
Au Nanoparticle-Based Amplified DNA Detection on Poly-l-lysine Monolayer-Functionalized Electrodes

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
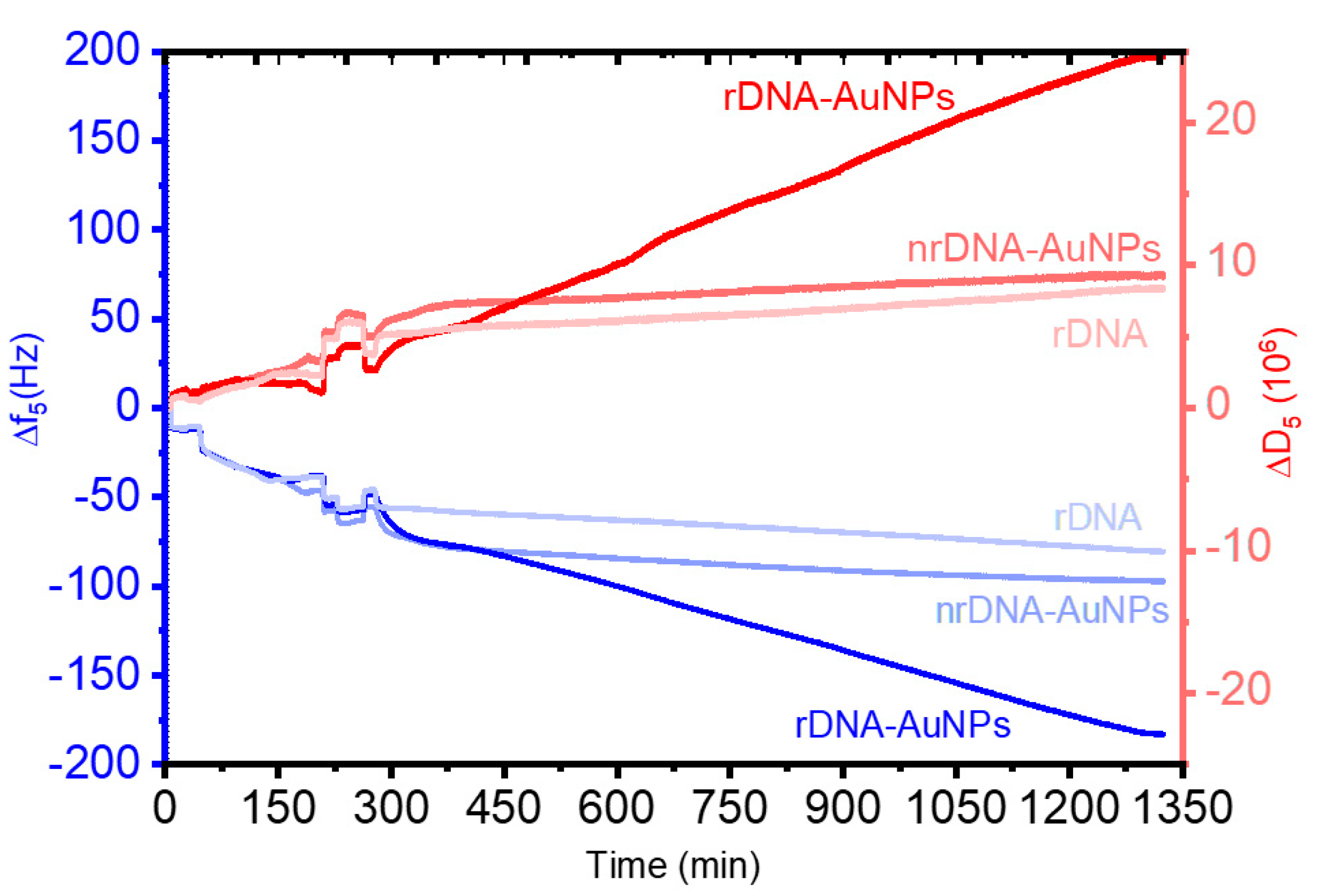
2.2.1. Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D)
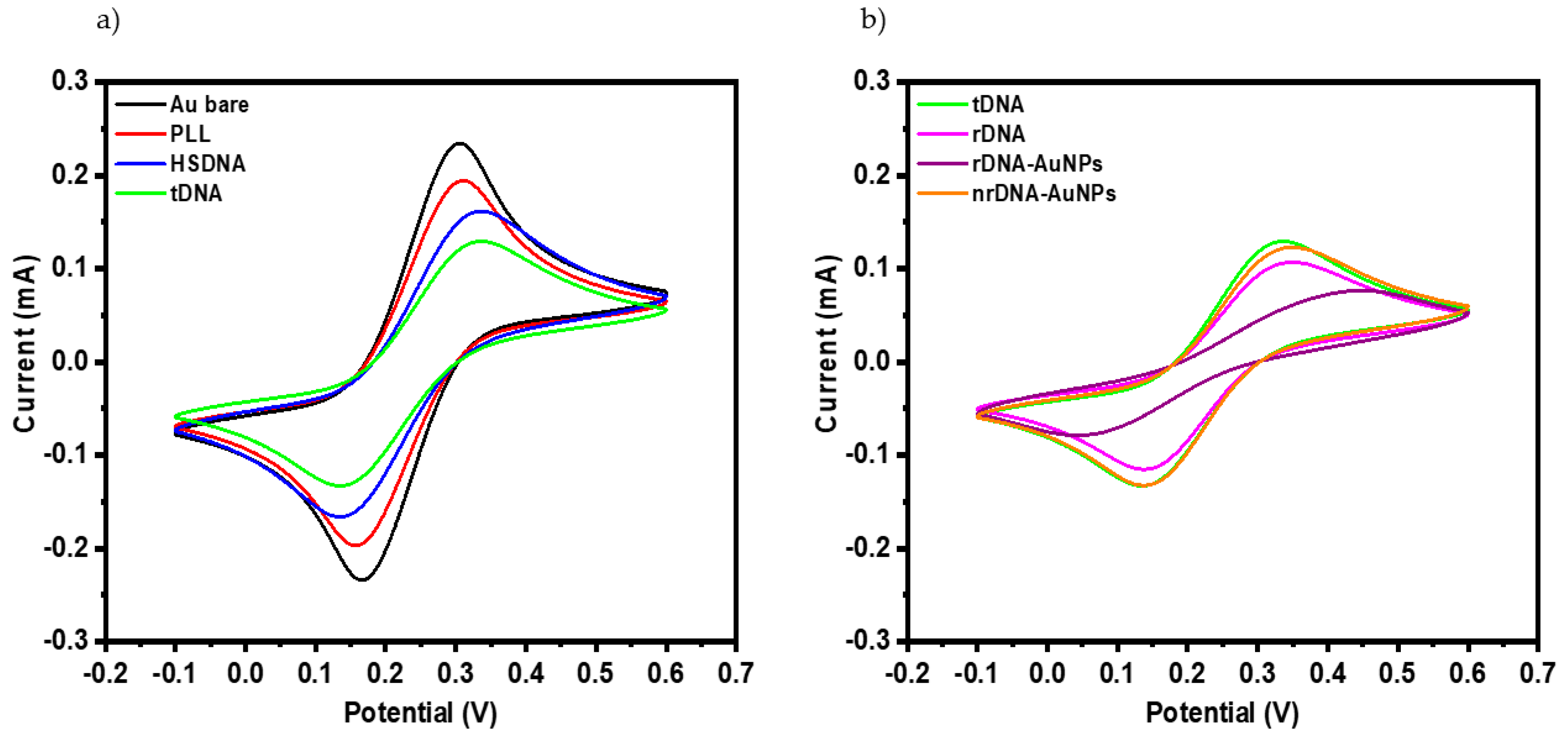
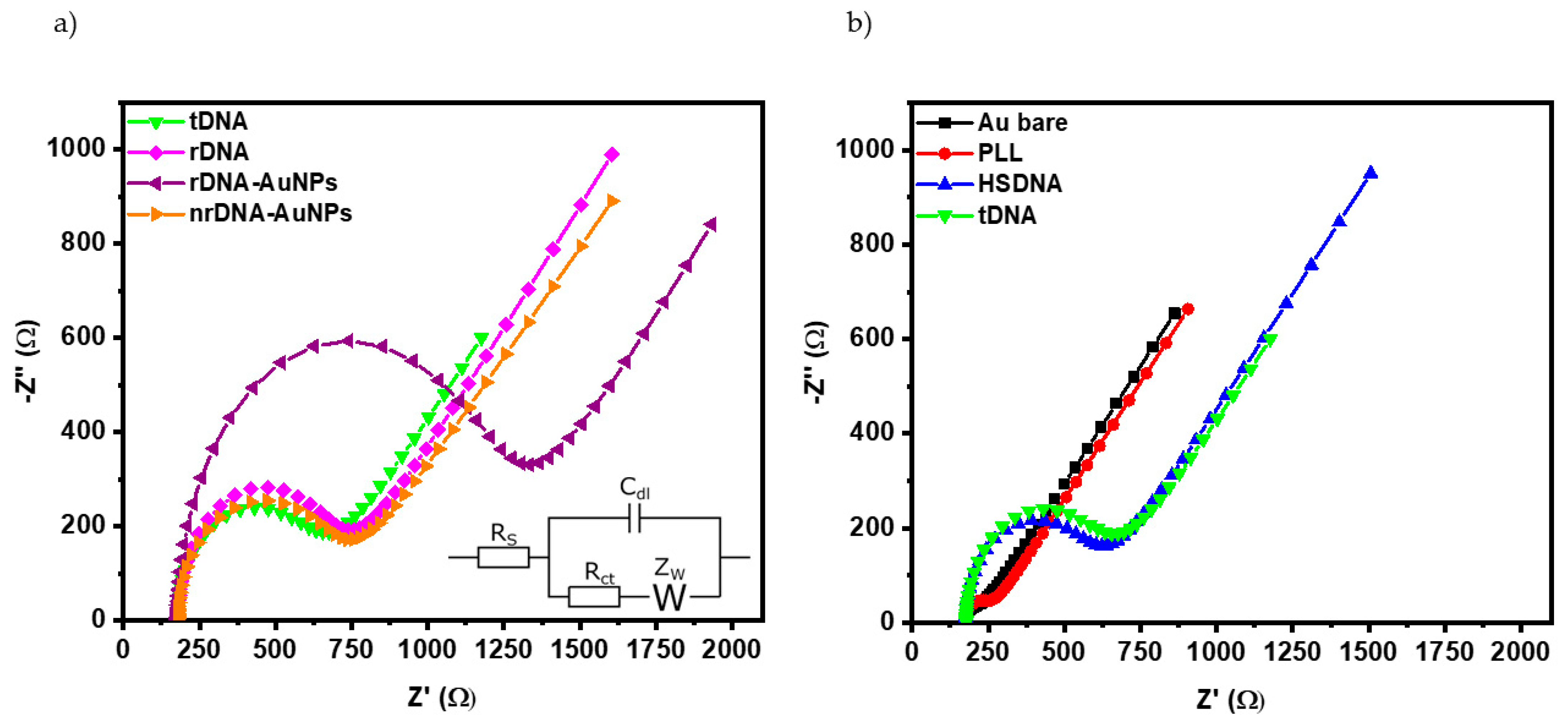
2.2.2. Electrochemical Measurements
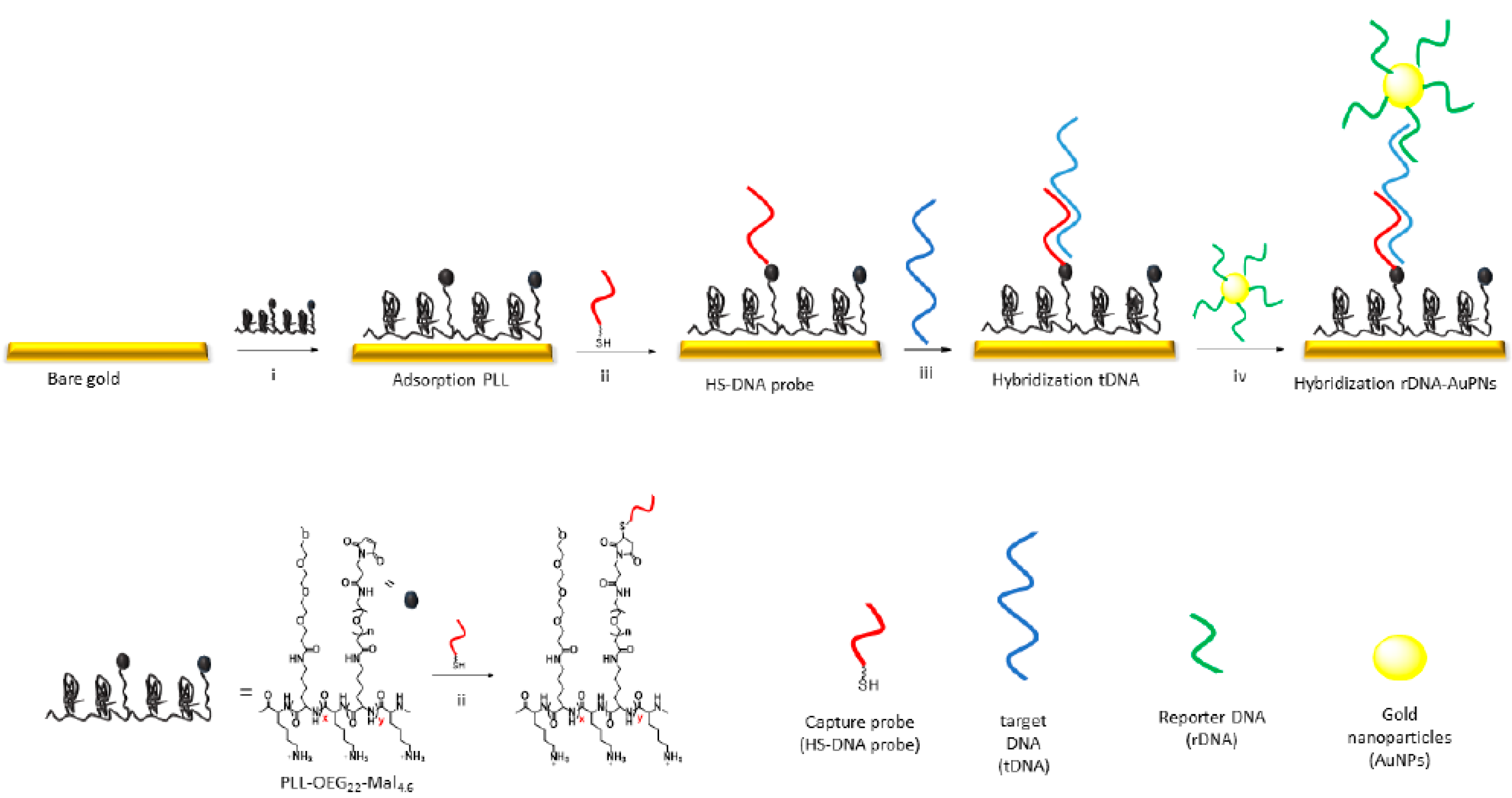
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, A.P.F. Biosensors: Sense and sensibility. Chem. Soc. Rev. 2013, 42, 3184–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooding, J.J.; Darwish, N. The rise of self-assembled monolayers for fabricating electrochemical biosensors-an interfacial perspective. Chem. Rec. 2012, 12, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Srisombat, L.; Jamison, A.C.; Lee, T.R. Stability: A key issue for self-assembled monolayers on gold as thin-film coatings and nanoparticle protectants. Coll. Surf. A Physicochem. Eng. Asp. 2011, 390, 1–19. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challenges for a well-known system. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Shivashankar, M.; Vinodini, V.R.; Mishra, P.; Uma, K. Applications of implantable medical sensors for heart failure: A review. Int. J. Pharm. Pharm. Sci. 2014, 6, 1–5. [Google Scholar]
