
Effects of Mg Doping at Different Positions in Li-Rich Mn-Based Cathode Material on Electrochemical Performance

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Methods
2.3. Electrochemical Measurements
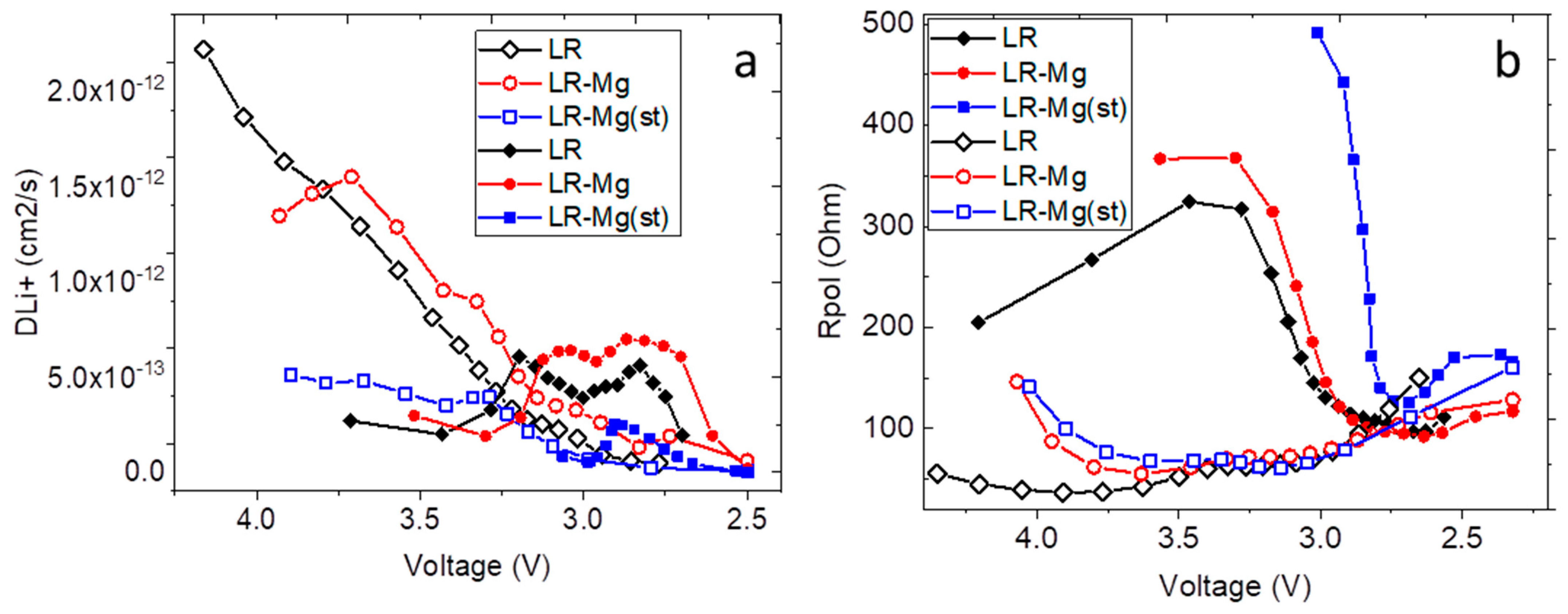
- DLi+—lithium-ion diffusion coefficient (cm2/s);
- τ—charge/discharge time of one GITT step (s);
- m—the active material weight (g);
- Vm—the molar volume of the sample (cm3/mol);
- M—the atomic weight (g/mol);
- S—the area of the sample-electrolyte interface (cm2);
- ∆Es—the voltage difference between the initial discharge voltage and the voltage after relaxation;
- ∆Et—the voltage change during one GITT step;
- L—the sample thickness (cm).
- I—discharge current (A).
