
Cargo-Dependent Targeted Cellular Uptake Using Quaternized Starch as a Carrier



Abstract
:1. Introduction
2. Materials and Methods
2.1. Starch Quaternization
2.2. Quaternized Starch Labeling
2.3. Quaternized Starch Chemical Analysis
2.4. Q-Starch/Cargo Complex Preparation
2.5. Q-Starch/Cargo Complex Characterization
2.6. Cell Culture Handling
2.7. Determination of Intracellular Paths of Complexes
2.8. Biological Activity Evaluation
2.8.1. pDNA: pGFP Transfection
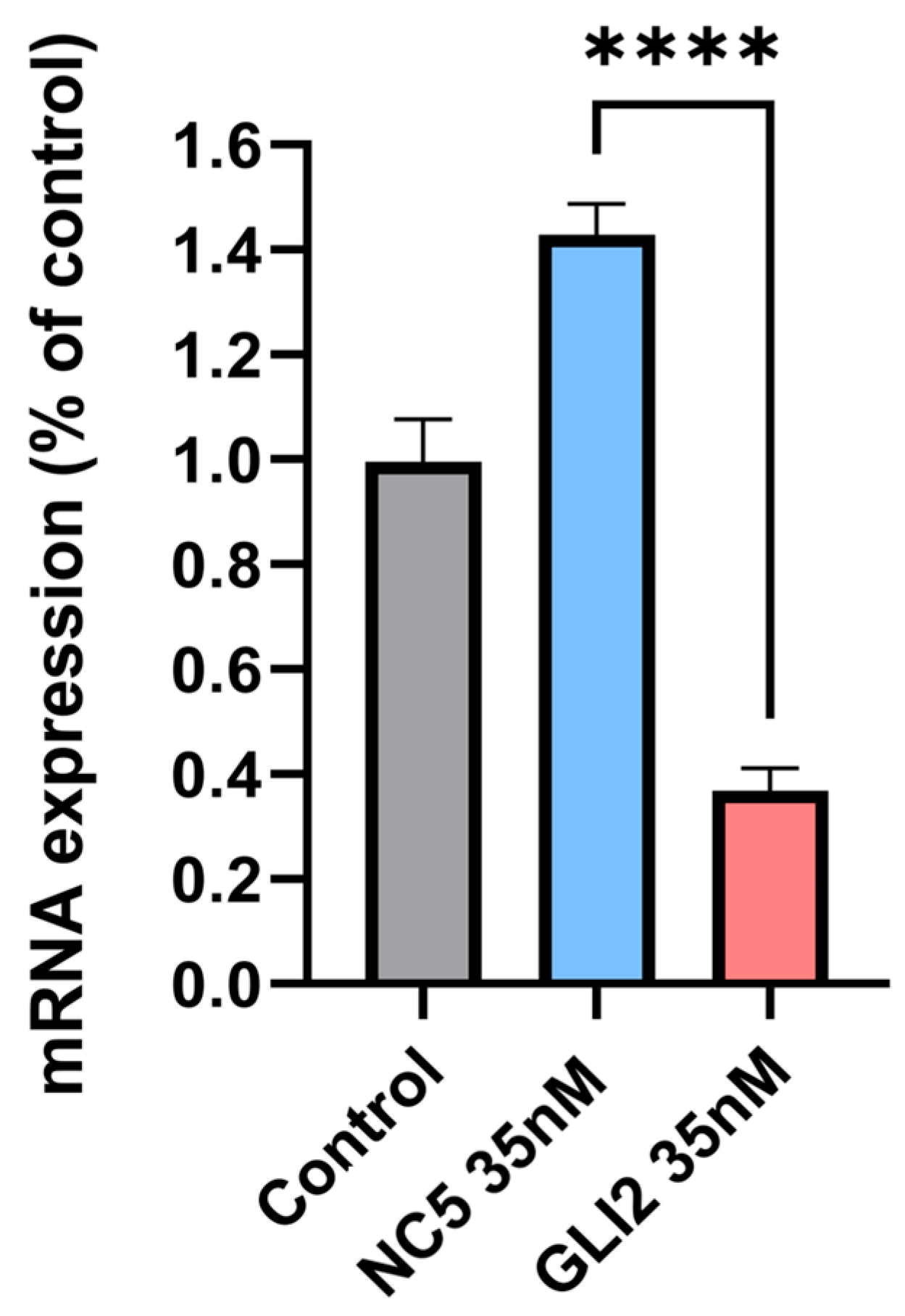
2.8.2. siRNA: GLI2 Gene Silencing
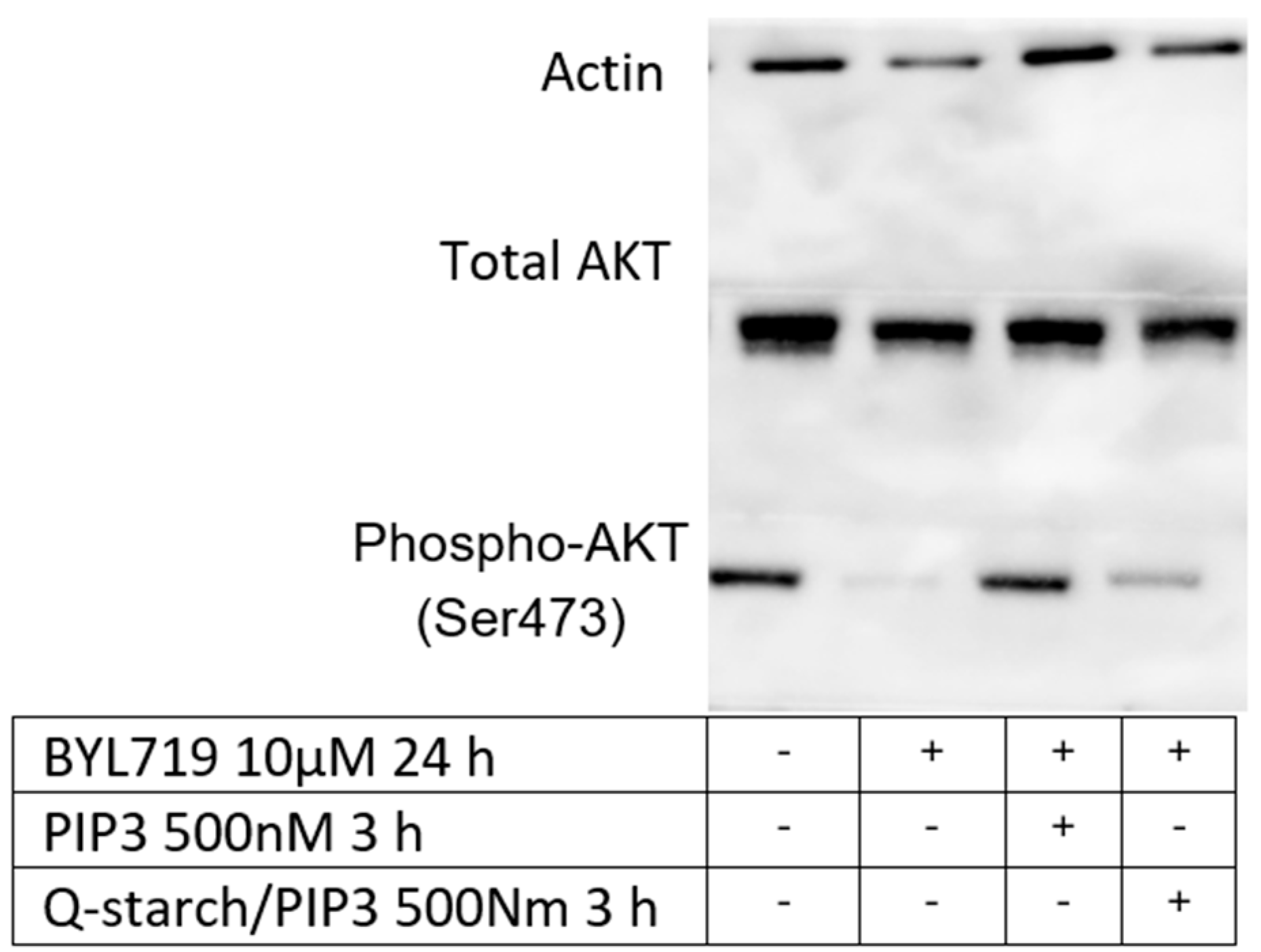
2.8.3. PIP3: PI3K Protein Activation by Q-Starch/PIP3 Complexes Assessed by Western Blot
3. Results and Discussion
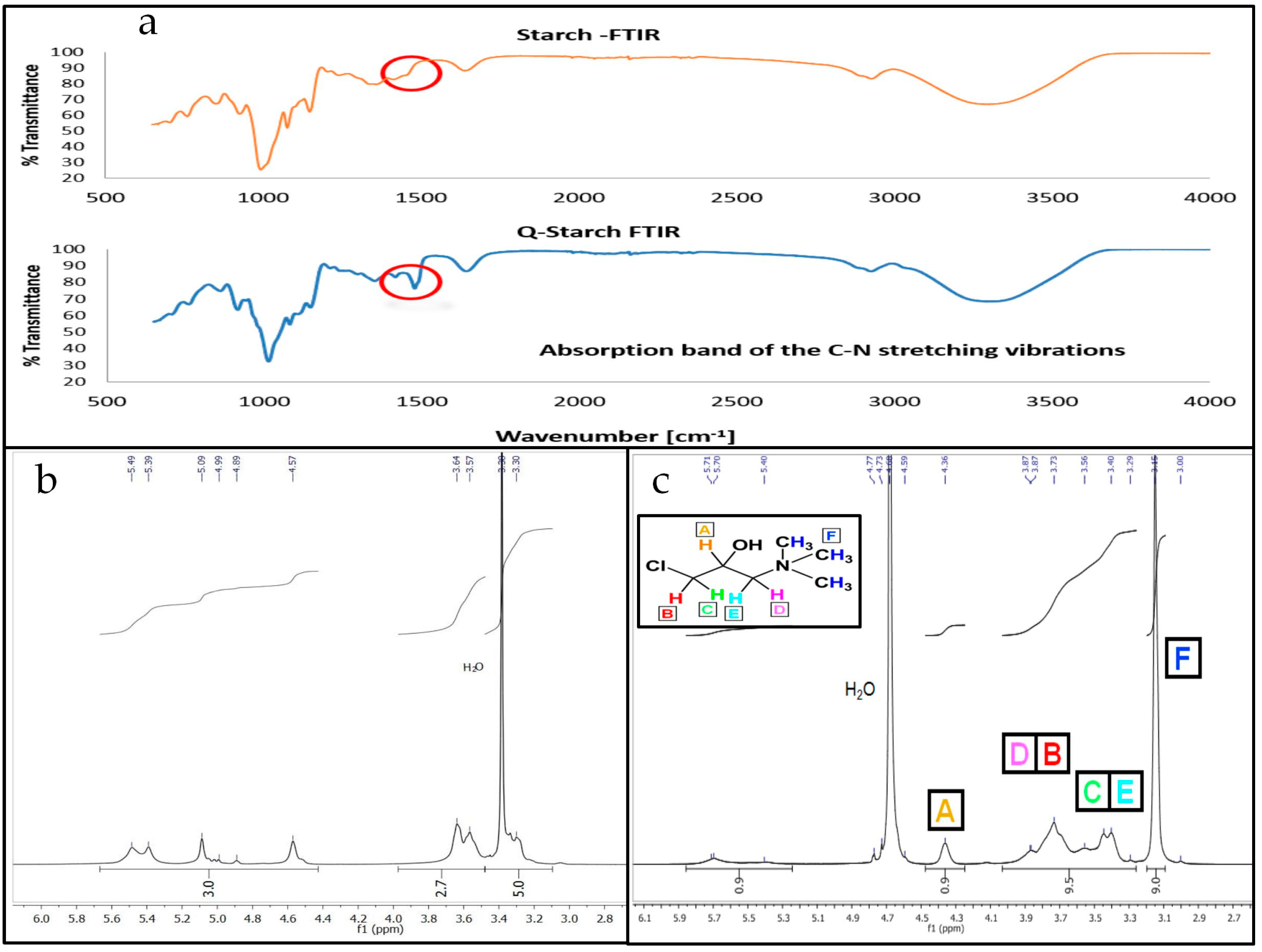
3.1. Q-Starch Synthesis and Chemical Characterization
3.2. Q-Starch/Cargo Complex Characterization
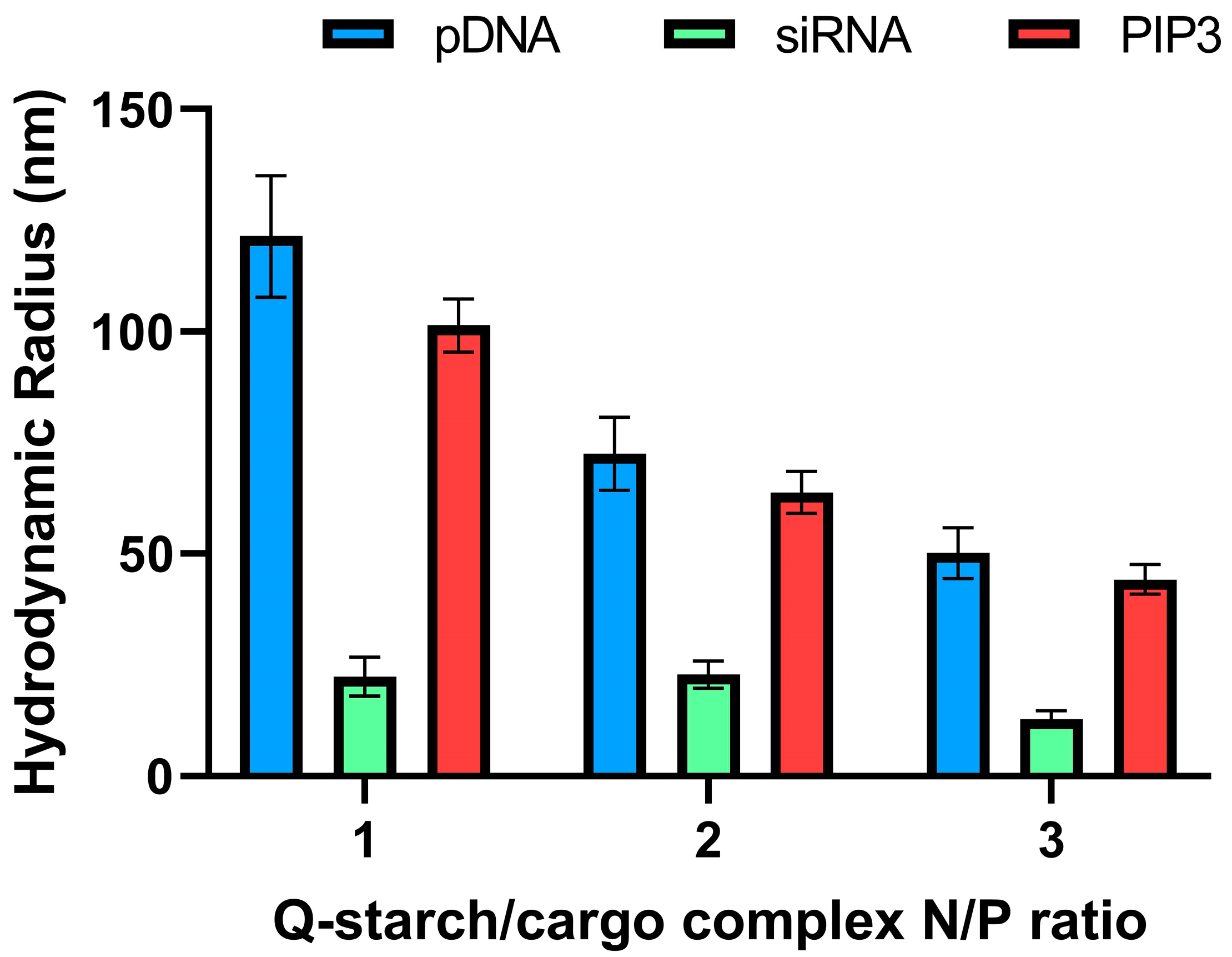
3.2.1. Size
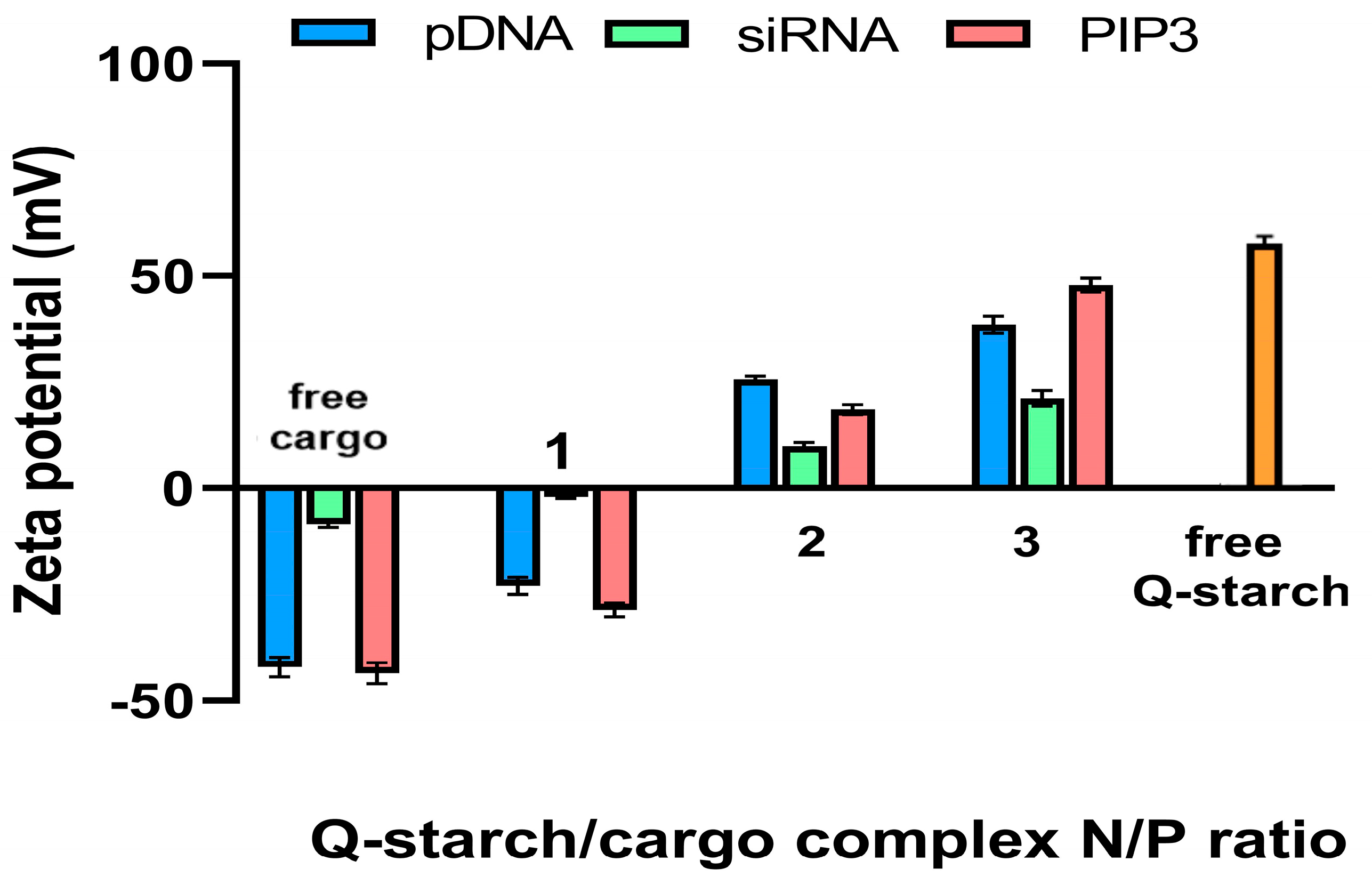
3.2.2. Zeta Potential
3.2.3. Morphology
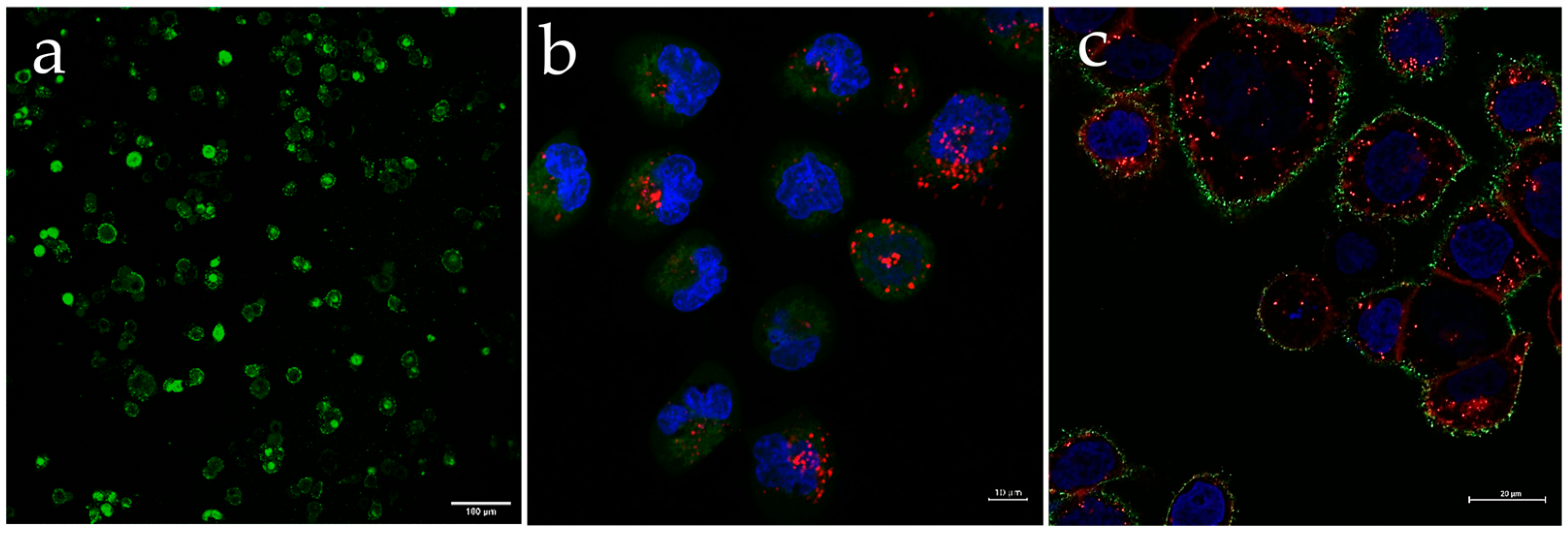
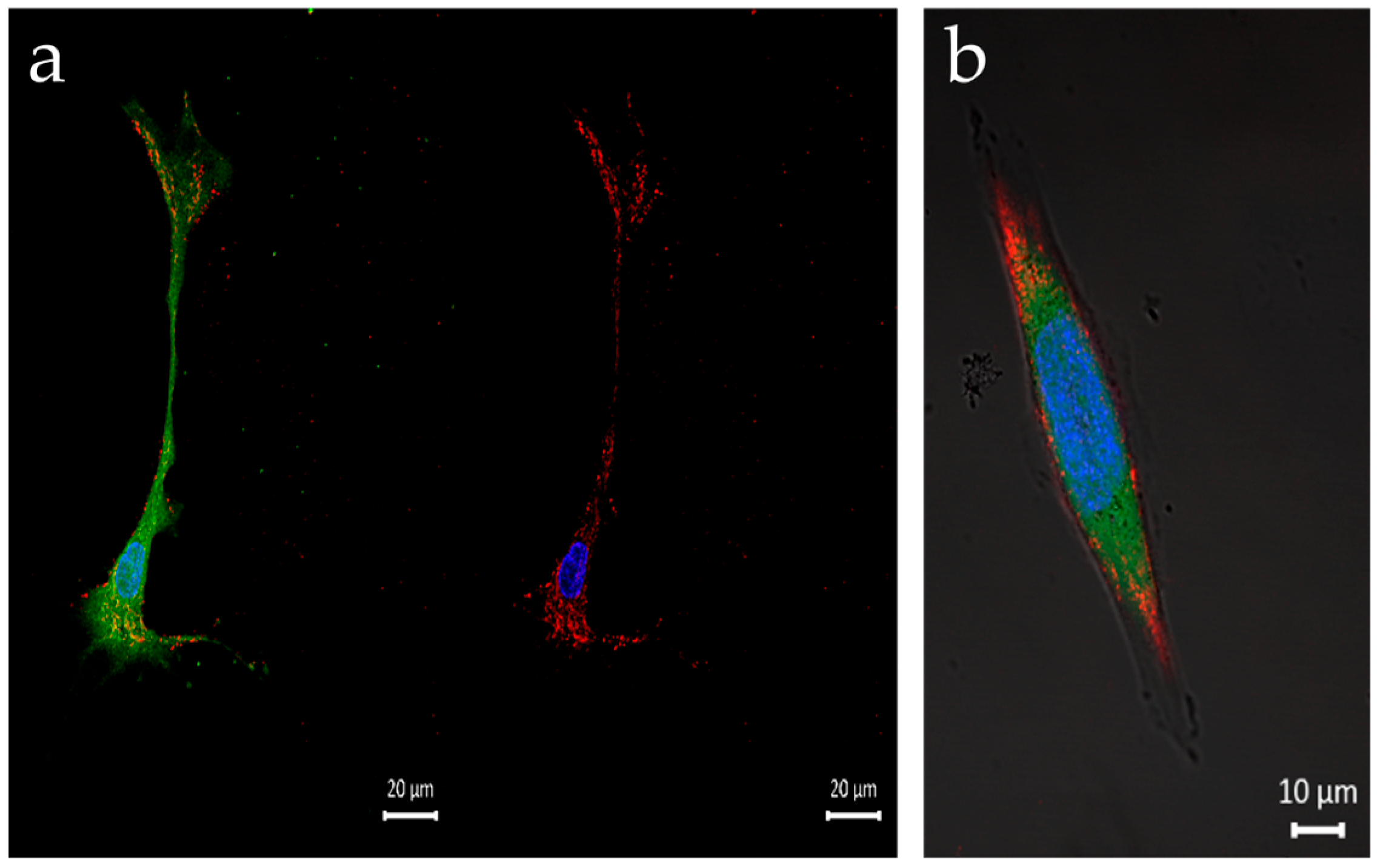
3.3. Q-Starch/Cargo Complexes’ Cellular Localization
3.4. Biological Activity
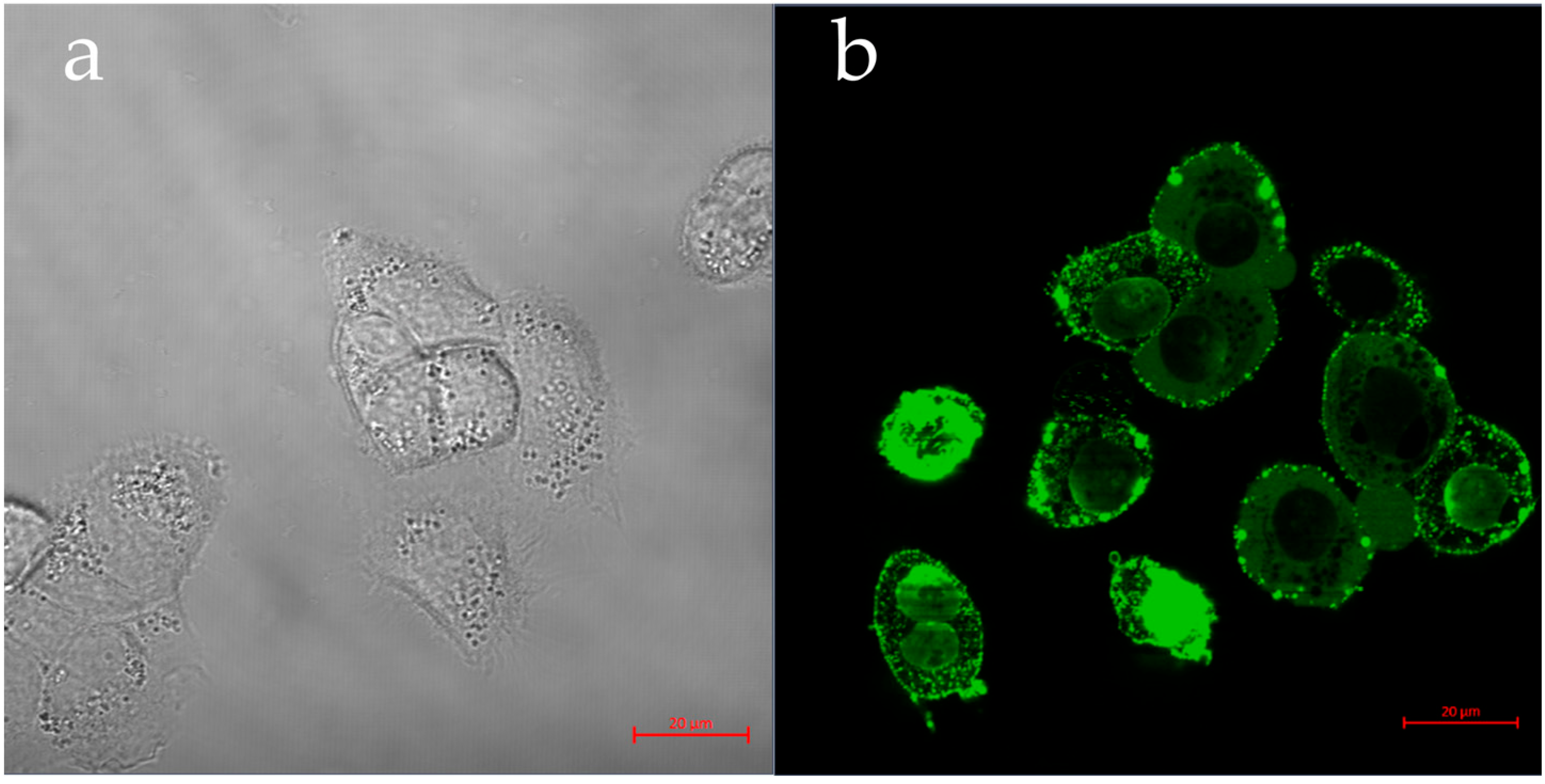
3.4.1. pDNA—Transfection GFP Assay
3.4.2. siRNA—Gene Silencing
3.4.3. PIP3: Western Blot
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.J.; Xu, S.; Wang, H.M.; Ling, Y.; Dong, J.; Xia, R.D.; Sun, X.H. Nanoparticles: Oral Delivery for Protein and Peptide Drugs. AAPS PharmSciTech 2019, 20, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.M.; Choi, Y.H.; Tu, M.J. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef]
- Ma, C.C.; Wang, Z.L.; Xu, T.; He, Z.Y.; Wei, Y.Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Đorđević, S.; Gonzalez, M.M.; Conejos-Sánchez, I.; Carreira, B.; Pozzi, S.; Acúrcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv. Transl. Res. 2021, 12, 500–525. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Prather, K.J.; Prazeres, D.M.F.; Monteiro, G.A. Structural instability of plasmid biopharmaceuticals: Challenges and implications. Trends Biotechnol. 2009, 27, 503–511. [Google Scholar] [CrossRef]
- Elmowafy, M.; Shalaby, K.; Elkomy, M.H.; Alsaidan, O.A.; Gomaa, H.A.M.; Abdelgawad, M.A.; Mostafa, E.M. Polymeric Nanoparticles for Delivery of Natural Bioactive Agents: Recent Advances and Challenges. Polymers 2023, 15, 1123. [Google Scholar] [CrossRef]
