
Liposomal PHD2 Inhibitors and the Enhanced Efficacy in Stabilizing HIF-1α

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Liposomes with Transmembrane Calcium Acetate Gradients
2.3. Remote Loading of PHD2 Inhibitors into Liposomes
2.4. Quantification of Liposome-Encapsulated Drugs
2.5. Cryo-EM Imaging of Drug-Loaded Liposomes
2.6. Preparation and Characterization of IOX2-Loaded Liposomes for Cell Experiments
2.7. Liposomal IOX2 Treatment in Cell Experiments
2.8. Liposomal IOX2-Induced HIF-1α Stabilization Evaluated by Western Blotting
3. Results and Discussion
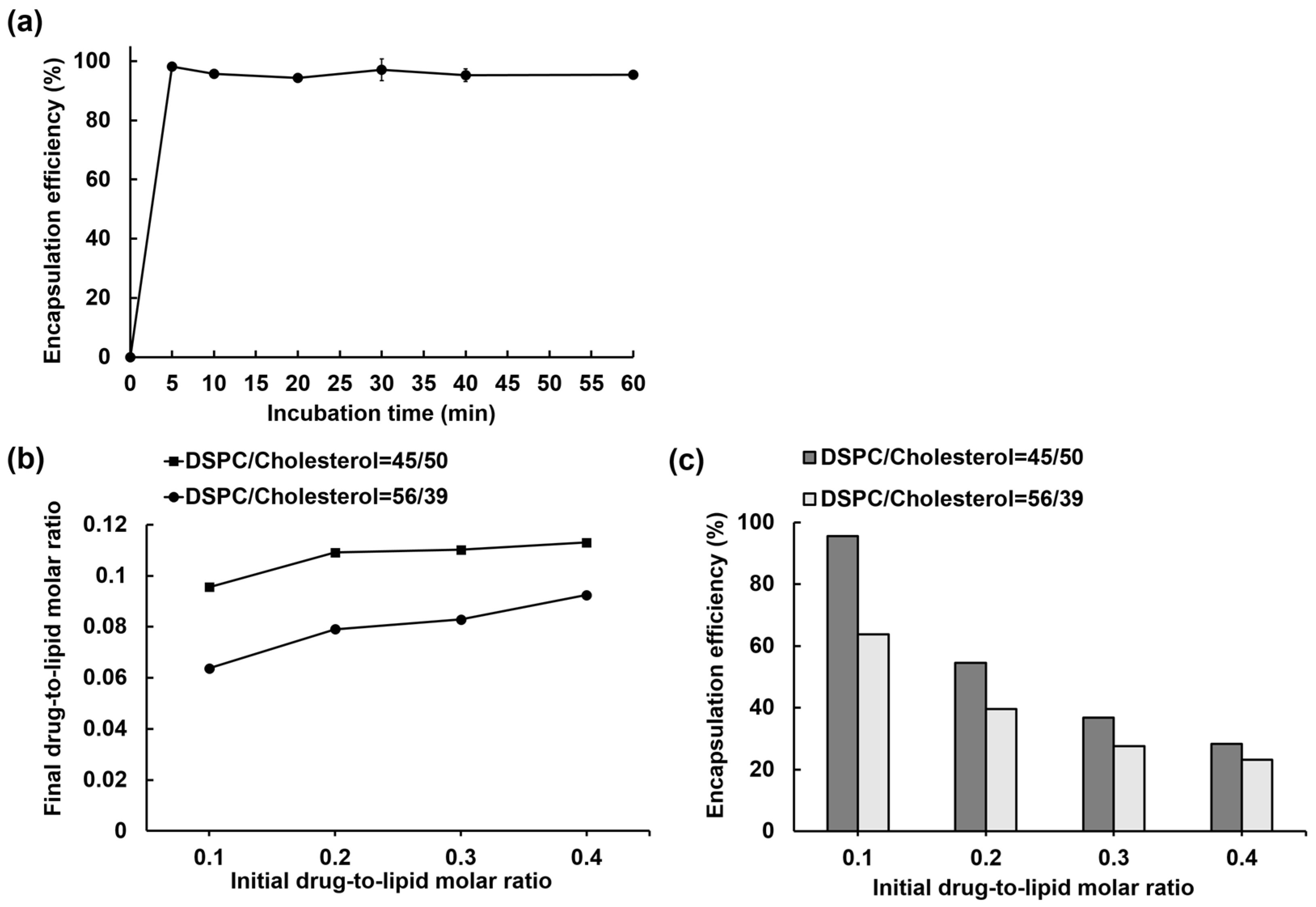
3.1. IOX2 Loading into Liposomes via a Transmembrane Calcium Acetate Gradient and the Optimization for the Encapsulation Efficiency
3.2. Liposome Loading Kinetics and Capacity of IOX2 via a Calcium Acetate Gradient
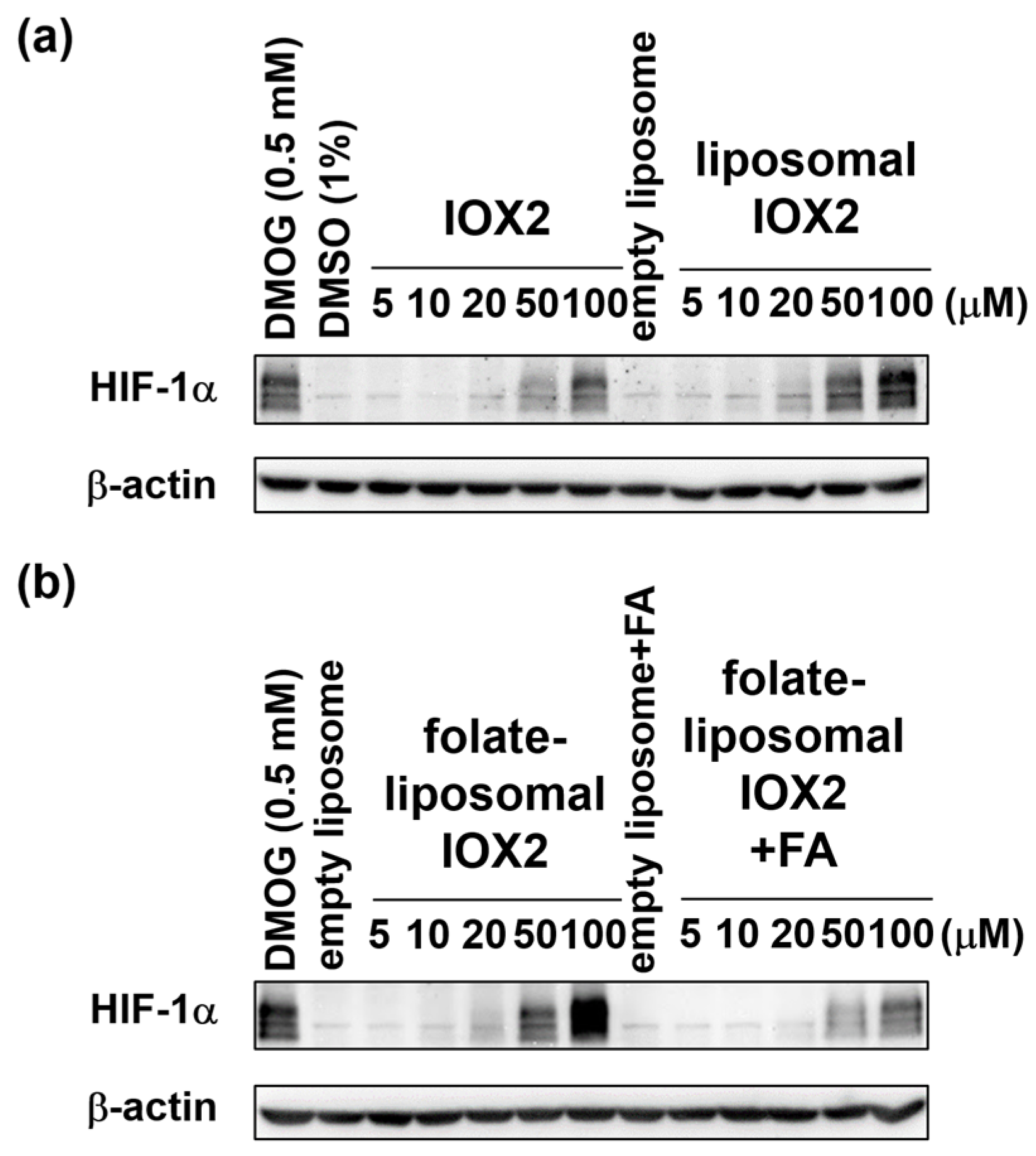
3.3. Comparison of Free IOX2 and Liposomal IOX2-Induced HIF-1α Stabilization in Cells
3.4. Remote Loading of other PHD2 Inhibitors into Liposomes via a Calcium Acetate Gradient
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Majmundar, A.J.; Wong, W.H.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Eckardt, K.U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor-1 Is a Basic-Helix-Loop-Helix-Pas Heterodimer Regulated by Cellular O-2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1 alpha and HIF-2 alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.F.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia- inducible factor 1. Annu. Rev. Cell. Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Bruick Richard, K.; McKnight Steven, L. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, N.; Lei, Y.H.; Hu, T.H.; You, Q.D.; Zhang, X.J. Small-molecule inhibitors of HIF-PHD2: A valid strategy to renal anemia treatment in clinical therapy. Medchemcomm 2016, 7, 1271–1284. [Google Scholar] [CrossRef]
- Joharapurkar, A.A.; Pandya, V.B.; Patel, V.J.; Desai, R.C.; Jain, M.R. Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases. J. Med. Chem. 2018, 61, 6964–6982. [Google Scholar] [CrossRef]