- Sassolas, A.; Leca-Bouvier, B.D.; Blum, L.J. DNA biosensors and microarrays. Chem. Rev. 2008, 108, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Kerman, K.; Nagatani, N.; Takamura, Y.; Tamiya, E. Label-free detection of peptide nucleic acid-DNA hybridization using localized surface plasmon resonance based optical biosensor. Anal. Chem. 2005, 77, 6976–6984. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, F.; Tombelli, S.; Minunni, M.; Marrazza, G.; Mascini, M. Electrochemical and piezoelectric DNA biosensors for hybridization detection. Anal. Chim. Acta 2008, 609, 139–159. [Google Scholar] [CrossRef]
- Squires, T.M.; Messinger, R.J.; Manalis, S.R. Making it stick: Convection, reaction and diffusion in surface-based biosensors. Nat. Biotechnol. 2008, 26, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Ravan, H.; Kashanian, S.; Sanadgol, N.; Badoei-Dalfard, A.; Karami, Z. Strategies for optimizing DNA hybridization on surfaces. Anal. Biochem. 2014, 444, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bellassai, N.; Marti, A.; Spoto, G.; Huskens, J. Low-fouling, mixed-charge poly-l-lysine polymers with anionic oligopeptide side-chains. J. Mater. Chem. B 2018, 6, 