- Pekker, M.; Shneider, M.N. The surface charge of a cell lipid membrane. J. Phys. Chem. Biophys. 2014, 5, 2161-0398. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers break barriers in drug delivery: Endocytosis and endosomal escape of gene delivery vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef] [Green Version]
- Finbloom, J.A.; Sousa, F.; Stevens, M.M.; Desai, T.A. Engineering the drug carrier biointerface to overcome biological barriers to drug delivery. Adv. Drug Deliv. Rev. 2020, 167, 89–108. [Google Scholar] [CrossRef]
- Rajeev, A.; Siby, A.; Koottungal, M.J.; George, J.; John, F. Knocking Down Barriers: Advances in siRNA Delivery. ChemistrySelect 2021, 6, 13350–13362. [Google Scholar] [CrossRef]
- Woodley, J. Enzymatic barriers. In Oral Delivery of Macromolecular Drugs; Springer: New York, NY, USA, 2009; pp. 1–19. [Google Scholar] [CrossRef]
- Shete, H.K.; Prabhu, R.H.; Patravale, V.B. Endosomal Escape: A Bottleneck in Intracellular Delivery. J. Nanosci. Nanotechnol. 2014, 14, 460–474. [Google Scholar] [CrossRef]
- Stewart, M.P.; Lorenz, A.; Dahlman, J.; Sahay, G. Challenges in carrier-mediated intracellular delivery: Moving beyond endosomal barriers. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 465–478. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Chen, F. pH-Responsive Drug-Delivery Systems. Chem.–Asian J. 2015, 10, 284–305. [Google Scholar] [CrossRef]
- Hu, Q.; Katti, P.S.; Gu, Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale 2014, 6, 12273–12286. [Google Scholar] [CrossRef]
- Guo, X.; Cheng, Y.; Zhao, X.; Luo, Y.; Chen, J.; Yuan, W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 74. [Google Scholar] [CrossRef] [Green Version]
- Shahriari, M.; Zahiri, M.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Enzyme responsive drug delivery systems in cancer treatment. J. Control. Release 2019, 308, 172–189. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Mackiewicz, M.; Yazdimamaghani, M. Recent advances in the redox-responsive drug delivery nanoplatforms: A chemical structure and physical property perspective. Mater. Sci. Eng. C 2021, 118, 111536. [Google Scholar] [CrossRef]
- Sensini, A.; Gualandi, C.; Cristofolini, L.; Tozzi, G.; Dicarlo, M.; Teti, G.; Mattioli-Belmonte, M.; Letizia Focarete, M. Biofabrication of bundles of poly(lactic acid)-collagen blends mimicking the fascicles of the human Achille tendon. Biofabrication 2017, 9, 015025. [Google Scholar] [CrossRef]