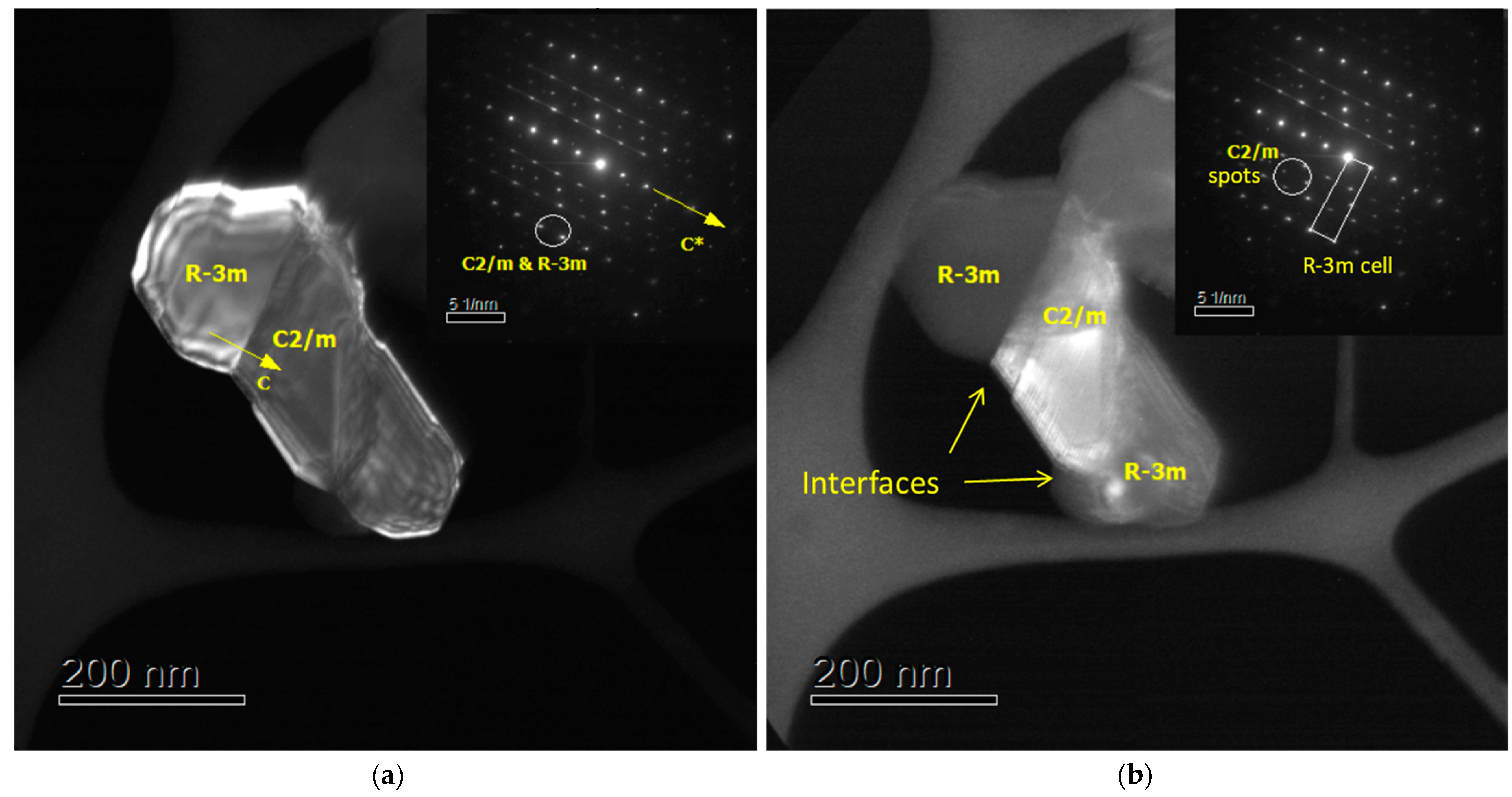