- Yeh, T.L.; Leissing, T.M.; Abboud, M.I.; Thinnes, C.C.; Atasoylu, O.; Holt-Martyn, J.P.; Zhang, D.; Tumber, A.; Lippl, K.; Lohans, C.T.; et al. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 2017, 8, 7651–7668. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Jamadarkhana, P.; Chaudhary, A.; Chhipa, L.; Dubey, A.; Mohanan, A.; Gupta, R.; Deshpande, S. Treatment with a Novel Hypoxia-Inducible Factor Hydroxylase Inhibitor (TRC160334) Ameliorates Ischemic Acute Kidney Injury. Am. J. Nephrol. 2012, 36, 208–218. [Google Scholar] [CrossRef]
- Zhou, J.; Li, J.; Rosenbaum, D.M.; Zhuang, J.; Poon, C.; Qin, P.; Rivera, K.; Lepore, J.; Willette, R.N.; Hu, E.; et al. The prolyl 4-hydroxylase inhibitor GSK360A decreases post-stroke brain injury and sensory, motor, and cognitive behavioral deficits. PLoS ONE 2017, 12, e0184049. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-S.; Lee, K.-J.; Chen, H.-C.; Prajnamitra Ray, P.; Hsu, C.-H.; Jian, C.-B.; Yu, X.-E.; Chueh, D.-Y.; Kuo Chiung, W.; Chiang, T.-C.; et al. Immune cell shuttle for precise delivery of nanotherapeutics for heart disease and cancer. Sci. Adv. 2021, 7, eabf2400. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ning, S.J.; Zhang, S.J. Tumor PHD2 Expression Is Correlated with Clinical Features and Prognosis of Patients with HCC Receiving Liver Resection. Medicine 2014, 93, e179. [Google Scholar]
- Kozlova, N.; Wottawa, M.; Katschinski, D.M.; Kristiansen, G.; Kietzmann, T. Hypoxia-inducible factor prolyl hydroxylase 2 (PHD2) is a direct regulator of epidermal growth factor receptor (EGFR) signaling in breast cancer. Oncotarget 2017, 8, 9885–9898. [Google Scholar] [CrossRef]
- Zhang, L.L.; Peng, S.; Dai, X.P.; Gan, W.J.; Nie, X.; Wei, W.Y.; Hu, G.Q.; Guo, J.P. Tumor suppressor SPOP ubiquitinates and degrades Eg1N2 to compromise growth of prostate cancer cells. Cancer Lett. 2017, 390, 11–20. [Google Scholar] [CrossRef]
- Klotzsche-von Ameln, A.; Muschter, A.; Mamlouk, S.; Kalucka, J.; Prade, I.; Franke, K.; Rezaei, M.; Poitz, D.M.; Breier, G.; Wielockx, B. Inhibition of HIF Prolyl Hydroxylase-2 Blocks Tumor Growth in Mice through the Antiproliferative Activity of TGF beta. Cancer Res. 2011, 71, 3306–3316. [Google Scholar] [CrossRef]
- Wottawa, M.; Leisering, P.; von Ahlen, M.; Schnelle, M.; Vogel, S.; Malz, C.; Bordoli, M.R.; Camenisch, G.; Hesse, A.; Napp, J.; et al. Knockdown of prolyl-4-hydroxylase domain 2 inhibits tumor growth of human breast cancer MDA-MB-231 cells by affecting TGF-1 processing. Int. J. Cancer 2013, 132, 2787–2798. [Google Scholar] [CrossRef]
