

Radiofluorination of an Anionic, Azide-Functionalized Teroligomer by Copper-Catalyzed Azide-Alkyne Cycloaddition

 ,
,  , , ,
, , ,  , , , and
, , , and 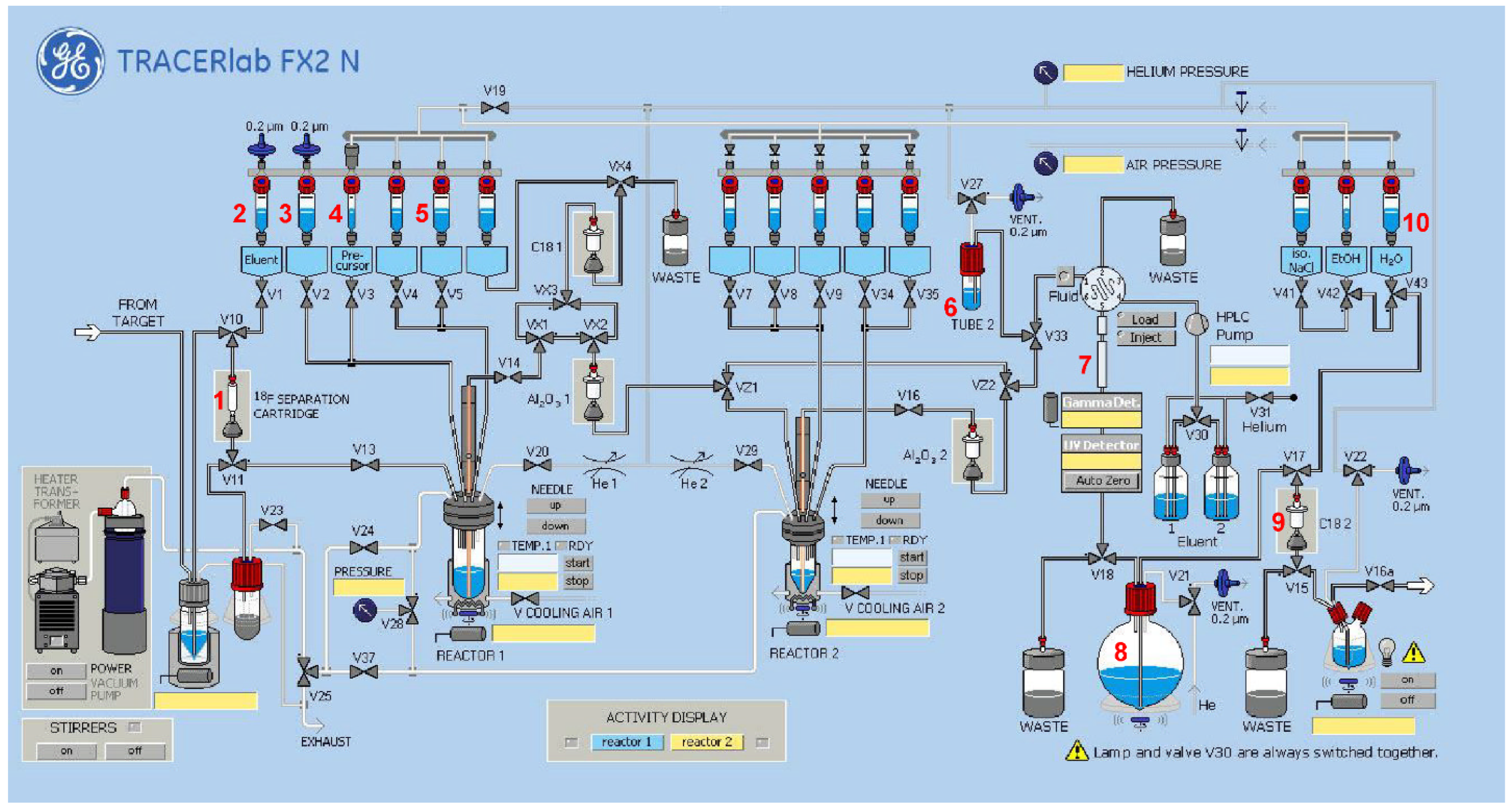{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Oligomer Synthesis
2.1.1. Materials
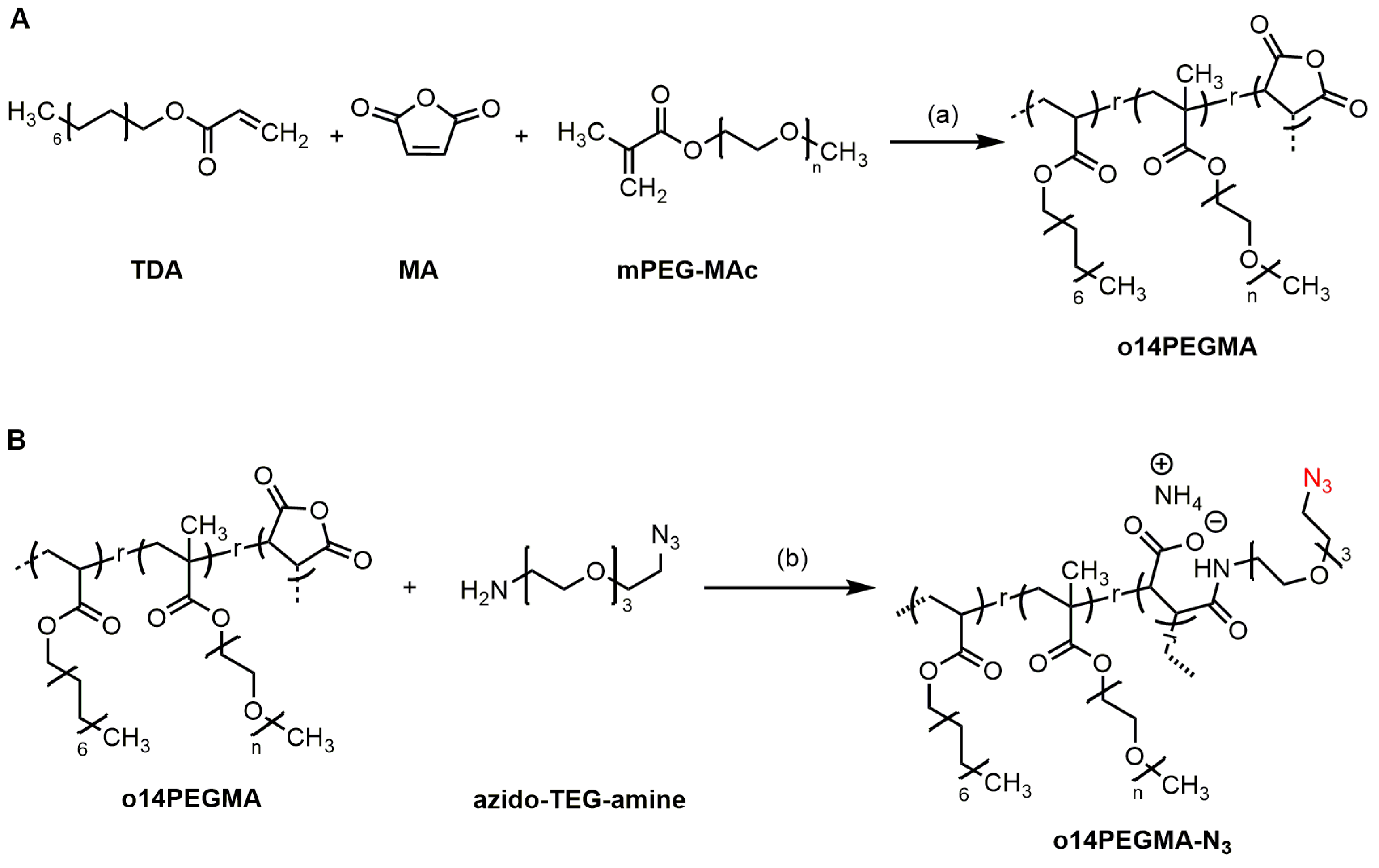
2.1.2. Synthesis of Pristine Teroligomer o14PEGMA (1:1:2.5)
2.1.3. Synthesis of Azide-Modified Teroligomer and Isolation as Ammonium Salt (o14PEGMA-N3)
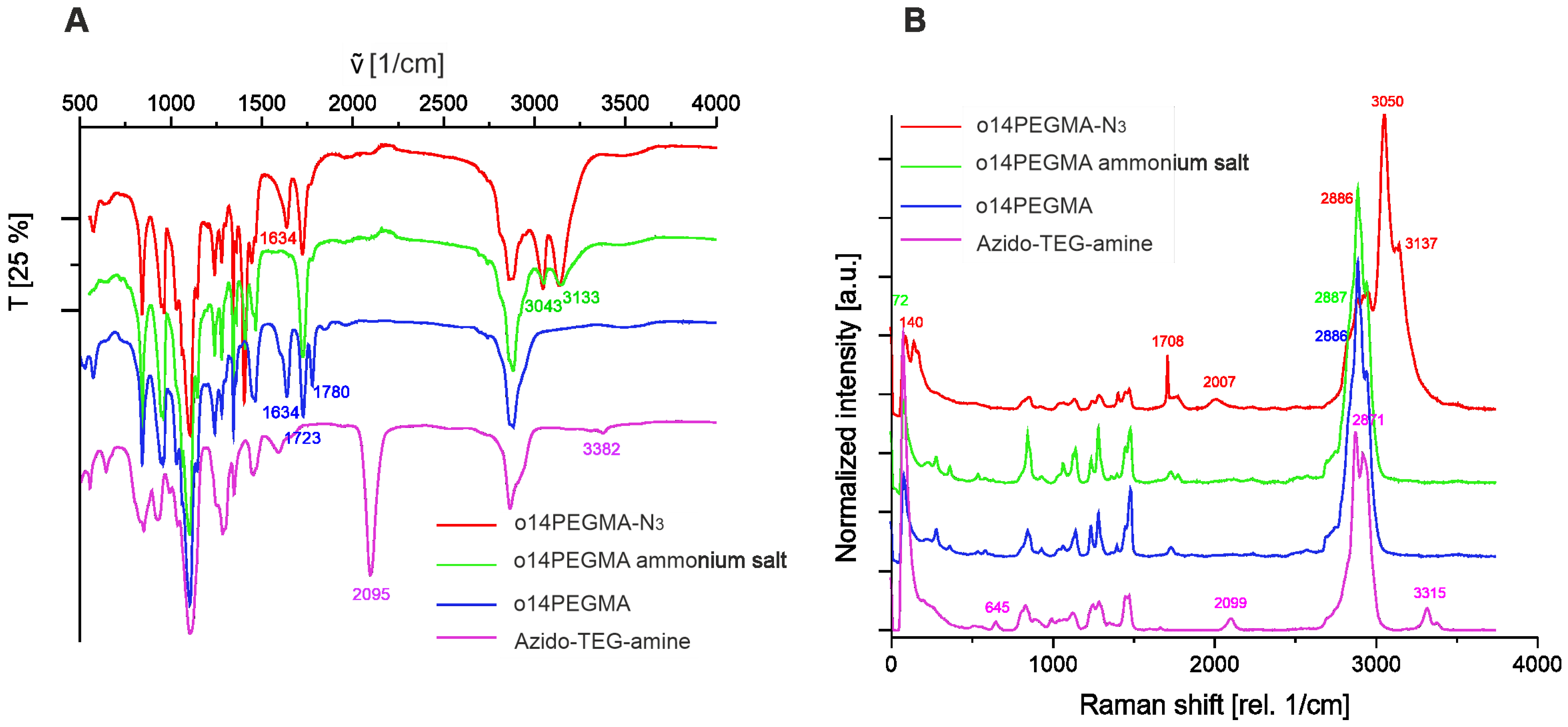
2.1.4. Fourier Transform Infrared Spectroscopy (FT-IR) and Confocal Raman Spectroscopy
2.2. Radiochemistry
2.2.1. Synthesis of Non-Radioactive Reference and Precursor
2.2.2. Analytics
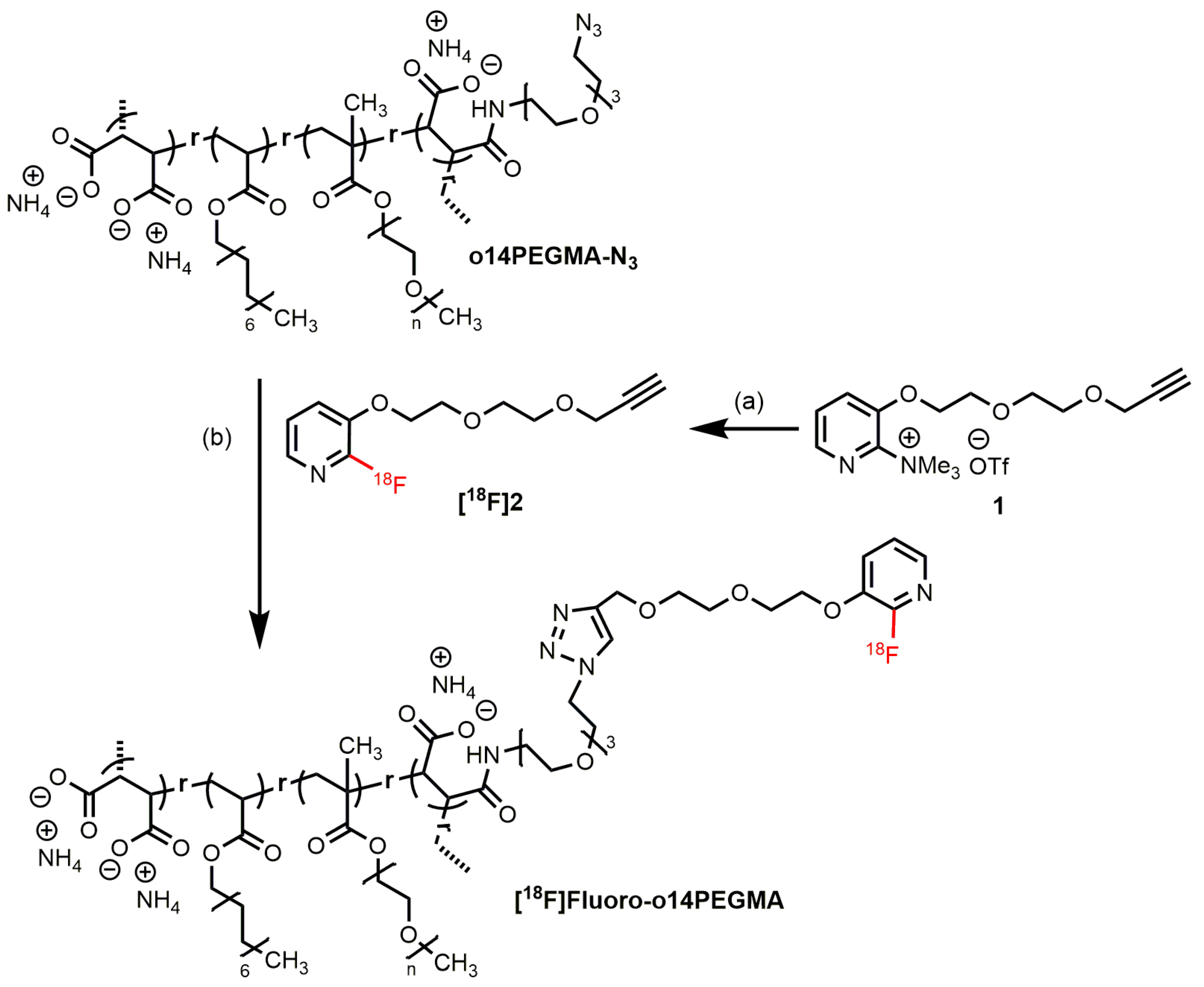
2.2.3. Radiosynthesis of [18F]2
2.2.4. Radiosynthesis of the Teroligomer [18F]fluoro-o14PEGMA by Click-Conjugation of [18F]2
3. Results and Discussion
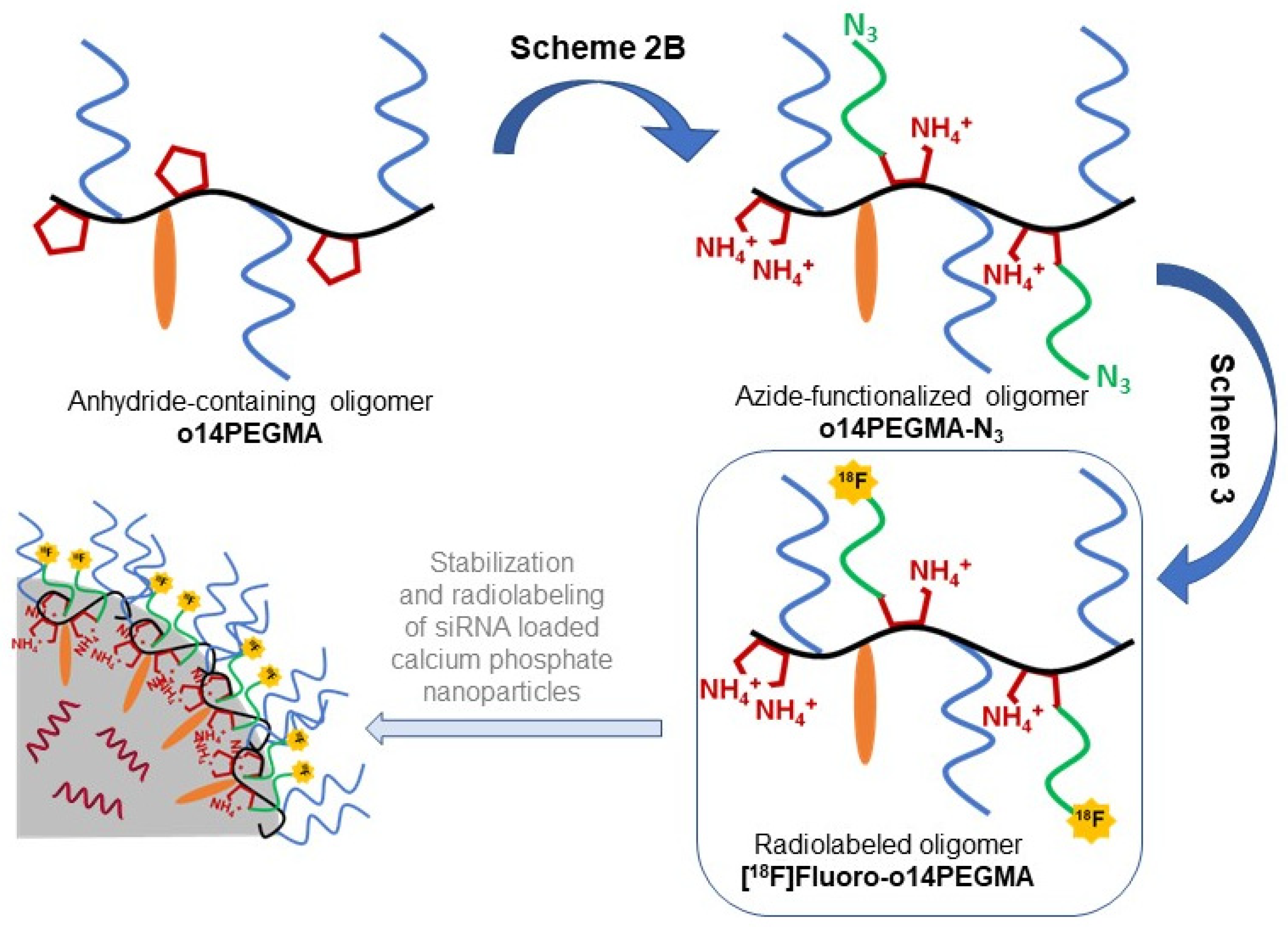
3.1. Polymer Synthesis and Characterization

3.2. Linker Modification
3.3. Radiochemistry
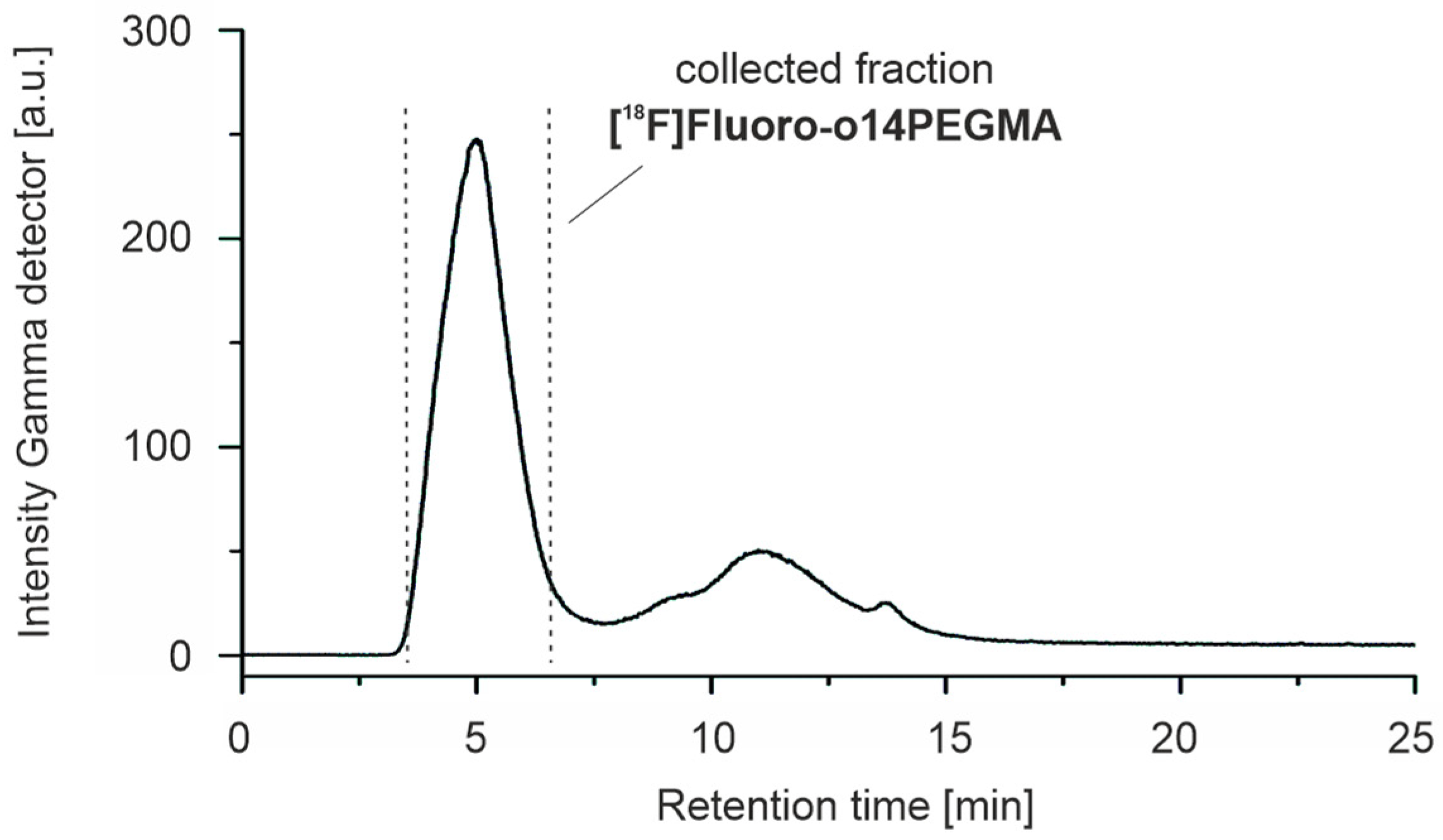
3.3.1. Radiosynthesis of the Alkyne-Substituted Heteroaromatic Group [18F]2
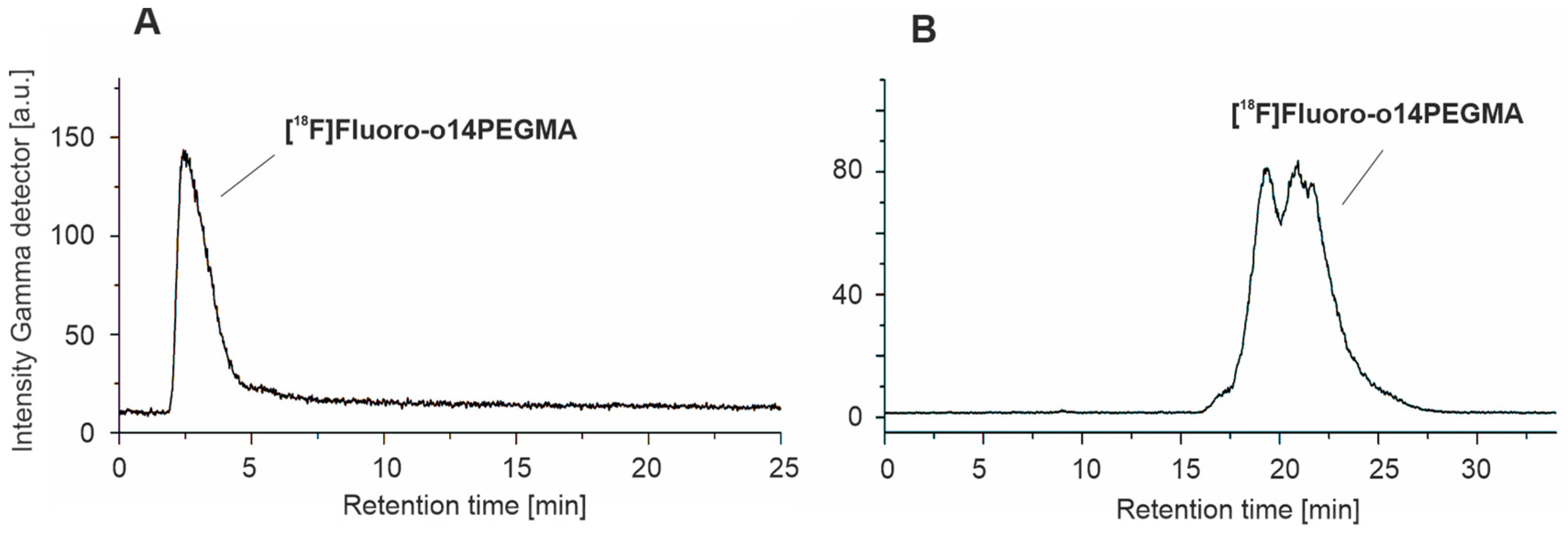
3.3.2. Radiosynthesis of the Teroligomer [18F]fluoro-o14PEGMA by Conjugation of o14PEGMA-N3 with [18F]2