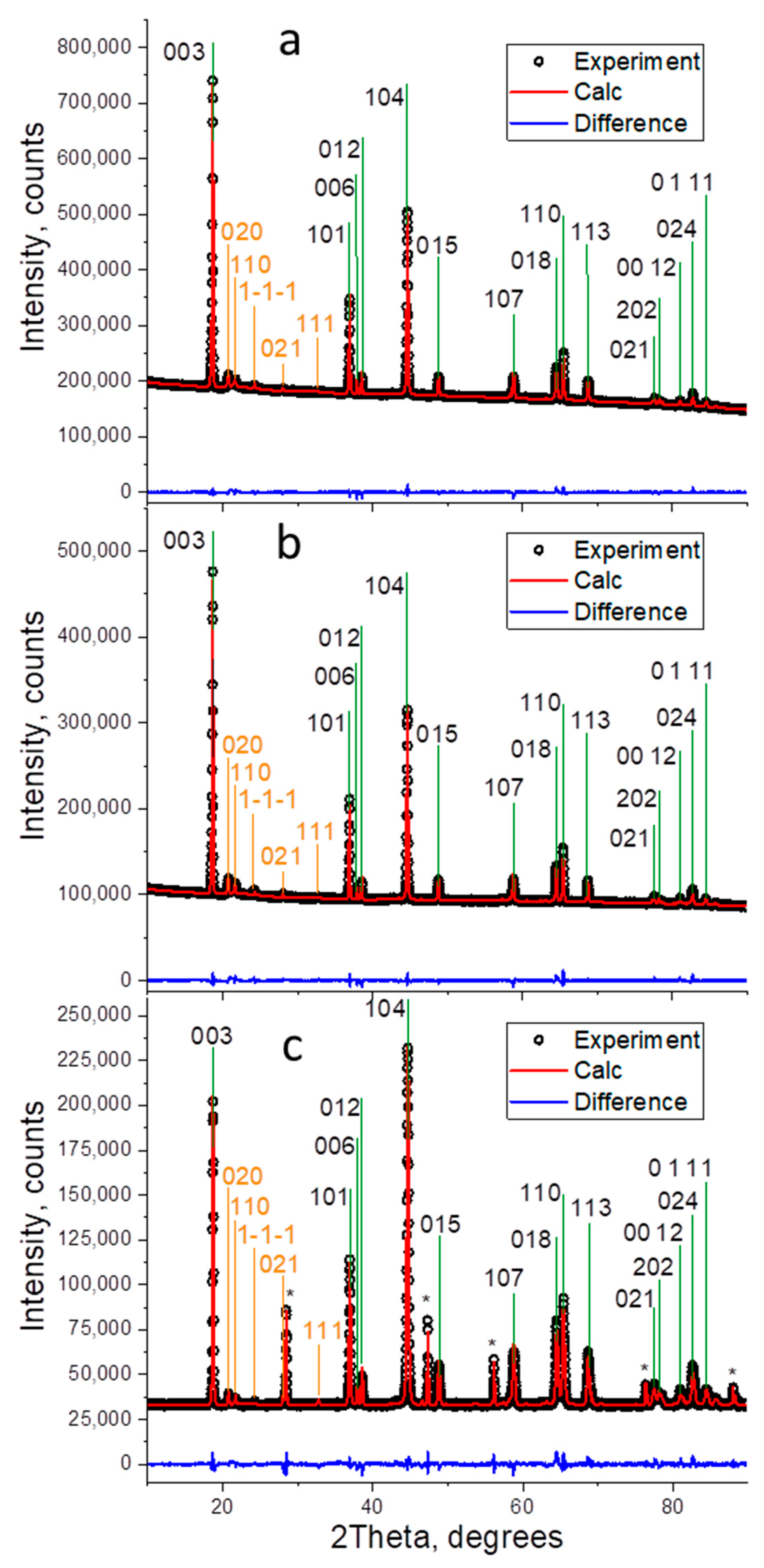