- Kachamakova-Trojanowska, N.; Podkalicka, P.; Bogacz, T.; Barwacz, S.; Jozkowicz, A.; Dulak, J.; Loboda, A. HIF-1 stabilization exerts anticancer effects in breast cancer cells in vitro and in vivo. Biochem. Pharmacol. 2020, 175, 113922. [Google Scholar] [CrossRef]
- Koyama, S.; Matsunaga, S.; Imanishi, M.; Maekawa, Y.; Kitano, H.; Takeuchi, H.; Tomita, S. Tumour blood vessel normalisation by prolyl hydroxylase inhibitor repaired sensitivity to chemotherapy in a tumour mouse model. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Nishide, S.; Matsunaga, S.; Shiota, M.; Yamaguchi, T.; Kitajima, S.; Maekawa, Y.; Takeda, N.; Tomura, M.; Uchida, J.; Miura, K.; et al. Controlling the Phenotype of Tumor-Infiltrating Macrophages via the PHD-HIF Axis Inhibits Tumor Growth in a Mouse Model. iScience 2019, 19, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Nishide, S.; Uchida, J.; Matsunaga, S.; Tokudome, K.; Yamaguchi, T.; Kabei, K.; Moriya, T.; Miura, K.; Nakatani, T.; Tomita, S. Prolyl-hydroxylase inhibitors reconstitute tumor blood vessels in mice. J. Pharmacol. Sci. 2020, 143, 122–126. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.L.; Deschoemaeker, S.; Henze, A.T.; Debackere, K.; Finisguerra, V.; Takeda, Y.; Roncal, C.; Dettori, D.; Tack, E.; Jonsson, Y.; et al. Gene-Targeting of Phd2 Improves Tumor Response to Chemotherapy and Prevents Side-Toxicity. Cancer Cell 2012, 22, 263–277. [Google Scholar]
- Kiriakidis, S.; Hoer, S.S.; Burrows, N.; Biddlecome, G.; Khan, M.N.; Thinnes, C.C.; Schofield, C.J.; Rogers, N.; Botto, M.; Paleolog, E.; et al. Complement C1q is hydroxylated by collagen prolyl 4 hydroxylase and is sensitive to off-target inhibition by prolyl hydroxylase domain inhibitors that stabilize hypoxia-inducible factor. Kidney Int. 2017, 92, 900–908. [Google Scholar] [CrossRef]
- Nagano, N. Selectivity of HIF-PH inhibitors: Concerns regarding possible off-target effects. Clin. Exp. Nephrol. 2021, 25, 1047–1048. [Google Scholar] [CrossRef]
- Minamishima, Y.A.; Moslehi, J.; Bardeesy, N.; Cullen, D.; Bronson, R.T.; Kaelin, W.G. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 2008, 111, 3236–3244. [Google Scholar] [CrossRef]