- Bordat, A.; Boissenot, T.; Nicolas, J.; Tsapis, N. Thermoresponsive polymer nanocarriers for biomedical applications. Adv. Drug Deliv. Rev. 2019, 138, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M.; Capasso Palmiero, U.; Moscatelli, D. Thermo-responsive polymers: Applications of smart materials in drug delivery and tissue engineering. Mater. Sci. Eng. C 2019, 102, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Bami, M.S.; Raeisi Estabragh, M.A.; Khazaeli, P.; Ohadi, M.; Dehghannoudeh, G. pH-responsive drug delivery systems as intelligent carriers for targeted drug therapy: Brief history, properties, synthesis, mechanism and application. J. Drug Deliv. Sci. Technol. 2022, 70, 102987. [Google Scholar] [CrossRef]
- Díaz-Montes, E. Polysaccharides: Sources, Characteristics, Properties, and Their Application in Biodegradable Films. Polysaccharides 2022, 3, 480–501. [Google Scholar] [CrossRef]
- Ullah, S.; Khalil, A.A.; Shaukat, F.; Song, Y. Sources, Extraction and Biomedical Properties of Polysaccharides. Foods 2019, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Auriemma, G.; Russo, P.; Del Gaudio, P.; García-González, C.A.; Landín, M.; Aquino, R.P. Technologies and Formulation Design of Polysaccharide-Based Hydrogels for Drug Delivery. Molecules 2020, 25, 3156. [Google Scholar] [CrossRef]
- Barclay, T.G.; Day, C.M.; Petrovsky, N.; Garg, S. Review of polysaccharide particle-based functional drug delivery. Carbohydr. Polym. 2019, 221, 94–112. [Google Scholar] [CrossRef]
- Zhang, M.; Ma, H.; Wang, X.; Yu, B.; Cong, H.; Shen, Y. Polysaccharide-based nanocarriers for efficient transvascular drug delivery. J. Control. Release 2023, 354, 167–187. [Google Scholar] [CrossRef]
- Dattilo, M.; Patitucci, F.; Prete, S.; Parisi, O.I.; Puoci, F. Polysaccharide-Based Hydrogels and Their Application as Drug Delivery Systems in Cancer Treatment: A Review. J. Funct. Biomater. 2023, 14, 55. [Google Scholar] [CrossRef]
- Hou, C.; Chen, L.; Yang, L.; Ji, X. An insight into anti-inflammatory effects of natural polysaccharides. Int. J. Biol. Macromol. 2020, 153, 248–255. [Google Scholar] [CrossRef]
- Jurak, M.; Wiącek, A.E.; Ładniak, A.; Przykaza, K.; Szafran, K. What affects the biocompatibility of polymers? Adv. Colloid Interface Sci. 2021, 294, 102451. [Google Scholar] [CrossRef]