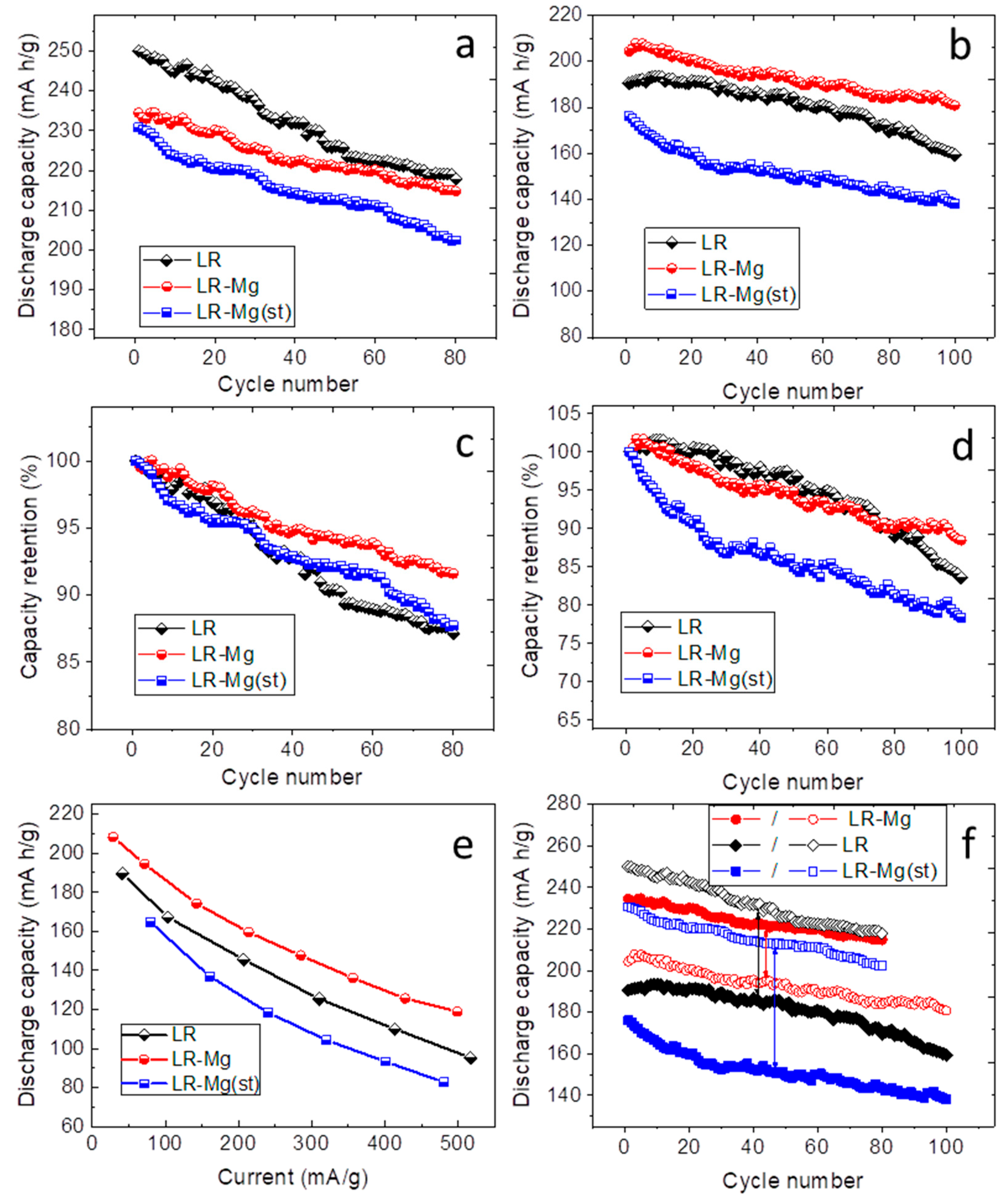
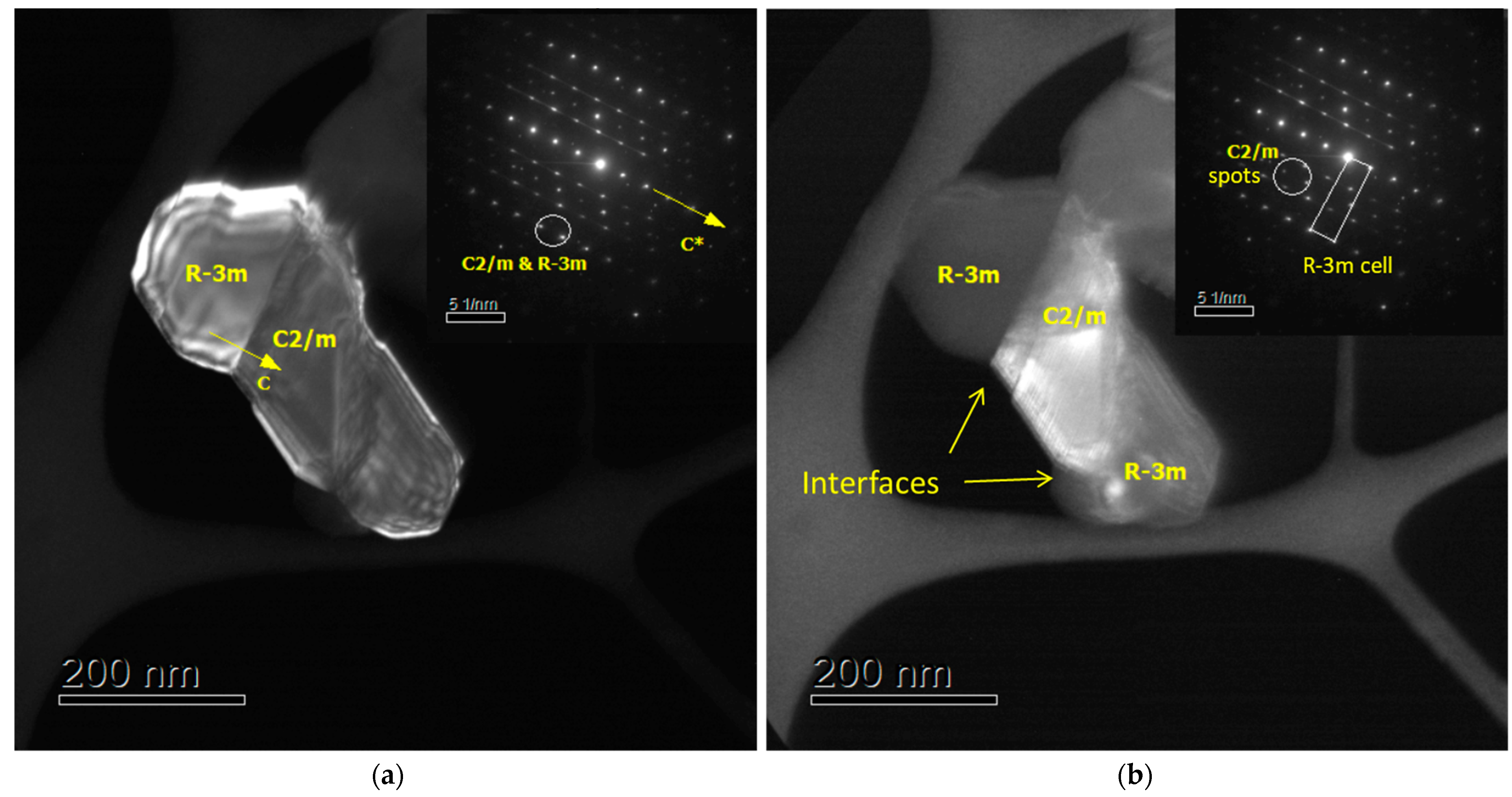