- Moslehi, J.; Minamishima, Y.A.; Shi, J.R.; Neuberg, D.; Charytan, D.M.; Padera, R.F.; Signoretti, S.; Liao, R.; Kaelin, W.G. Loss of Hypoxia-Inducible Factor Prolyl Hydroxylase Activity in Cardiomyocytes Phenocopies Ischemic Cardiomyopathy. Circulation 2010, 122, 1004–1016. [Google Scholar] [CrossRef]
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.K.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non-Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Walters, W.P. Acidic and Basic Drugs in Medicinal Chemistry: A Perspective. J. Med. Chem. 2014, 57, 9701–9717. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, F.M.; van Wijk, D.F.; Lobatto, M.E.; Verberne, H.J.; Storm, G.; Willems, M.C.M.; Legemate, D.A.; Nederveen, A.J.; Calcago, C.; Mani, V.; et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1039–1046. [Google Scholar] [CrossRef]
- Clark, A.J.; Wiley, D.T.; Zuckerman, J.E.; Webster, P.; Chao, J.; Lin, J.; Yen, Y.; Davis, M.E. CRLX101 nanoparticles localize in human tumors and not in adjacent, nonneoplastic tissue after intravenous dosing. Proc. Natl. Acad. Sci. USA 2016, 113, 3850–3854. [Google Scholar] [CrossRef]
- Xie, D.; Wang, J.; Hu, G.; Chen, C.; Yang, H.; Ritter, J.K.; Qu, Y.; Li, N. Kidney-Targeted Delivery of Prolyl Hydroxylase Domain Protein 2 Small Interfering RNA with Nanoparticles Alleviated Renal Ischemia/Reperfusion Injury. J. Pharmacol. Exp. Ther. 2021, 378, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal doxorubicin—Review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Del. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into Active Targeting of Nanoparticles in Drug Delivery: Advances in Clinical Studies and Design Considerations for Cancer Nanomedicine. Bioconj. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef]
- Shin, J.H.; Shum, P.; Grey, J.; Fujiwara, S.; Malhotra, G.S.; Gonzalez-Bonet, A.; Hyun, S.H.; Moase, E.; Allen, T.M.; Thompson, D.H. Acid-Labile mPEG-Vinyl Ether-1,2-Dioleylglycerol Lipids with Tunable pH Sensitivity: Synthesis and Structural Effects on Hydrolysis Rates, DOPE Liposome Release Performance, and Pharmacokinetics. Mol. Pharm. 2012, 9, 3266–3276. [Google Scholar] [CrossRef]