7662–7673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yang, X.; Yuan, R.; Chai, Y. Switchable target-responsive 3D DNA hydrogels as a signal amplification strategy combining with SERS technique for ultrasensitive detection of miRNA 155. Anal. Chem. 2017, 89, 8538–8544. [Google Scholar] [CrossRef] [PubMed]
- Malecka, K.; Grabowska, I.; Radecki, J.; Stachyra, A.; Góra-Sochacka, A.; Sirko, A.; Radecka, H. Voltammetric Detection of a Specific DNA Sequence of Avian Influenza Virus H5N1 Using HS-ssDNA Probe Deposited onto Gold Electrode. Electroanalysis 2012, 2, 439–446. [Google Scholar] [CrossRef]
- Morisset, D.; Stebih, D.; Cankar, K.; Zel, J.; Gruden, K. Alternative DNA amplification methods to PCR and their application in GMO detection: A review. Eur. Food Res. Technol. 2008, 227, 1287–1297. [Google Scholar] [CrossRef]
- Kurzątkowska, K.; Sirko, A.; Zagórski-Ostoja, W.; Dehaen, W.; Radecka, H.; Radecki, J. Electrochemical label-free and reagentless genosensor based on an ion barrier switch-off system for DNA sequence-specific detection of the avian influenza virus. Anal. Chem. 2015, 87, 9702–9709. [Google Scholar] [CrossRef] [PubMed]