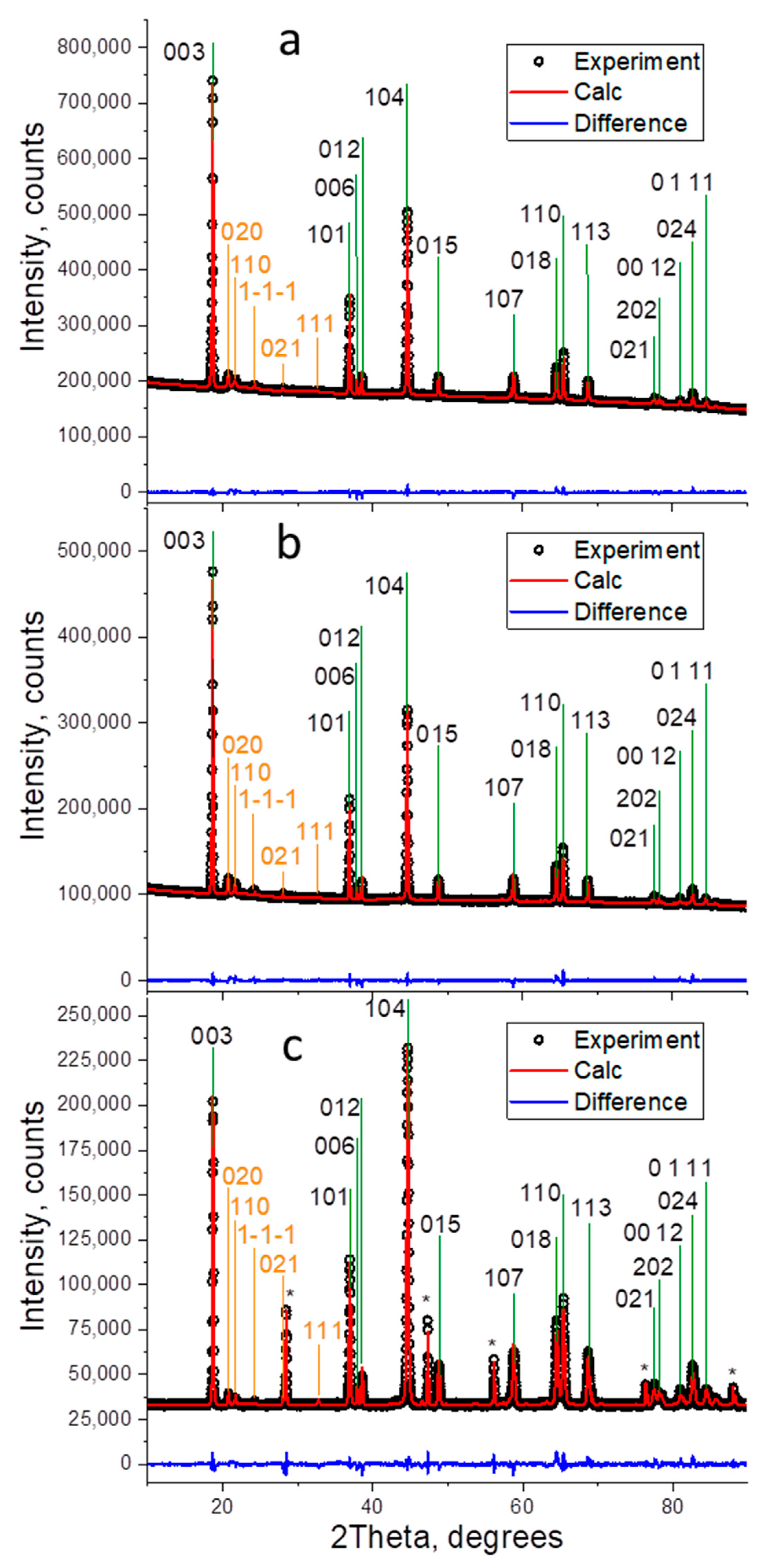