- Naveed, M.; Phil, L.; Sohail, M.; Hasnat, M.; Baig, M.M.F.A.; Ihsan, A.U.; Shumzaid, M.; Kakar, M.U.; Mehmood Khan, T.; Akabar, M.D.; et al. Chitosan oligosaccharide (COS): An overview. Int. J. Biol. Macromol. 2019, 129, 827–843. [Google Scholar] [CrossRef]
- Benedini, L.; Laiuppa, J.; Santillán, G.; Baldini, M.; Messina, P. Antibacterial alginate/nano-hydroxyapatite composites for bone tissue engineering: Assessment of their bioactivity, biocompatibility, and antibacterial activity. Mater. Sci. Eng. C 2020, 115, 111101. [Google Scholar] [CrossRef]
- Zhang, W.; Ren, G.; Xu, H.; Zhang, J.; Liu, H.; Mu, S.; Cai, X.; Wu, T. Genipin cross-linked chitosan hydrogel for the controlled release of tetracycline with controlled release property, lower cytotoxicity, and long-term bioactivity. J. Polym. Res. 2016, 23, 156. [Google Scholar] [CrossRef]
- Campelo, P.H.; Sant’Ana, A.S.; Pedrosa Silva Clerici, M.T. Starch nanoparticles: Production methods, structure, and properties for food applications. Curr. Opin. Food Sci. 2020, 33, 136–140. [Google Scholar] [CrossRef]
- Li, S.; Xiong, Q.; Lai, X.; Li, X.; Wan, M.; Zhang, J.; Yan, Y.; Cao, M.; Lu, L.; Guan, J.; et al. Molecular Modification of Polysaccharides and Resulting Bioactivities. Compr. Rev. Food Sci. Food Saf. 2016, 15, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Wiacek, A.E.; Dul, K. Effect of surface modification on starch/phospholipid wettability. Colloids Surf. A Physicochem. Eng. Asp. 2015, 480, 351–359. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M.; Mehta, M.; Satija, S.; Aljabali, A.A.; Tambuwala, M.M.; Anand, K.; Sharma, N.; Dureja, H.; Jha, N.K.; et al. Current-status and applications of polysaccharides in drug delivery systems. Colloid Interface Sci. Commun. 2021, 42, 100418. [Google Scholar] [CrossRef]
- Simsek, M.; Asiyanbi-Hammed, T.T.; Rasaq, N.; Hammed, A.M. Progress in Bioactive Polysaccharide-Derivatives: A Review. Food Rev. Int. 2021, 39, 1612–1627. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, X.; Wu, J.; Zhao, J. Modifications of polysaccharide-based biomaterials under structure-property relationship for biomedical applications. Carbohydr. Polym. 2021, 266, 118097. [Google Scholar] [CrossRef]