- Bezinge, L.; Suea-Ngam, A.; deMello, A.J.; Shih, C.J. Nanomaterials for molecular signal amplification in electrochemical nucleic acid biosensing: Recent advances and future prospects for point-of-care diagnostics. Mol. Syst. Des. Eng. 2020, 5, 49–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Nanomaterial-based amplified transduction of biomolecular interactions. Small 2005, 1, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Marín, S.; Merkoçi, A. Nanomaterials based electrochemical sensing applications for safety and security. Electroanalysis 2012, 24, 459–469. [Google Scholar] [CrossRef]
- Zanoli, L.M.; D’Agata, R.; Spoto, G. Functionalized gold nanoparticles for ultrasensitive DNA detection. Anal. Bioanal. Chem. 2012, 402, 1759–1771. [Google Scholar] [CrossRef]
- Bellassai, N.; D’Agata, R.; Marti, A.; Rozzi, A.; Allegretti, M.; Corradini, R.; Giacomini, P.; Huskens, J.; Spoto, G. Straightforward detection of tumor DNA in whole human plasma with a dual-functional low-fouling PLL-based surface layer and nanoparticle-enhanced plasmonic biosensing. ACS Sens. 2021, 6, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, D.; Marti, A.; Koeman, S.; Huskens, J. Clickable poly-l-lysine for the formation of biorecognition surfaces. RSC Adv. 2019, 9, 35608–35613. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Song, W.; Guo, X.; Wang, Z. Ultrasensitive detection of nucleic acids and proteins using quartz crystal microbalance and surface plasmon resonance sensors based on target-triggering multiple signal amplification strategy. Anal. Chim. Acta 2017, 978, 42–47. [Google Scholar] [CrossRef]
- Movilli, J.; Rozzi, A.; Ricciardi, R.; Corradini, R.; Huskens, J. Control of Probe Density at DNA Biosensor Surfaces Using Poly(l-lysine) with Appended Reactive Groups. Bioconjug. Chem. 2018, 29, 4110–4118. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Cao, X.; Wang, L.; Lan, T.; Li, Y.; Xie, G. DNA-AuNPs based signal amplification for highly sensitive detection of DNA methylation, methyltransferase activity and inhibitor screening. Biosens. Bioelectron. 2014, 58, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Castro, R.; Santos-Álvarez, N.D.L.; Lobo-Castañón, M.J. Understanding the factors affecting the analytical performance of sandwich-hybridization genosensors on gold electrodes. Electroanalysis 2018, 30, 1229–1240. [Google Scholar] [CrossRef]
- He, P.; Liu, L.; Qiao, W.; Zhang, S. Ultrasensitive detection of thrombin using surface plasmon resonance and quartz crystal microbalance sensors by aptamer-based rolling circle amplification and nanoparticle signal enhancement. Chem. Commun. 2014, 50, 1481–1484. [Google Scholar] [CrossRef]
