
The First-Principle Study on Tuning Optical Properties of MA2Z4 by Cr Replacement of Mo Atoms in MoSi2N4

 ,
,
Abstract
:1. Introduction
2. Computation Details
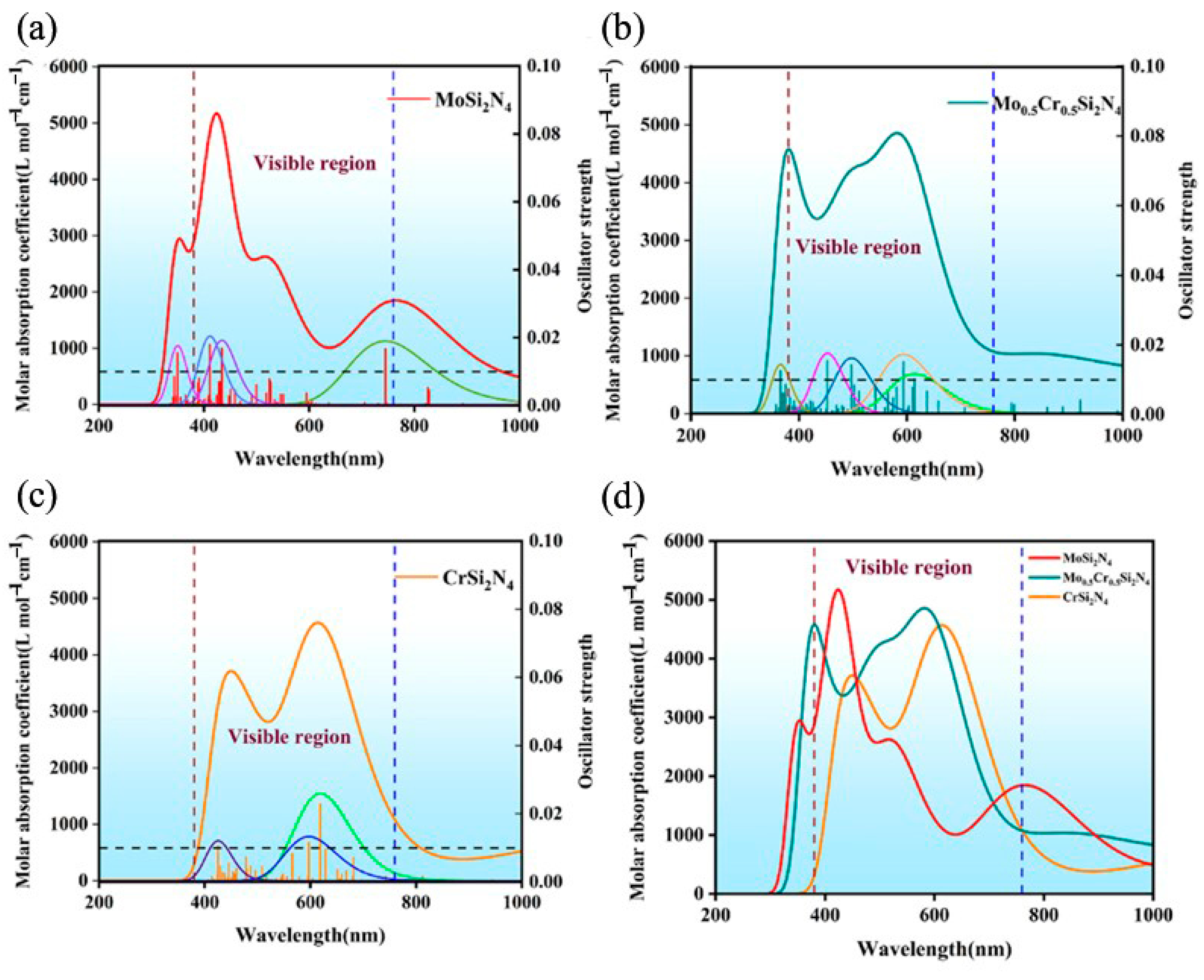
3. Results, Analysis and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hashimoto, A.; Suenaga, K.; Gloter, A.; Urita, K.; Iijima, S. Direct evidence for atomic defects in graphene layers. Nature 2004, 430, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, X.; Liu, B.; Deng, S.; Xie, D.; Liu, Q.; Wang, Y.; Wu, J.; Wang, X.; Tu, J. Multiscale graphene-based materials for applications in sodium ion batteries. Adv. Energy Mater. 2019, 9, 1803342. [Google Scholar] [CrossRef]
- Panda, P.K.; Dash, P.; Yang, J.M.; Chang, Y. Development of chitosan, graphene oxide, and cerium oxide composite blended films: Structural, physical, and functional properties. Cellulose 2022, 29, 2399–2411. [Google Scholar] [CrossRef]
- Sw, A.; Yla, B.; Xin, W.A.; Zi, G.; Zhou, C.; Liu, B.; Liu, G.; Wang, L.; Huang, W. One-step supramolecular preorganization constructed crinkly graphitic carbon nitride nanosheets with enhanced photocatalytic activity. J. Mater. Sci. Technol. 2021, 104, 155–162. [Google Scholar]
- Geng, P.; Zheng, S.; Tang, H.; Zhu, R.; Zhang, L.; Cao, S.; Xue, H.; Pang, H. Transition Metal Sulfides Based on Graphene for Electrochemical Energy Storage. Adv. Energy Mater. 2018, 8, 1703259. [Google Scholar] [CrossRef]
- Sun, Z.; Liao, T.; Dou, Y.; Hwang, S.; Park, M.; Jiang, L.; Kim, J.; Dou, S. Generalized self-assembly of scalable two-dimensional transition metal oxide nanosheets. Nat. Commun. 2014, 5, 3813. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Z.; Xu, Y.; Tang, L.; Xu, S.; Li, D.; Zhu, J.; Jiang, D. Bimetallic Co-Mo Nitride Nanosheet Arraysas High-Performance Bifunctional Electrocatalysts for Overall Water Splitting. Chem. Eng. J. 2021, 411, 128433. [Google Scholar] [CrossRef]
