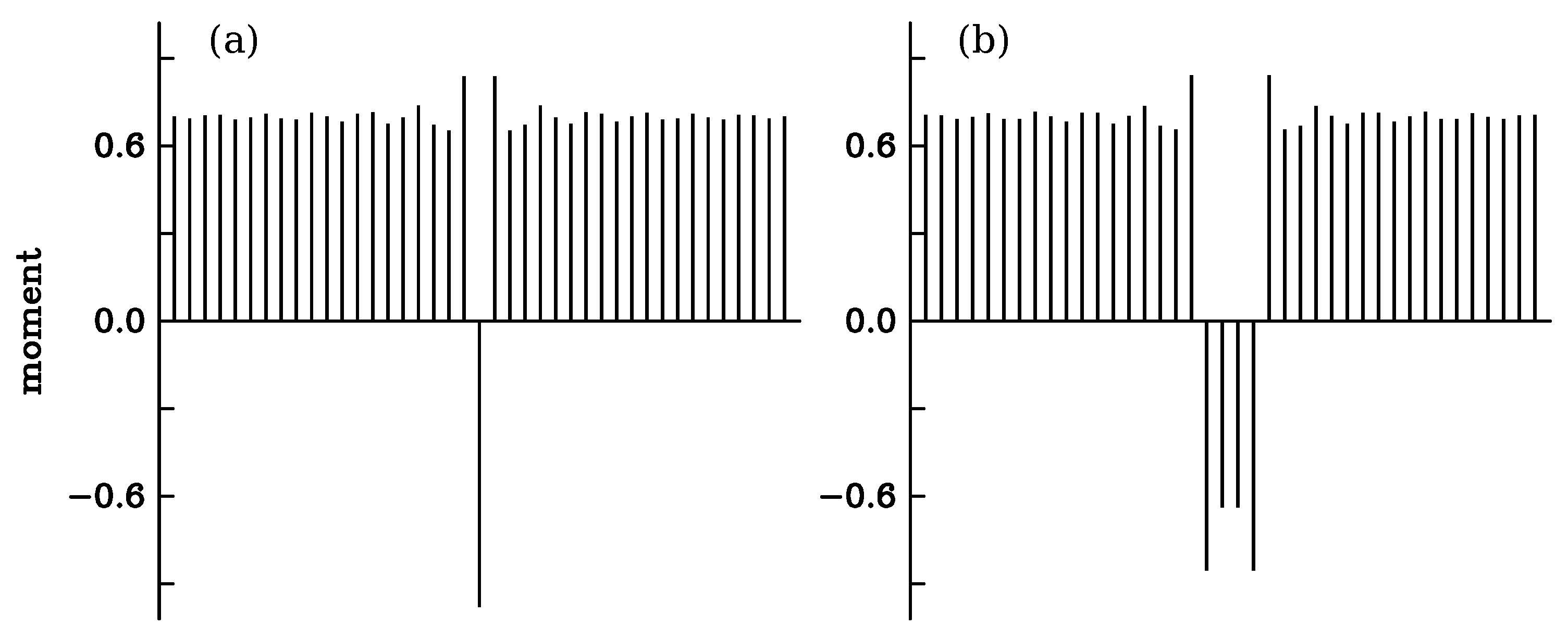


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| - | Mo Atom | S Atom | S Dimer |
|---|---|---|---|
| Mo edge | −2.03, 1.42, 1.42, −4.03, 0.51 | 0.28, −8.28, −12.24 | −0.55, −5.28, −8.24 |
| S edge | −2.03, 4.30, −0.80, −12.03, −2.60 | −1.90, 0.18, −6.50 | - |
| (eV) | (eV) | U (eV) | (eV) | |
|---|---|---|---|---|
| −0.8397 | −0.04595 | 0.65 | 0.7428 | −1.3225 |
| Structure | Non-Magnetic | Ferromagnetic | Antiferromagnetic |
|---|---|---|---|
| (meV/atom) | 0 | −62 | −47 |
| 0.00 | 0.70 | ||
| (eV) | −0.7980 | −0.6337 | −0.5874 |
| Size | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| (meV) | 7.5 | 14.9 | 11.6 | 9.8 | 11.9 | 12.9 |
| −0.46 | −0.75 | −0.09 | −0.42 | −0.72 | −0.01 |
| Size | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| = 0.025 eV, = 1.5 | ||||||
| (meV) | −7.8 | −0.6 | −0.4 | +8.8 | +12.4 | +11.0 |
| = 0.05 eV, = 1.5 | ||||||
| (meV) | −11.1 | −8.3 | −7.4 | −2.4 | +1.0 | +8.3 |
| = 0.075 eV, = 1.5 | ||||||
| (meV) | −11.3 | −10.4 | −9.4 | −6.5 | −3.0 | +2.7 |
| = 0.05 eV, = 2.0 | ||||||
| (meV) | −11.3 | −9.7 | −9.3 | −6.4 | −4.2 | +0.6 |
| = 0.05 eV, = 2.5 | ||||||
| (meV) | −11.4 | −10.4 | −10.2 | −8.4 | −7.1 | −4.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castenetto, P.; Lambin, P.; Vancsó, P. Edge Magnetism in MoS2 Nanoribbons: Insights from a Simple One-Dimensional Model. Nanomaterials 2023, 13, 3086. https://doi.org/10.3390/nano13243086
Castenetto P, Lambin P, Vancsó P. Edge Magnetism in MoS2 Nanoribbons: Insights from a Simple One-Dimensional Model. Nanomaterials. 2023; 13(24):3086. https://doi.org/10.3390/nano13243086
Chicago/Turabian StyleCastenetto, Pauline, Philippe Lambin, and Péter Vancsó. 2023. "Edge Magnetism in MoS2 Nanoribbons: Insights from a Simple One-Dimensional Model" Nanomaterials 13, no. 24: 3086. https://doi.org/10.3390/nano13243086
APA StyleCastenetto, P., Lambin, P., & Vancsó, P. (2023). Edge Magnetism in MoS2 Nanoribbons: Insights from a Simple One-Dimensional Model. Nanomaterials, 13(24), 3086. https://doi.org/10.3390/nano13243086