- Avnir, Y.; Ulmansky, R.; Wasserman, V.; Even-Chen, S.; Broyer, M.; Barenholz, Y.; Naparstek, Y. Amphipathic weak acid glucocorticoid Prodrugs remote-loaded into sterically stabilized nanoliposomes evaluated in arthritic rats and in a beagle dog. Arthritis Rheum. 2008, 58, 119–129. [Google Scholar] [CrossRef]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane Ammonium-Sulfate Gradients in Liposomes Produce Efficient and Stable Entrapment of Amphipathic Weak Bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Clerc, S.; Barenholz, Y. Loading of amphipathic weak acids into liposomes in response to transmembrane calcium acetate gradients. Biochim. Biophys. Acta Biomembr. 1995, 1240, 257–265. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.X.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef]
- Yang, W.Q.; Yang, Z.M.; Fu, J.R.; Guo, M.R.; Sun, B.J.; Wei, W.; Liu, D.; Liu, H.Z. The influence of trapping agents on the antitumor efficacy of irinotecan liposomes: Head-to-head comparison of ammonium sulfate, sulfobutylether--cyclodextrin and sucrose octasulfate. Biomater. Sci. 2019, 7, 419–428. [Google Scholar] [CrossRef]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef]
- Cern, A.; Golbraikh, A.; Sedykh, A.; Tropsha, A.; Barenholz, Y.; Goldblum, A. Quantitative structure—Property relationship modeling of remote liposome loading of drugs. J. Control. Release 2012, 160, 147–157. [Google Scholar] [CrossRef]
- McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.R.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. USA 2006, 103, 9814–9819. [Google Scholar] [CrossRef]
- Chowdhury, R.; Candela-Lena, J.I.; Chan, M.C.; Greenald, D.J.; Yeoh, K.K.; Tian, Y.M.; McDonough, M.A.; Tumber, A.; Rose, N.R.; Conejo-Garcia, A.; et al. Selective Small Molecule Probes for the Hypoxia Inducible Factor (HIF) Prolyl Hydroxylases. ACS Chem. Biol. 2013, 8, 1488–1496. [Google Scholar] [CrossRef]
- Chan, M.C.; Atasoylu, O.; Hodson, E.; Tumber, A.; Leung, I.K.H.; Chowdhury, R.; Gomez-Perez, V.; Demetriades, M.; Rydzik, A.M.; Holt-Martyn, J.; et al. Potent and Selective Triazole-Based Inhibitors of the Hypoxia-Inducible Factor Prolyl-Hydroxylases with Activity in the Murine Brain. PLoS ONE 2015, 10, e0132004. [Google Scholar] [CrossRef]
- Deppe, J.; Popp, T.; Egea, V.; Steinritz, D.; Schmidt, A.; Thiermann, H.; Weber, C.; Ries, C. Impairment of hypoxia-induced HIF-1 alpha signaling in keratinocytes and fibroblasts by sulfur mustard is counteracted by a selective PHD-2 inhibitor. Arch. Toxicol. 2016, 90, 1141–1150. [Google Scholar] [CrossRef]