- Tian, H.; Liu, M.; Zheng, W. Constructing 2D graphitic carbon nitride nanosheets/layered MoS2/graphene ternarynanojunction with enhanced photocatalytic activity. Appl. Catal. B Environ. 2018, 225, 468–476. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, H.; Tu, W.; Liu, Y.; Wu, S.; Tan, Y.; Chew, J. Construction of hierarchical 2D-2D Zn 3 In 2 S 6/fluorinated polymeric carbon nitride nanosheets photocatalyst for boosting photocatalytic degradation andhydrogen production performance. Appl. Catal. B Environ. 2018, 233, 58–69. [Google Scholar] [CrossRef]
- Liu, H.; Hu, K.; Yan, D.; Chen, R.; Zou, Y.; Liu, H.; Wang, S. Recent Advances on Black Phosphorus for Energy Storage, Catalysis, and Sensor Applications. Adv. Mater. 2018, 30, 1800295. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Liu, Z.; Chen, J.; Guo, J.; Kang, N.; Ma, X.; Cheng, H.; Ren, W. Large-area high-quality 2D ultrathin Mo2C superconducting crystals. Nat. Mater. 2015, 14, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, M.; Delbari, S.A.; Babapoo, A.; Namini, A.S.; Mohammadi, M.; Asl, M.S. Triplet carbide composites of TiC, WC, and SiC. Ceram. Int. 2020, 46, 9070–9078. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, L.; Li, L. Preparation and properties of 2D C/SiC-ZrB2-TaC composites. Ceram. Int. 2011, 37, 891–896. [Google Scholar]