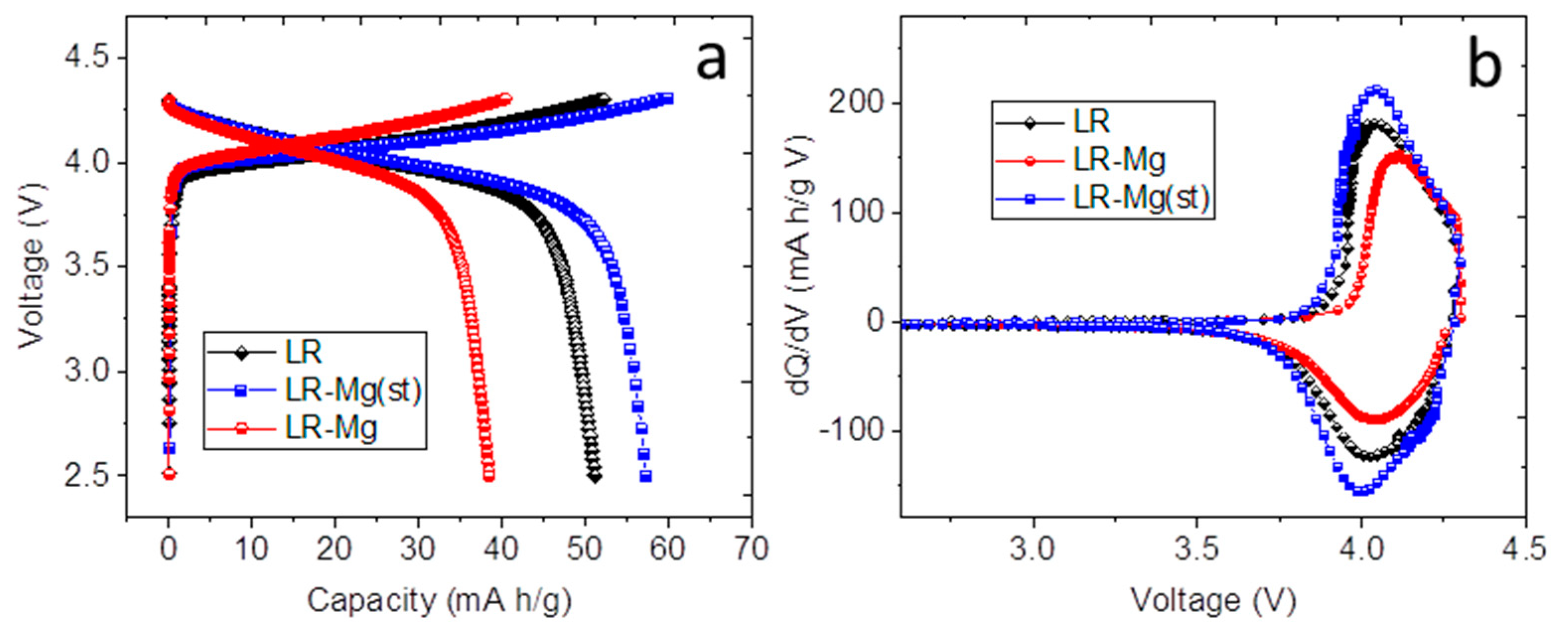