- Abraham, S.A.; Edwards, K.; Karlsson, G.; MacIntosh, S.; Mayer, L.D.; McKenzie, C.; Bally, M.B. Formation of transition metal–doxorubicin complexes inside liposomes. Biochim. Biophys. Acta 2002, 1565, 41–54. [Google Scholar] [CrossRef]
- Taggar, A.S.; Alnajim, J.; Anantha, M.; Thomas, A.; Webb, M.; Ramsay, E.; Bally, M.B. Copper-topotecan complexation mediates drug accumulation into liposomes. J. Control. Release 2006, 114, 78–88. [Google Scholar] [CrossRef]
- Maurer, N.; Wong, K.F.; Hope, M.J.; Cullis, P.R. Anomalous solubility behavior of the antibiotic ciprofloxacin encapsulated in liposomes: A H-1-NMR study. Biochim. Biophys. Acta Biomembr. 1998, 1374, 9–20. [Google Scholar] [CrossRef][Green Version]
- Wehbe, M.; Malhotra, A.; Anantha, M.; Roosendaal, J.; Leung, A.W.Y.; Plackett, D.; Edwards, K.; Gilabert-Oriol, R.; Bally, M.B. A simple passive equilibration method for loading carboplatin into pre-formed liposomes incubated with ethanol as a temperature dependent permeability enhancer. J. Control. Release 2017, 252, 50–61. [Google Scholar] [CrossRef]
- Tang, W.L.; Tang, W.H.; Szeitz, A.; Kulkarni, J.; Cullis, P.; Li, S.D. Systemic study of solvent-assisted active loading of gambogic acid into liposomes and its formulation optimization for improved delivery. Biomaterials 2018, 166, 13–26. [Google Scholar] [CrossRef]
- Dos Santos, N.; Mayer, L.D.; Abraham, S.A.; Gallagher, R.C.; Cox, R.A.K.; Tardi, P.G.; Bally, M.B. Improved retention of idarubicin after intravenous injection obtained for cholesterol-free liposomes. Biochim. Biophys. Acta Biomembr. 2002, 1561, 188–201. [Google Scholar] [CrossRef]
- Dos Santos, N.; Cox, K.A.; McKenzie, C.A.; van Baarda, F.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Mayer, L.D.; Allen, C.; Bally, M.B. pH gradient loading of anthracyclines into cholesterol-free liposomes: Enhancing drug loading rates through use of ethanol. Biochim. Biophys. Acta Biomembr. 2004, 1661, 47–60. [Google Scholar] [CrossRef]
- Javanainen, M.; Martinez-Seara, H.; Vattulainen, I. Nanoscale Membrane Domain Formation Driven by Cholesterol. Sci. Rep. 2017, 7, 1143. [Google Scholar] [CrossRef]
- Kaddah, S.; Khreich, N.; Kaddah, F.; Charcosset, C.; Greige-Gerges, H. Cholesterol modulates the liposome membrane fluidity and permeability for a hydrophilic molecule. Food Chem. Toxicol. 2018, 113, 40–48. [Google Scholar] [CrossRef]