- Dong, Z.M.; Jin, X.; Zhao, G.C. Amplified QCM biosensor for type IV collagenase based on collagenase-cleavage of gold nanoparticles functionalized peptide. Biosens. Bioelectron. 2018, 106, 111–116. [Google Scholar] [CrossRef]
- Liu, L.S.; Wu, C.; Zhang, S. Ultrasensitive detection of DNA and Ramos cell using in situ selective crystallization based Quartz Crystal Microbalance. Anal. Chem. 2017, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Farka, Z.; Juřík, T.; Pastucha, M.; Skládal, P. Enzymatic precipitation enhanced surface plasmon resonance immunosensor for the detection of Salmonella in powdered milk. Anal. Chem. 2016, 88, 11830–11836. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, P.; Wang, D.; Liu, Y.; Wei, W.; Zhang, Y.; Liu, S. Quartz Crystal Microbalance detection of poly(ADP-ribose) Polymerase-1 based on gold nanorods signal amplification. Anal. Chem. 2019, 91, 11038–11044. [Google Scholar] [CrossRef] [PubMed]
- Höök, F.; Ray, A.; Nordén, B.; Kasemo, B. Characterization of PNA and DNA immobilization and subsequent hybridization with DNA using acoustic-shear-wave attenuation measurements. Langmuir 2001, 17, 8305–8312. [Google Scholar] [CrossRef]