- Khan, T.; Date, A.; Chawda, H.; Patel, K. Polysaccharides as potential anticancer agents—A review of their progress. Carbohydr. Polym. 2019, 210, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Minzanova, S.T.; Mironov, V.F.; Arkhipova, D.M.; Khabibullina, A.V.; Mironova, L.G.; Zakirova, Y.M.; Milyukov, V.A. Biological Activity and Pharmacological Application of Pectic Polysaccharides: A Review. Polymers 2018, 10, 1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonia, T.A.; Sharma, C.P. Chitosan and its derivatives for drug delivery perspective. Adv. Polym. Sci. 2011, 243, 23–54. [Google Scholar] [CrossRef]
- Serrano-Sevilla, I.; Artiga, Á.; Mitchell, S.G.; De Matteis, L.; de la Fuente, J.M. Natural polysaccharides for siRNA delivery: Nanocarriers based on chitosan, hyaluronic acid, and their derivatives. Molecules 2019, 24, 2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrion, C.C.; Nasrollahzadeh, M.; Sajjadi, M.; Jaleh, B.; Soufi, G.J.; Iravani, S. Lignin, lipid, protein, hyaluronic acid, starch, cellulose, gum, pectin, alginate and chitosan-based nanomaterials for cancer nanotherapy: Challenges and opportunities. Int. J. Biol. Macromol. 2021, 178, 193–228. [Google Scholar] [CrossRef]
- Mo, Y.; Wang, H.; Liu, J.; Lan, Y.; Guo, R.; Zhang, Y.; Xue, W.; Zhang, Y. Controlled release and targeted delivery to cancer cells of doxorubicin from polysaccharide-functionalised single-walled carbon nanotubes. J. Mater. Chem. B 2015, 3, 1846–1855. [Google Scholar] [CrossRef]
- Jafernik, K.; Ładniak, A.; Blicharska, E.; Czarnek, K.; Ekiert, H.; Wiącek, A.E.; Szopa, A. Chitosan-Based Nanoparticles as Effective Drug Delivery Systems—A review. Molecules 2023, 28, 1963. [Google Scholar] [CrossRef]
- Sun, Y.; Jing, X.; Ma, X.; Feng, Y.; Hu, H. Versatile Types of Polysaccharide-Based Drug Delivery Systems: From Strategic Design to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 9159. [Google Scholar] [CrossRef]
- Meng, Q.; Zhong, S.; Gao, Y.; Cui, X. Advances in polysaccharide-based nano/microcapsules for biomedical applications: A review. Int. J. Biol. Macromol. 2022, 220, 878–891. [Google Scholar] [CrossRef]
- Du, C.; Zhao, J.; Fei, J.; Cui, Y.; Li, J. Assembled Microcapsules by Doxorubicin and Polysaccharide as High Effective Anticancer Drug Carriers. Adv. Healthc. Mater. 2013, 2, 1246–1251. [Google Scholar] [CrossRef]
- Ma, G.; Liu, Y.; Fang, D.; Chen, J.; Peng, C.; Fei, X.; Nie, J. Hyaluronic acid/chitosan polyelectrolyte complexes nanofibers prepared by electrospinning. Mater. Lett. 2012, 74, 78–80. [Google Scholar] [CrossRef]