- Hong, Y.L.; Liu, Z.B.; Wang, L.; Zhou, T.Y.; Ma, W.; Xu, C.; Feng, S.; Chen, L.; Chen, M.L.; Sun, D.M.; et al. Chemical vapor deposition of layered two-dimensional MoSi2N4 materials. Science 2020, 369, 670–674. [Google Scholar] [CrossRef]
- Sibatov, R.; Meftakhutdinov, R.; Kochaev, A. Asymmetric XMoSiN2 (X = S, Se, Te) monolayers as novel promising 2D materials for nanoelectronics and photovoltaics. Appl. Surf. Sci. 2022, 585, 152465. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Ang, Y.S.; Nguyen, S.T.; Hoang, N.V.; Hung, N.M.; Nguyen, C.V. Tunable type-II band alignment and electronic structure of C3N4/MoSi2N4 heterostructure: Interlayer coupling and electric field. Phys. Rev. B 2022, 105, 045303. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, C.; Wang, Q.; Li, J.; Yu, Y.; Xu, F.; Wang, B.; Wei, Y. Novel Two-Dimensional Layered MoSi2Z4 (Z = P, As): New Promising Optoelectronic Materials. Nanomaterials 2021, 11, 559. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Q.; Sun, Z.; Mwankemwa, N.; Zhang, W.-b.; Yang, W.X. First-principles investigations of electronic, optical, and photocatalytic properties of Au-adsorbed MoSi2N4 monolayer. J. Phys. Chem. Solids 2022, 162, 110494. [Google Scholar] [CrossRef]
- Ray, A.; Tyagi, S.; Singh, N.; Schwingenschlgl, U. Inducing Half-Metallicity in Monolayer MoSi2N4. ACS Omega 2021, 6, 30371–30375. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, Z.; Zhu, Y.; Lin, L.; Yu, W.; Wang, Q.; Yang, X.; Jia, X.; Jia, Y. A two-dimensional MoSe2/MoSi2N4 van der Waals heterostructure with high carrier mobility and diversified regulation of its electronic properties. J. Mater. Chem. C 2021, 9, 10073–10083. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Stampfl, C.; Sarsari, I.A.; Ghergherehchi, M. Band-gap engineering, magnetic behavior and Dirac-semimetal character in the MoSi2N4 nanoribbon with armchair and zigzag edges. Mater. Sci. 2021, 55, 035301. [Google Scholar] [CrossRef]