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Vicent, M.J. Polymer therapeutics-prospects for 21st century: The end of the beginning. Adv. Drug Deliv. Rev. 2013, 65, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Sedlacek, O.; Nadar, R.A.; Hruby, M.; Stephan, H. Radiolabelled Polymeric Materials for Imaging and Treatment of Cancer: Quo Vadis? Adv. Healthc. Mater. 2017, 6, 1601115. [Google Scholar] [CrossRef]
- Stockhofe, K.; Postema, J.M.; Schieferstein, H.; Ross, T.L. Radiolabeling of Nanoparticles and Polymers for PET Imaging. Pharmaceuticals 2014, 7, 392–418. [Google Scholar] [CrossRef] [PubMed]
- Delplace, V.; Couvreur, P.; Nicolas, J. Recent trends in the design of anticancer polymer prodrug nanocarriers. Polym. Chem. 2014, 5, 1529–1544. [Google Scholar] [CrossRef]
- Parveen, S.; Arjmand, F.; Tabassum, S. Clinical developments of antitumor polymer therapeutics. RSC Adv. 2019, 9, 24699–24721. [Google Scholar] [CrossRef] [PubMed]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar] [CrossRef]
- Mitrach, F.; Schmid, M.; Toussaint, M.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Franke, H.; Ewe, A.; Aigner, A.; Wölk, C.; Brust, P.; et al. Amphiphilic Anionic Oligomer-Stabilized Calcium Phosphate Nanoparticles with Prospects in siRNA Delivery via Convection-Enhanced Delivery. Pharmaceutics 2022, 14, 326. [Google Scholar] [CrossRef]
- Herth, M.M.; Barz, M.; Moderegger, D.; Allmeroth, M.; Jahn, M.; Thews, O.; Zentel, R.; Rösch, F. Radioactive Labeling of Defined HPMA-Based Polymeric Structures Using [18F]FETos for In Vivo Imaging by Positron Emission Tomography. Biomacromolecules 2009, 10, 1697–1703. [Google Scholar] [CrossRef]