- Lifshiz Zimon, R.; Lerman, G.; Elharrar, E.; Meningher, T.; Barzilai, A.; Masalha, M.; Chintakunta, R.; Hollander, E.; Goldbart, R.; Traitel, T.; et al. Ultrasound targeting of Q-starch/miR-197 complexes for topical treatment of psoriasis. J. Control. Release 2018, 284, 103–111. [Google Scholar] [CrossRef]
- Sieradzki, R.; Traitel, T.; Goldbart, R.; Geresh, S.; Kost, J. Tailoring quaternized starch as a non-viral carrier for gene delivery applications. Polym. Adv. Technol. 2014, 25, 552–561. [Google Scholar] [CrossRef]
- Amar-Lewis, E.; Azagury, A.; Chintakunta, R.; Goldbart, R.; Traitel, T.; Prestwood, J.; Landesman-Milo, D.; Peer, D.; Kost, J. Quaternized starch-based carrier for siRNA delivery: From cellular uptake to gene silencing. J. Control. Release 2014, 185, 109–120. [Google Scholar] [CrossRef]
- Zheng, Y.; Bai, L.; Zhou, Y.; Tong, R.; Zeng, M.; Li, X.; Shi, J. Polysaccharides from Chinese herbal medicine for anti-diabetes recent advances. Int. J. Biol. Macromol. 2019, 121, 1240–1253. [Google Scholar] [CrossRef]
- Faggio, C.; Morabito, M.; Minicante, S.A.; Lo Piano, G.; Pagano, M.; Genovese, G. Potential use of polysaccharides from the brown alga undaria pinnatifida as anticoagulants. Braz. Arch. Biol. Technol. 2015, 58, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Caprio, C.; Varricchio, S.; Bilio, M.; Feo, F.; Ferrentino, R.; Russo, D.; Staibano, S.; Alfano, D.; Missero, C.; Ilardi, G.; et al. TBX1 and Basal Cell Carcinoma: Expression and Interactions with Gli2 and Dvl2 Signaling. Int. J. Mol. Sci. 2020, 21, 607. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, Y.S.; Lee, C.; Shin, M.S.; Kim, J.W.; Jang, B.G. Expression profile of sonic hedgehog signaling-related molecules in basal cell carcinoma. PLoS ONE 2019, 14, e0225511. [Google Scholar] [CrossRef] [Green Version]
- Gobin, B.; Huin, M.B.; Lamoureux, F.; Ory, B.; Charrier, C.; Lanel, R.; Battaglia, S.; Redini, F.; Lezot, F.; Blanchard, F.; et al. BYL719, a new α-specific PI3K inhibitor: Single administration and in combination with conventional chemotherapy for the treatment of osteosarcoma. Int. J. Cancer 2015, 136, 784–796. [Google Scholar] [CrossRef]
- Keam, B.; Kim, S.; Ahn, Y.O.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Heo, D.S. In Vitro Anticancer Activity of PI3K Alpha Selective Inhibitor BYL719 in Head and Neck Cancer. Anticancer Res. 2015, 35, 175–182. [Google Scholar]