- Abdelati, M.A.; Maarouf, A.A.; Fadlallah, M.M. Substitutional transition metal doping in MoSi2N4 monolayer: Structural, electronic and magnetic properties. Phys. Chem. Chem. Phys. 2022, 24, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Chowdhury, S. Comment on ‘MoSi2N4 single-layer: A novel two-dimensional material with outstanding mechanical, thermal, electronic and optical properties’. J. Phys. D Appl. Phys. 2021, 55, 068001. [Google Scholar] [CrossRef]
- Wang, Z.; Kuang, X.; Yu, G.; Zhao, P.; Zhong, H.; Yuan, S. Electronic properties and quasiparticle model of monolayer MoSi2N4. Phys. Rev. B 2021, 104, 155110. [Google Scholar] [CrossRef]
- Ghobadi, N.; Hosseini, M.; Touski, S.B. Field-Effect Transistor Based on MoSi2N4 and WSi2N4 Monolayers Under Biaxial Strain: A Computational Study of the Electronic Properties. IEEE Trans. Electron Devices 2022, 69, 863–869. [Google Scholar] [CrossRef]
- Kang, L.; Lin, Z. Second Harmonic Generation of MoSi2N4-type Layers. Phys. Rev. B 2021, 103, 195404. [Google Scholar] [CrossRef]
- Touski, S.B.; Ghobadi, N. Vertical Strain-Induced Modification of the Electrical and Spin Properties of Monolayer MoSi2X4 (X = N, P, As and Sb). J. Phys. D Appl. Phys. 2021, 54, 485302. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, L.; Liang, S.; Wu, W.; Wang, G.; Lee, C.H.; Ong, W.L.; Yang, H.Y.; Ang, L.K.; Yang, S.A.; et al. Efficient Ohmic contacts and built-in atomic sublayer protection in MoSi2N4 and WSi2N4 monolayers. NPJ 2D Mater. Appl. 2021, 5, 71. [Google Scholar] [CrossRef]