- Allmeroth, M.; Moderegger, D.; Gündel, D.; Buchholz, H.G.; Mohr, N.; Koynov, K.; Rösch, F.; Thews, O.; Zentel, R. PEGylation of HPMA-based block copolymers enhances tumor accumulation in vivo: A quantitative study using radiolabeling and positron emission tomography. J. Control Release 2013, 172, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Schieferstein, H.; Kelsch, A.; Reibel, A.; Koynov, K.; Barz, M.; Buchholz, H.G.; Bausbacher, N.; Thews, O.; Zentel, R.; Ross, T.L. 18F-Radiolabeling, Preliminary Evaluation of Folate-pHPMA Conjugates via PET. Macromol. Biosci. 2014, 14, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, P.P.; Gomez-Vallejo, V.; Maldonado, Z.B.; Roig, J.L.; Borros, S. Novel 18F-Labeling Strategy for Polyester-Based NPs for In Vivo PET-CT Imaging. Bioconjug. Chem. 2015, 26, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Reibel, A.T.; Müller, S.S.; Pektor, S.; Bausbacher, N.; Miederer, M.; Frey, H.; Rösch, F. Fate of Linear and Branched Polyether-Lipids In Vivo in Comparison to Their Liposomal Formulations by 18F-Radiolabeling and Positron Emission Tomography. Biomacromolecules 2015, 16, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Wagener, K.; Worm, M.; Pektor, S.; Schinnerer, M.; Thiermann, R.; Miederer, M.; Frey, H.; Rösch, F. Comparison of Linear and Hyperbranched Polyether Lipids for Liposome Shielding by 18F-Radiolabeling and Positron Emission Tomography. Biomacromolecules 2018, 19, 2506–2516. [Google Scholar] [CrossRef]