- Lu, Y.J.; Low, P.S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Del. Rev. 2002, 54, 675–693. [Google Scholar] [CrossRef]
- Ai, X.Z.; Ho, C.J.H.; Aw, J.; Attia, A.B.E.; Mu, J.; Wang, Y.; Wang, X.Y.; Wang, Y.; Liu, X.G.; Chen, H.B.; et al. In vivo covalent cross-linking of photon-converted rare-earth nanostructures for tumour localization and theranostics. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Ross, J.F.; Chaudhuri, P.K.; Ratnam, M. Differential Regulation of Folate Receptor Isoforms in Normal and Malignant-Tissues in-Vivo and in Established Cell-Lines—Physiological and Clinical Implications. Cancer 1994, 73, 2432–2443. [Google Scholar] [CrossRef]
- Modi, S.; Xiang, T.X.; Anderson, B.D. Enhanced active liposomal loading of a poorly soluble ionizable drug using supersaturated drug solutions. J. Control. Release 2012, 162, 330–339. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Mechanisms of Disease: Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intraliposomal Trapping Solution | Extraliposomal Solution | Encapsulation Efficiency (%) |
|---|---|---|
| 150 mM NaCl | 150 mM NaCl | 1.8 |
| 100 mM Ca(OAc)2 | 10 mM NaCl | 43.0 |
| 46.4 * | ||
| 10 mM HEPES (pH 7.4) | 84.8 | |
| 10 mM HEPES (pH 7.4) 1 mM EDTA | 96.8 | |
| 150 mM NaCl 10 mM HEPES (pH 7.4) 1 mM EDTA | ~100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, C.-B.; Yu, X.-E.; Gao, H.-D.; Chen, H.-A.; Jheng, R.-H.; Chen, C.-Y.; Lee, H.-M. Liposomal PHD2 Inhibitors and the Enhanced Efficacy in Stabilizing HIF-1α. Nanomaterials 2022, 12, 163. https://doi.org/10.3390/nano12010163
Jian C-B, Yu X-E, Gao H-D, Chen H-A, Jheng R-H, Chen C-Y, Lee H-M. Liposomal PHD2 Inhibitors and the Enhanced Efficacy in Stabilizing HIF-1α. Nanomaterials. 2022; 12(1):163. https://doi.org/10.3390/nano12010163
Chicago/Turabian StyleJian, Cheng-Bang, Xu-En Yu, Hua-De Gao, Huai-An Chen, Ren-Hua Jheng, Chong-Yan Chen, and Hsien-Ming Lee. 2022. "Liposomal PHD2 Inhibitors and the Enhanced Efficacy in Stabilizing HIF-1α" Nanomaterials 12, no. 1: 163. https://doi.org/10.3390/nano12010163
APA StyleJian, C.-B., Yu, X.-E., Gao, H.-D., Chen, H.-A., Jheng, R.-H., Chen, C.-Y., & Lee, H.-M. (2022). Liposomal PHD2 Inhibitors and the Enhanced Efficacy in Stabilizing HIF-1α. Nanomaterials, 12(1), 163. https://doi.org/10.3390/nano12010163