- Xiao, C.W.; Ma, Z.; Sa, R.; Cui, Z.; Gao, S.; Du, W.; Sun, X.; Li, Q. Adsorption Behavior of Environmental Gas Molecules on Pristine and Defective MoSi2N4: Possible Application as Highly Sensitive and Reusable Gas Sensors. ACS Omega 2022, 7, 8706–8716. [Google Scholar] [CrossRef]
- Shen, C.; Wang, L.; Wei, D.; Zhang, Y.; Qin, G.; Chen, X.Q.; Zhang, H. Two-dimensional layered MSi2N4 (M = Mo, W) as promising thermal management materials: A comparative study. Phys. Chem. Chem. Phys. 2022, 24, 3086–3093. [Google Scholar] [CrossRef]
- Nandan, K.; Ghosh, B.; Agarwal, A.; Bhowmick, S.; Chauhan, Y.S. Two-dimensional MoSi2N4: An Excellent 2D Semiconductor for Field-Effect Transistors. IEEE Trans. Electron Devices 2022, 69, 406–413. [Google Scholar] [CrossRef]
- Huang, J.; Li, P.; Ren, X.; Guo, Z. Promising Properties of Sub-5-nm Monolayer MoSi2N4 Transistor. Phys. Rev. Appl. 2021, 16, 044022. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Lou, F.; Guo, K.; Yu, Z. Non-precious metal activated MoSi2N4 monolayers for high-performance OER and ORR electrocatalysts: A first-principles study. Appl. Surf. Sci. 2022, 579, 152234. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y.; He, H.; Zhou, P.; Liang, Y.; Frauenheim, T. Stacking Engineering: A Boosting Strategy for 2D Photocatalysts. J. Phys. Chem. Lett. 2021, 12, 10190–10196. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, J.; Guo, Z.; Sun, Z. Novel Two-Dimensional Janus MoSiGeN4 and WSiGeN4 as Highly Efficient Photocatalysts for Spontaneous Overall Water Splitting. ACS Appl. Mater. Interfaces 2021, 13, 28090–28097. [Google Scholar] [CrossRef]