- Glassner, M.; Palmieri, L.; Monnery, B.D.; Verbrugghen, T.; Deleye, S.; Stroobants, S.; Staelens, S.; Wyffels, L.; Hoogenboom, R. The Label Matters: µPET Imaging of the Biodistribution of Low Molar Mass 89Zr and 18F-Labeled Poly(2-ethyl-2-oxazoline). Biomacromolecules 2017, 18, 96–102. [Google Scholar] [CrossRef]
- Inkster, J.; Lin, K.S.; Ait-Mohand, S.; Gosselin, S.; Benard, F.; Guerin, B.; Pourghiasian, M.; Ruth, T.; Schaffer, P.; Storr, T. 2-Fluoropyridine prosthetic compounds for the 18F-labeling of bombesin analogues. Bioorganic Med. Chem. Lett. 2013, 23, 3920–3926. [Google Scholar] [CrossRef]
- Kuhnast, B.; Damont, A.; Hinnen, F.; Huss, C.; Dolle, F. PEG-[18F] FPyZIDE and PEG-[18F] FPyKYNE, Two New Fluoropyridine-Based Reagents for the Fluorine-18 Labeling of Macromolecules Using Click Chemistry. J. Label. Compd. Radiopharm. 2009, 52, S184. [Google Scholar]
- Brown, A.; Fujimori, K. A Method for the Determination of Maleic-Anhydride Content in Copolymers. Polym. Bull. 1986, 16, 441–444. [Google Scholar] [CrossRef]
- Endo, R.; Hinokuma, T.; Takeda, M. Studies of Solution Properties of Copolymers. 2. Copolymer of Maleic Anhydride and Styrene. J. Polym. Sci. Part A-2 Polym. Phys. 1968, 6, 665–673. [Google Scholar] [CrossRef]