- Larsson, C.; Rodahl, M.; Höök, F. Characterization of DNA immobilization and subsequent hybridization on a 2D arrangement of streptavidin on a biotin-modified lipid bilayer supported on SiO2. Anal. Chem. 2003, 75, 5080–5087. [Google Scholar] [CrossRef]
- Su, X.; Wu, Y.J.; Knoll, W. Comparison of surface plasmon resonance spectroscopy and quartz crystal microbalance techniques for studying DNA assembly and hybridization. Biosens. Bioelectron. 2005, 21, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Moazzenzade, T.; Huskens, J.; Lemay, S.G. Stochastic electrochemistry at ultralow concentrations: The case for digital sensors. Analyst 2020, 145, 750–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanni, A.; Del Valle, M. Use of nanomaterials for impedimetric DNA sensors: A review. Anal. Chim. Acta 2010, 678, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steel, A.B.; Herne, T.M.; Tarlov, M.J. Electrochemical quantitation of DNA immobilized on gold. Anal. Chem. 1998, 70, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Lao, R.; Song, S.; Wu, H.; Wang, L.; Zhang, Z.; He, L.; Fan, C. Electrochemical interrogation of DNA monolayers on gold surfaces. Anal. Chem. 2005, 77, 6475–6480. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Liu, Y.; Tabata, M.; Zhu, H.; Miyahara, Y. Sensitive detection of microRNA by chronocoulometry and rolling circle amplification on a gold electrode. Chem. Commun. 2014, 50, 9704–9706. [Google Scholar] [CrossRef]