- Chen, X.; Han, W.; Jia, M.; Ren, F.; Peng, C.; Gu, Q.; Wang, B.; Yin, H. A direct Z-scheme MoSi2N4/BlueP vdW heterostructure for photocatalytic overall water splitting. J. Phys. D Appl. Phys. 2022, 55, 215502. [Google Scholar] [CrossRef]
- Ding, W.; Xue, Z.; Li, J.; Li, M.; Bai, L.; Zhou, Q.; Zhou, X.; Peng, Y.; Miao, L. Excited State Properties of Layered Two-Dimensional MSi2N4 (M = Mo, Cr, and W) Materials from First-Principles Calculations. ECS J. Solid State Sci. Technol. 2022, 11, 016001. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M.; Erratum, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Li, L.; Hu, J.; Shi, X.; Ruan, W.; Luo, J.; Wei, X. Theoretical Studies on Structures, Properties and Dominant Debromination Pathways for Selected Polybrominated Diphenyl Ethers. Int. J. Mol. Sci. 2016, 17, 927. [Google Scholar] [CrossRef]
- Bruhn, T.; Pescitelli. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 2016, 28, 466–474. [Google Scholar]
- Yi, W.; Tang, G.; Chen, X.; Yang, B.; Liu, X. qvasp: A flexible toolkit for VASP users in materials simulations. Comput. Phys. Commun. 2020, 257, 107535. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A Pre- and Post-Processing Program for VASP code. Comput. Sci. 2019. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Isravel, A.D.; Jeyaraj, J.K.; Thangasamy, S.; Jone, W.J. DFT, NBO, HOMO-LUMO, NCI, Stability, Fukui Function and Hole—Electron analyses of Tolcapone. Comput. Theor. Chem. 2021, 1202, 113296. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Hoat, D.M.; Fadlallah, M.; Shahrokhi, M.; Shojaei, F.; Gogova, D.; Ghergherehchi, M. MoSi2N4 single-layer: A novel two-dimensional material with outstanding mechanical, thermal, electronic, optical, and photocatalytic properties. J. Phys. D Appl. Phys. 2021, 54, 155303. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Abdollahzadeh.; Ziabari, A.; Fadlallah, M.M.; Nguyen, C.V.; Ghergherehchi, M.; Feghhi, S.A.H. A van der Waals heterostructure of MoS2/MoSi2N4: A first-principles study. New J. Chem. 2021, 45, 8291–8296. [Google Scholar] [CrossRef]
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.H.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 2019, 18, 827–832. [Google Scholar] [CrossRef]
- Mortazavi, B.; Javvaji, B.; Shojaei, F.; Rabczuk, T.; Shapeev, A.; Zhuang, X.Y. Exceptional piezoelectricity, high thermal conductivity and stiffness and promising photocatalysis in two-dimensional MoSi2N4 family confirmed by first-principles. Nano Energy 2021, 82, 105716. [Google Scholar] [CrossRef]
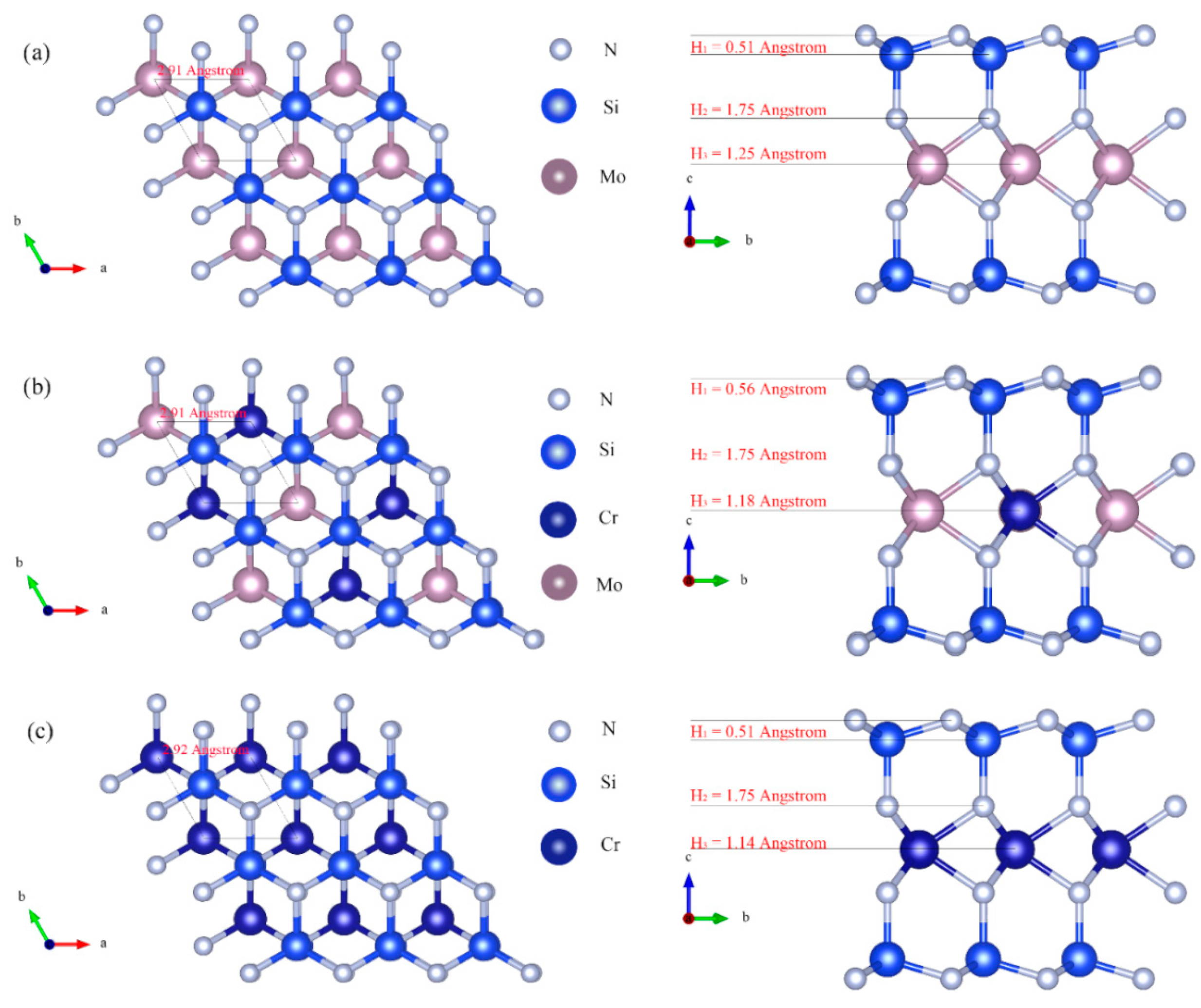
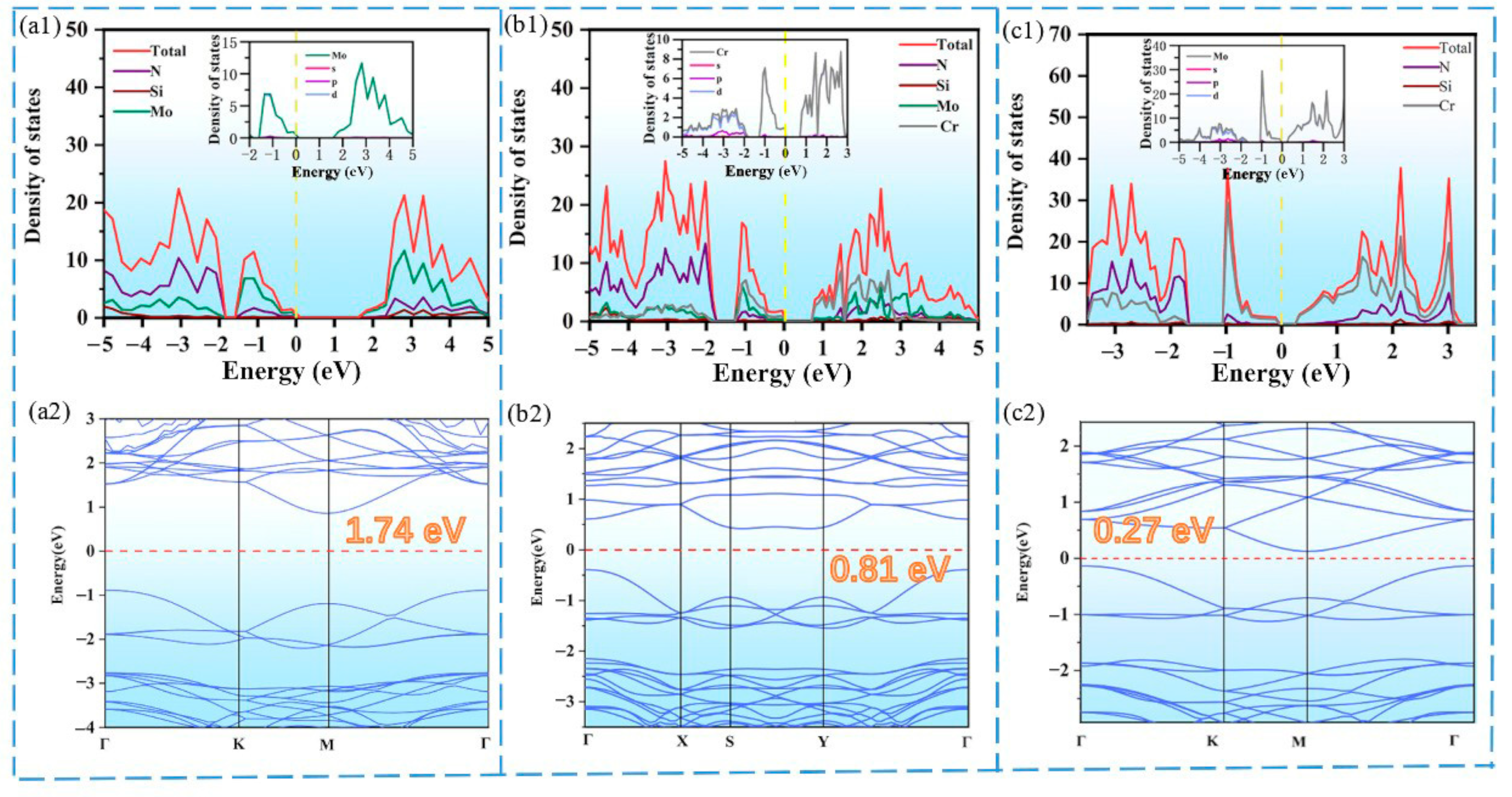
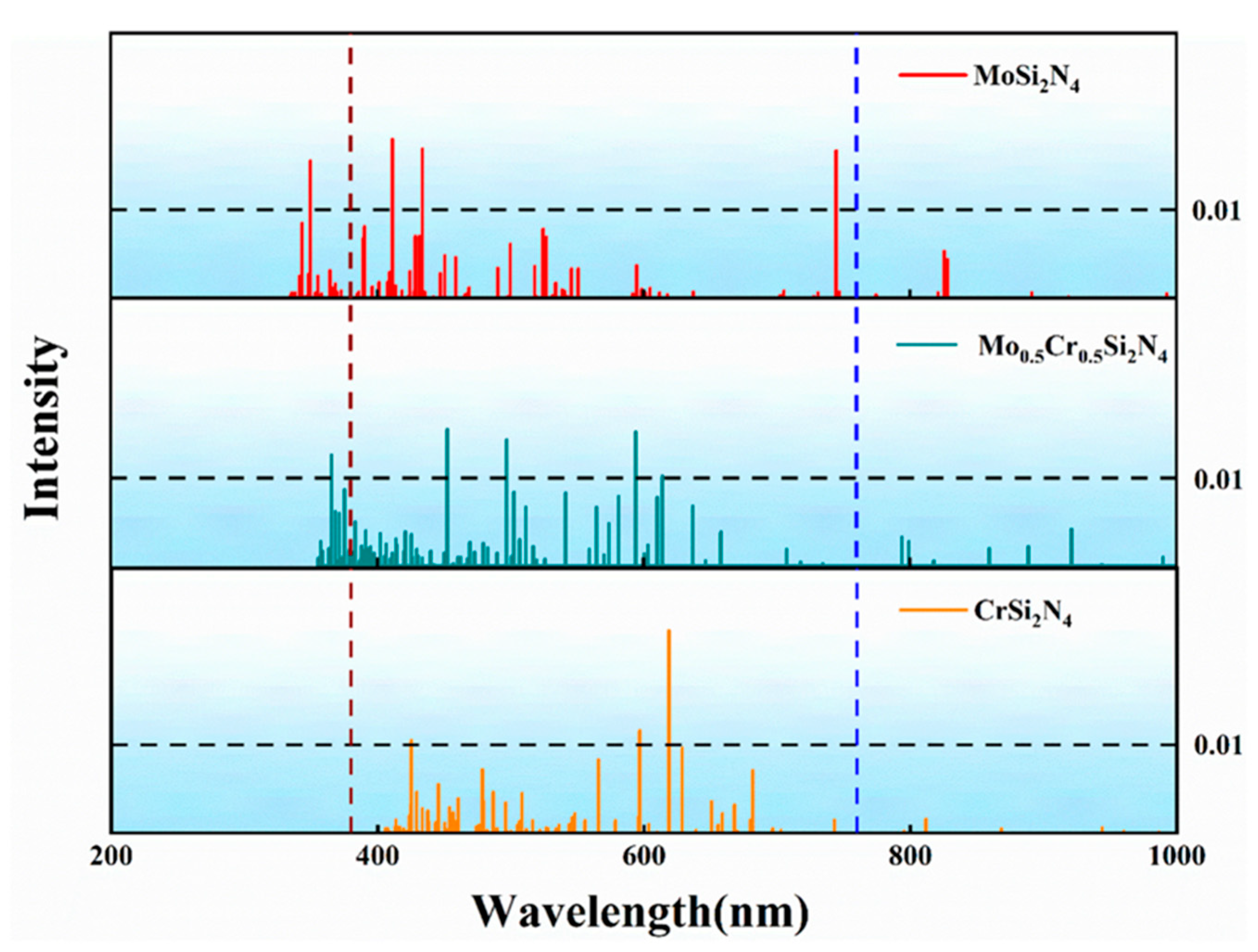

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | MoSi2N4 | Mo0.5Cr0.5Si2N4 | CrSi2N4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Excited state | #19 | #54 | #63 | #28 | #32 | #46 | #57 | #37 | #40 | #88 |
| The excitation energy (eV) | 1.6650 | 2.8571 | 3.0137 | 2.0184 | 2.0876 | 2.4937 | 2.7388 | 2.0038 | 2.0775 | 2.9155 |
| Corresponding wavelength(n m) | 744.65 | 433.95 | 411.40 | 614.27 | 593.91 | 497.19 | 452.70 | 618.75 | 596.80 | 425.26 |
| Orbital contribution | H − 1 > L + 2 74.4% H > L + 2 18.1% | H − 3 > L + 4 51.8% H − 8 > L + 2 16.6% H − 6 > L + 2 16.3% | H − 17 > L + 1 42.6% H − 13 > L + 1 28.9% | H − 3 > L + 2 24.8% H − 2 > L + 2 15.2% H − 12 > L 11.9% H − 6 > L + 1 11.7% H − 4 > L + 2 10.3%, H − 7 > L 5.5% | H − 10 > L 19.8% H − 13 > L 17.8% H − 12 > L 16.0% H − 4 > L + 2 10.0% H − 10 > L + 1 5.2% | H − 1 > L + 4 19.3% H − 15 > L 18.0% H − 1 > L + 6 9.2% H − 1 > L + 5 6.3% H − 2 > L + 3 5.5% | H − 1 > L + 6 43.5% H − 1 > L + 4 13.2% H − 1 > L + 8 7.8% H − 17 > L 7.4% | H − 4 > L + 2 38.9% H − 5 > L + 1 12.9% H > L + 6 9.0% H − 4 > L + 4 8.4% | H − 1 > L + 7 35.1% H − 5 > L + 2 9.0% H > L + 8 8.7% H − 2 > L + 2 7.5% H − 2 > L + 8 7.3% | H − 18 > L 21.9% H − 22 > L 14.4% H − 15 > L 11.2% H − 3 > L + 7 5.2% H − 27 > L 5.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, J.; Wan, L.; Li, J.; Qu, H.; Ding, C.; Li, M.; Yu, D.; Fan, K.; Yao, H. The First-Principle Study on Tuning Optical Properties of MA2Z4 by Cr Replacement of Mo Atoms in MoSi2N4. Nanomaterials 2022, 12, 2822. https://doi.org/10.3390/nano12162822
Li Y, Li J, Wan L, Li J, Qu H, Ding C, Li M, Yu D, Fan K, Yao H. The First-Principle Study on Tuning Optical Properties of MA2Z4 by Cr Replacement of Mo Atoms in MoSi2N4. Nanomaterials. 2022; 12(16):2822. https://doi.org/10.3390/nano12162822
Chicago/Turabian StyleLi, Yongsheng, Jiawei Li, Lingyu Wan, Jiayu Li, Hang Qu, Cui Ding, Mingyang Li, Dan Yu, Kaidi Fan, and Huilu Yao. 2022. "The First-Principle Study on Tuning Optical Properties of MA2Z4 by Cr Replacement of Mo Atoms in MoSi2N4" Nanomaterials 12, no. 16: 2822. https://doi.org/10.3390/nano12162822
APA StyleLi, Y., Li, J., Wan, L., Li, J., Qu, H., Ding, C., Li, M., Yu, D., Fan, K., & Yao, H. (2022). The First-Principle Study on Tuning Optical Properties of MA2Z4 by Cr Replacement of Mo Atoms in MoSi2N4. Nanomaterials, 12(16), 2822. https://doi.org/10.3390/nano12162822