- Edward Semple, J.; Sullivan, B.; Vojkovsky, T.; Sill, K.N. Synthesis and facile end-group quantification of functionalized PEG azides. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2888–2895. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, H.A.; Schrock, K.; Schmid, M.; Krieghoff, J.; Maqsood, I.; Kascholke, C.; Kohn-Polster, C.; Schulz-Siegmund, M.; Hacker, M.C. Injectable oligomer-cross-linked gelatine hydrogels via anhydride-amine-conjugation. J. Mater. Chem. B 2021, 9, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Kascholke, C.; Loth, T.; Kohn-Polster, C.; Möller, S.; Bellstedt, P.; Schulz-Siegmund, M.; Schnabelrauch, M.; Hacker, M.C. Dual-Functional Hydrazide-Reactive and Anhydride-Containing Oligomeric Hydrogel Building Blocks. Biomacromolecules 2017, 18, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Wölk, C.; Janich, C.; Meister, A.; Drescher, S.; Langner, A.; Brezesinski, G.; Bakowsky, U. Investigation of Binary Lipid Mixtures of a Three-Chain Cationic Lipid with Phospholipids Suitable for Gene Delivery. Bioconjug. Chem. 2015, 26, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Olenick, L.L.; Troiano, J.M.; Vartanian, A.; Melby, E.S.; Mensch, A.C.; Zhang, L.L.; Hong, J.W.; Mesele, O.; Qiu, T.; Bozich, J.; et al. Lipid Corona Formation from Nanoparticle Interactions with Bilayers. Chem 2018, 4, 2709–2723. [Google Scholar] [CrossRef]