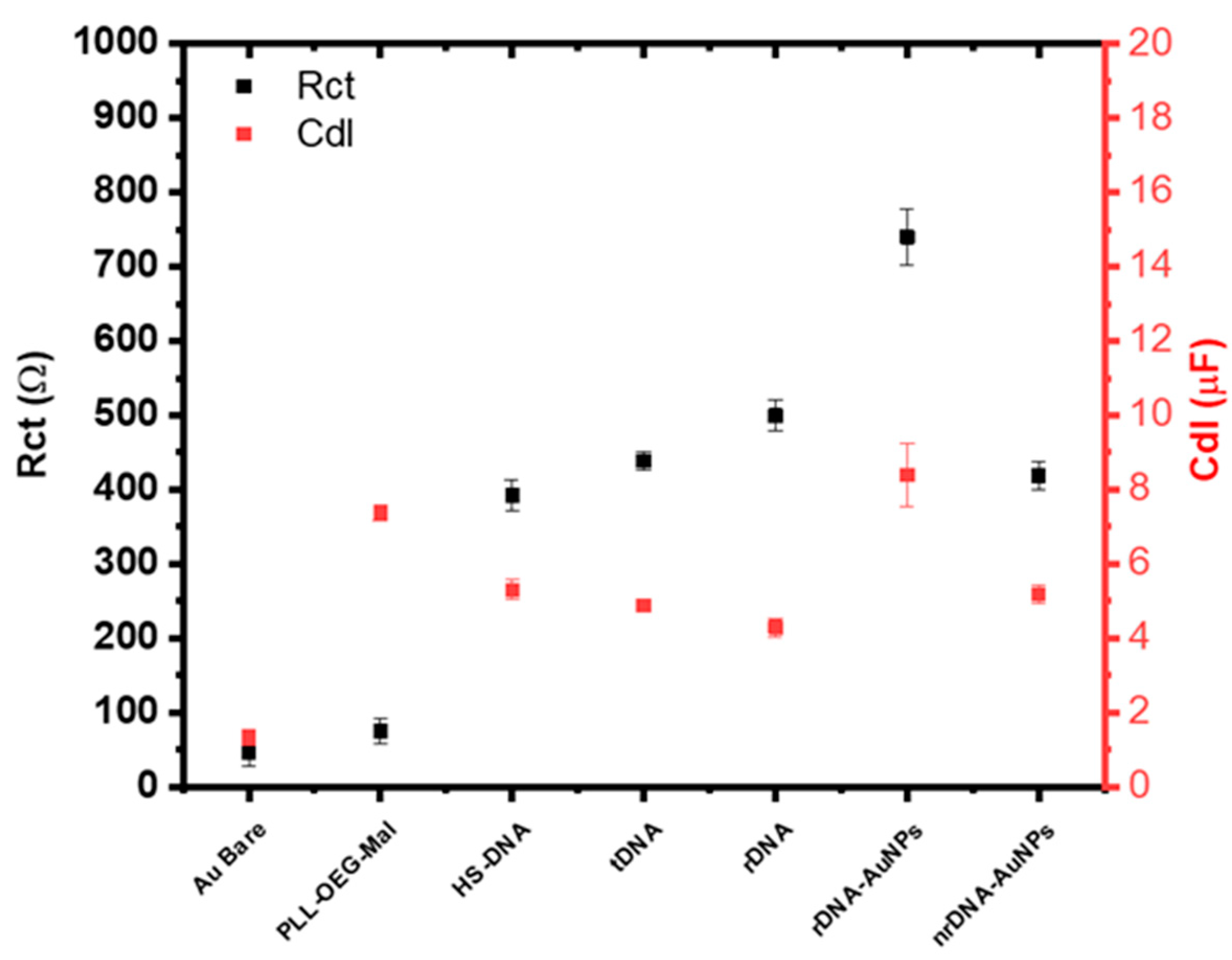
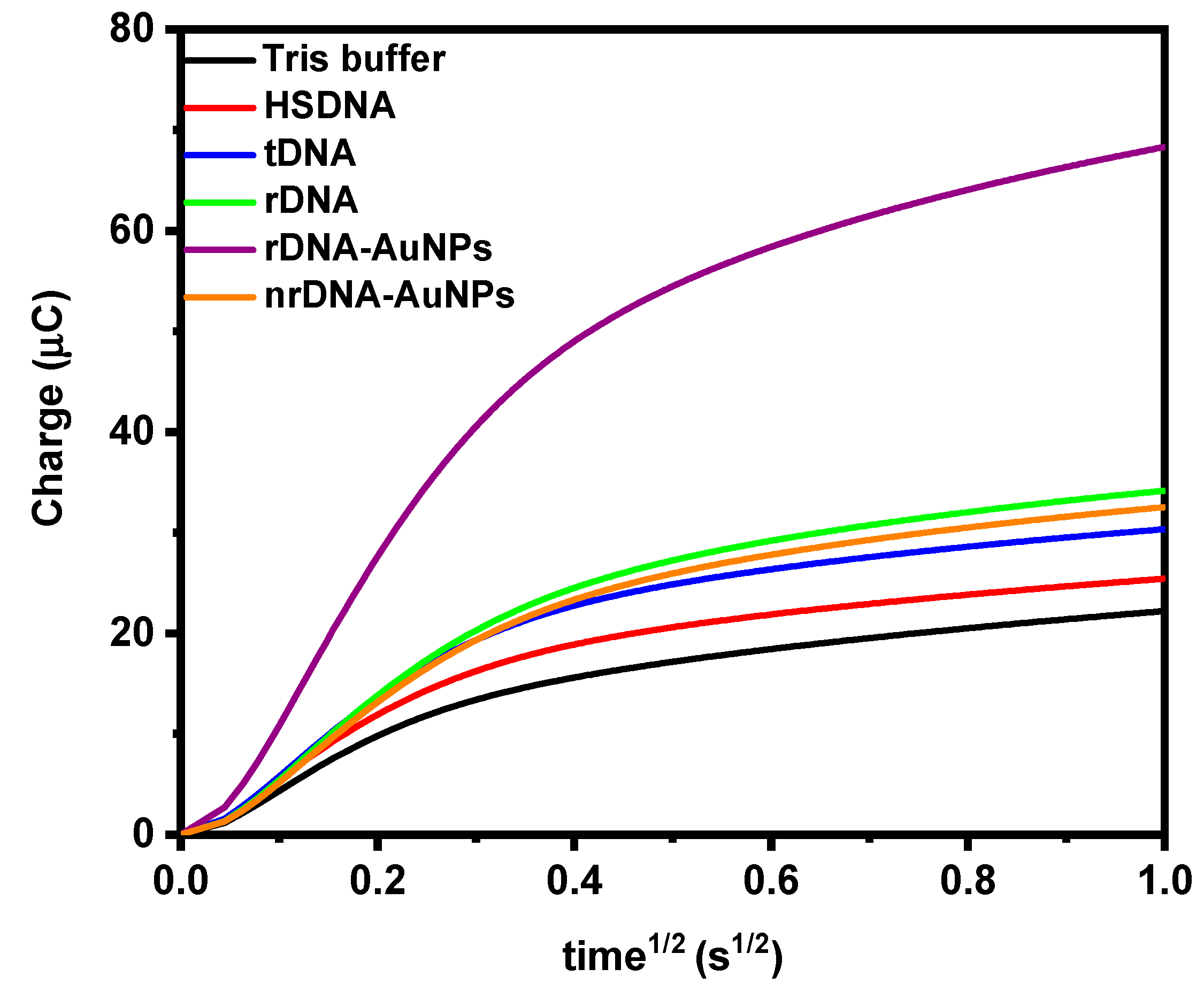

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybridization Step | ∆s/∆p ± σ 1 |
|---|---|
| tDNA | 1.15 ± 0.02 |
| rDNA | 1.17 ± 0.06 |
| rDNA-AuNPs | 1.57 ± 0.08 |
| nrDNA-AuNPs | 0.94 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marti, A.; Huskens, J. Au Nanoparticle-Based Amplified DNA Detection on Poly-l-lysine Monolayer-Functionalized Electrodes. Nanomaterials 2022, 12, 242. https://doi.org/10.3390/nano12020242
Marti A, Huskens J. Au Nanoparticle-Based Amplified DNA Detection on Poly-l-lysine Monolayer-Functionalized Electrodes. Nanomaterials. 2022; 12(2):242. https://doi.org/10.3390/nano12020242
Chicago/Turabian StyleMarti, Almudena, and Jurriaan Huskens. 2022. "Au Nanoparticle-Based Amplified DNA Detection on Poly-l-lysine Monolayer-Functionalized Electrodes" Nanomaterials 12, no. 2: 242. https://doi.org/10.3390/nano12020242
APA StyleMarti, A., & Huskens, J. (2022). Au Nanoparticle-Based Amplified DNA Detection on Poly-l-lysine Monolayer-Functionalized Electrodes. Nanomaterials, 12(2), 242. https://doi.org/10.3390/nano12020242