- Amar-Lewis, E.; Buaron, N.; Chintakunta, R.; Benafsha, C.; Goldbart, R.; Traitel, T.; Prasad, M.; Elkabets, M.; Kost, J. Quaternized Starch-Based Composite Nanoparticles for siRNA Delivery to Tumors. ACS Appl. Nano Mater. 2021, 4, 2218–2229. [Google Scholar] [CrossRef]
- Shang, L.; Nienhaus, K.; Nienhaus, G.U. Engineered nanoparticles interacting with cells: Size matters. J. Nanobiotechnol. 2014, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francia, V.; Reker-Smit, C.; Boel, G.; Salvati, A. Limits and challenges in using transport inhibitors to characterize how nano-sized drug carriers enter cells. Nanomedicine 2019, 14, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.P.; Dean, D.A. Progress and prospects: Nuclear import of nonviral vectors. Gene Ther. 2010, 17, 439–447. [Google Scholar] [CrossRef]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217. [Google Scholar] [CrossRef] [Green Version]
- Padda, I.S.; Mahtani, A.U.; Parmar, M. Small Interfering RNA (siRNA)-Based Therapy. In StatPearls; StatPearls: Tampa, FL, USA, 2023. [Google Scholar]
- Khelghati, N.; Mokhtarvand, J.S.; Mir, M.; Alemi, F.; Asemi, Z.; Sadeghpour, A.; Maleki, M.; Kafil, H.S.; Jadidi-niaragh, F.; Majidinia, M.; et al. The importance of co-delivery of nanoparticle-siRNA and anticancer agents in cancer therapy. Chem. Biol. Drug Des. 2021, 97, 997–1015. [Google Scholar] [CrossRef]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, S.; Stecca, B. Hedgehog-Gli signaling in basal cell carcinoma and other skin cancers: Prospects for therapy. Res. Rep. Biology. 2015, 6, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Kump, E.; Wernli, M.; Erb, P. Gene silencing of transcription factor Gli2 inhibits basal cell carcinomalike tumor growth in vivo. Int. J. Cancer. 2008, 122, 50–56. [Google Scholar] [CrossRef]
- Leavitt, E.; Lask, G.; Martin, S. Sonic Hedgehog Pathway Inhibition in the Treatment of Advanced Basal Cell Carcinoma. Curr. Treat. Options Oncol. 2019, 20, 84. [Google Scholar] [CrossRef]
- Grund-Gröschke, S.; Ortner, D.; Szenes-Nagy, A.B.; Zaborsky, N.; Weiss, R.; Neureiter, D.; Wipplinger, M.; Risch, A.; Hammerl, P.; Greil, R.; et al. Epidermal activation of Hedgehog signaling establishes an immunosuppressive microenvironment in basal cell carcinoma by modulating skin immunity. Mol. Oncol. 2020, 14, 1930–1946. [Google Scholar] [CrossRef]