- Loth, T.; Hennig, R.; Kascholke, C.; Hötzel, R.; Hacker, M.C. Reactive and stimuli-responsive maleic anhydride containing macromers—Multi-functional cross-linkers and building blocks for hydrogel fabrication. React. Funct. Polym. 2013, 73, 1480–1492. [Google Scholar] [CrossRef]
- Li, H.; Nawaz, H.A.; Masieri, F.F.; Vogel, S.; Hempel, U.; Bartella, A.K.; Zimmerer, R.; Simon, J.C.; Schulz-Siegmund, M.; Hacker, M.C.; et al. Osteogenic Potential of Mesenchymal Stem Cells from Adipose Tissue, Bone Marrow and Hair Follicle Outer Root Sheath in a 3D Crosslinked Gelatin-Based Hydrogel. Int. J. Mol. Sci. 2021, 22, 5404. [Google Scholar] [CrossRef]
- Seneca, S.; Simon, J.; Weber, C.; Ghazaryan, A.; Ethirajan, A.; Mailaender, V.; Morsbach, S.; Landfester, K. How Low Can You Go? Low Densities of Poly(ethylene glycol) Surfactants Attract Stealth Proteins. Macromol. Biosci. 2018, 18, e1800075. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert. Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Lagana, A. Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef]
- Dinari, A.; Moghadam, T.T.; Abdollahi, M.; Sadeghizadeh, M. Synthesis and Characterization of a Nano-Polyplex system of GNRs-PDMAEA-pDNA: An Inert Self-Catalyzed Degradable Carrier for Facile Gene Delivery. Sci. Rep. 2018, 8, 8112. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Li, Y.; Rungta, A.; Viswanath, A.; Gao, J.N.; Benicewicz, B.C.; Siegel, R.W.; Schadler, L.S. TiO2 nanocomposites with high refractive index and transparency. J. Mat. Chem. 2011, 21, 18623–18629. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Jain, P.; Peng, H.; Rahimi, K.; Singh, S.; Pich, A. Dual-Degradable Biohybrid Microgels by Direct Cross-Linking of Chitosan and Dextran Using Azide-Alkyne Cycloaddition. Biomacromolecules 2020, 21, 4933–4944. [Google Scholar] [CrossRef] [PubMed]
- Mudunkotuwa, I.A.; Minshid, A.A.; Grassian, V.H. ATR-FTIR spectroscopy as a tool to probe surface adsorption on nanoparticles at the liquid-solid interface in environmentally and biologically relevant media. Analyst 2014, 139, 870–881. [Google Scholar] [CrossRef]
- Kopf, A.H.; Koorengevel, M.C.; van Walree, C.A.; Dafforn, T.R.; Killian, J.A. A simple and convenient method for the hydrolysis of styrene-maleic anhydride copolymers to styrene-maleic acid copolymers. Chem. Phys. Lipids 2019, 218, 85–90. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef]
- Jiang, J.R.; Zhu, P.F.; Li, D.M.; Chen, Y.M.; Li, M.R.; Wang, X.L.; Liu, B.B.; Cui, Q.L.; Zhu, H.Y. High pressure studies of trimethyltin azide by Raman scattering, IR absorption, and synchrotron X-ray diffraction. RSC Adv. 2016, 6, 98921–98926. [Google Scholar] [CrossRef]
- Uliyanchenko, E.; van der Wal, S.; Schoenmakers, P.J. Challenges in polymer analysis by liquid chromatography. Polym. Chem. 2012, 3, 2313–2335. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenzel, B.; Schmid, M.; Teodoro, R.; Moldovan, R.-P.; Lai, T.H.; Mitrach, F.; Kopka, K.; Fischer, B.; Schulz-Siegmund, M.; Brust, P.; et al. Radiofluorination of an Anionic, Azide-Functionalized Teroligomer by Copper-Catalyzed Azide-Alkyne Cycloaddition. Nanomaterials 2023, 13, 2095. https://doi.org/10.3390/nano13142095
Wenzel B, Schmid M, Teodoro R, Moldovan R-P, Lai TH, Mitrach F, Kopka K, Fischer B, Schulz-Siegmund M, Brust P, et al. Radiofluorination of an Anionic, Azide-Functionalized Teroligomer by Copper-Catalyzed Azide-Alkyne Cycloaddition. Nanomaterials. 2023; 13(14):2095. https://doi.org/10.3390/nano13142095
Chicago/Turabian StyleWenzel, Barbara, Maximilian Schmid, Rodrigo Teodoro, Rareş-Petru Moldovan, Thu Hang Lai, Franziska Mitrach, Klaus Kopka, Björn Fischer, Michaela Schulz-Siegmund, Peter Brust, and et al. 2023. "Radiofluorination of an Anionic, Azide-Functionalized Teroligomer by Copper-Catalyzed Azide-Alkyne Cycloaddition" Nanomaterials 13, no. 14: 2095. https://doi.org/10.3390/nano13142095
APA StyleWenzel, B., Schmid, M., Teodoro, R., Moldovan, R.-P., Lai, T. H., Mitrach, F., Kopka, K., Fischer, B., Schulz-Siegmund, M., Brust, P., & Hacker, M. C. (2023). Radiofluorination of an Anionic, Azide-Functionalized Teroligomer by Copper-Catalyzed Azide-Alkyne Cycloaddition. Nanomaterials, 13(14), 2095. https://doi.org/10.3390/nano13142095