- Grund-Gröschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI signaling in tumor immunity—New therapeutic opportunities and clinical implications. Cell Commun. Signal. 2019, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef]
- Soares, J.A.P.; Leite, F.G.G.; Andrade, L.G.; Torres, A.A.; De Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.P.; Kroon, E.G.; Souto-Padrón, T.; et al. Activation of the PI3K/Akt Pathway Early during Vaccinia and Cowpox Virus Infections Is Required for both Host Survival and Viral Replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef] [Green Version]
- Walkowski, B.; Kleibert, M.; Majka, M.; Wojciechowska, M. Insight into the Role of the PI3K/Akt Pathway in Ischemic Injury and Post-Infarct Left Ventricular Remodeling in Normal and Diabetic Heart. Cells 2022, 11, 1553. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Q-StarchDTAF/pDNA | Q-Starch/siRNA NC5Cy5 | Q-StarchDTAF/PIP3 | |
|---|---|---|---|
| N/P ratio | 2 | 2 | 2 |
| Cargo concentration | 4 mg/mL (1 nM) | 100 nM | 500 nM |
| Incubation time | 4 h | 4 h | 4 h |
| Labeling | Q-starchDTAF/pDNA: green | Cell’s nucleus: blue; NucBlue Cell’s cytoplasm: green; Viafluor488 Q-starch/siRNA NC5Cy5: red | Cell’s nucleus: blue; NucBlue Cell’s cytoplasm: red; CellBrite Q-starchDTAF/PIP3: green |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blitsman, Y.; Benafsha, C.; Yarza, N.; Zorea, J.; Goldbart, R.; Traitel, T.; Elkabets, M.; Kost, J. Cargo-Dependent Targeted Cellular Uptake Using Quaternized Starch as a Carrier. Nanomaterials 2023, 13, 1988. https://doi.org/10.3390/nano13131988
Blitsman Y, Benafsha C, Yarza N, Zorea J, Goldbart R, Traitel T, Elkabets M, Kost J. Cargo-Dependent Targeted Cellular Uptake Using Quaternized Starch as a Carrier. Nanomaterials. 2023; 13(13):1988. https://doi.org/10.3390/nano13131988
Chicago/Turabian StyleBlitsman, Yossi, Chen Benafsha, Nir Yarza, Jonathan Zorea, Riki Goldbart, Tamar Traitel, Moshe Elkabets, and Joseph Kost. 2023. "Cargo-Dependent Targeted Cellular Uptake Using Quaternized Starch as a Carrier" Nanomaterials 13, no. 13: 1988. https://doi.org/10.3390/nano13131988
APA StyleBlitsman, Y., Benafsha, C., Yarza, N., Zorea, J., Goldbart, R., Traitel, T., Elkabets, M., & Kost, J. (2023). Cargo-Dependent Targeted Cellular Uptake Using Quaternized Starch as a Carrier. Nanomaterials, 13(13), 1988. https://doi.org/10.3390/nano13131988