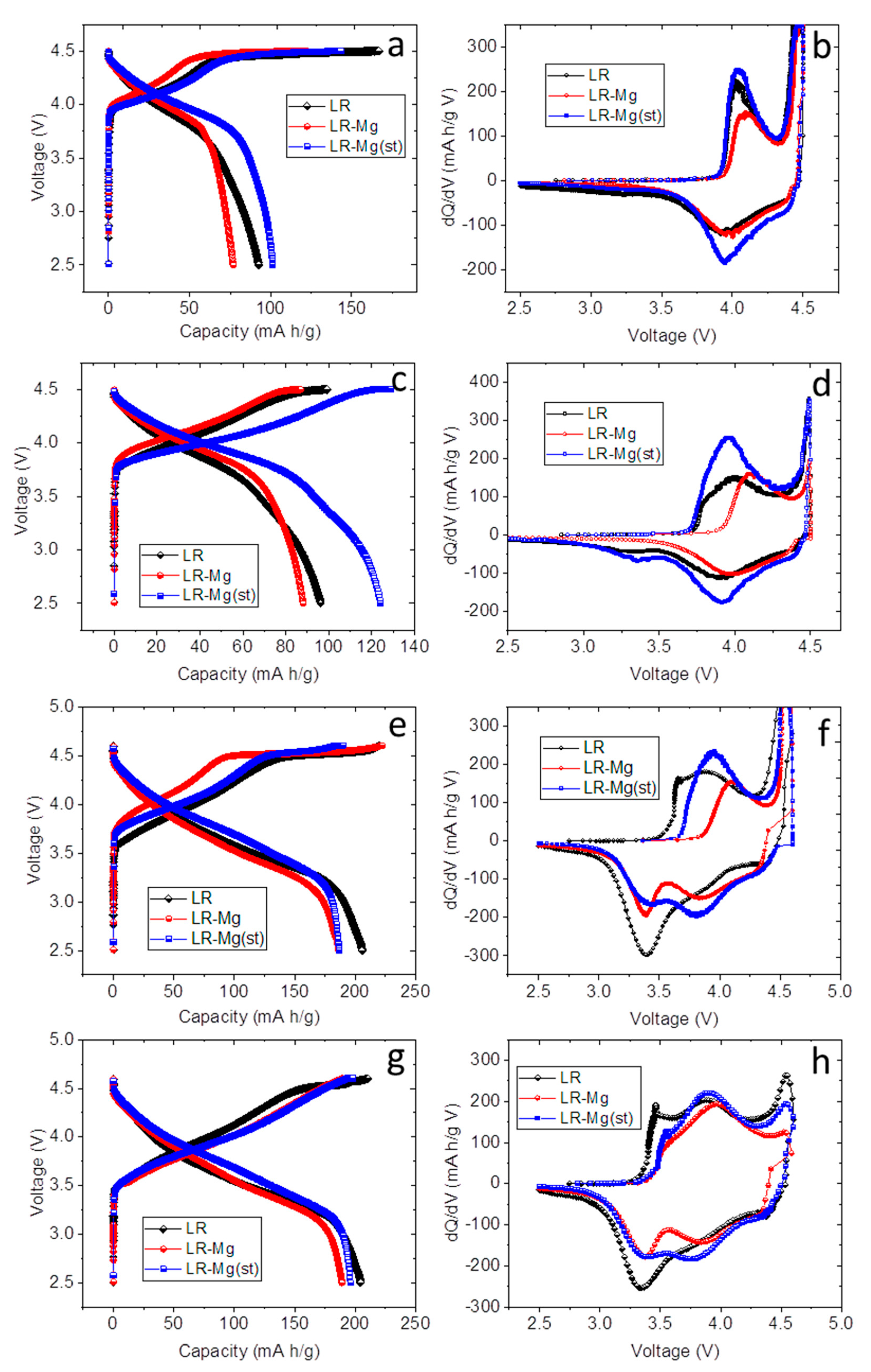
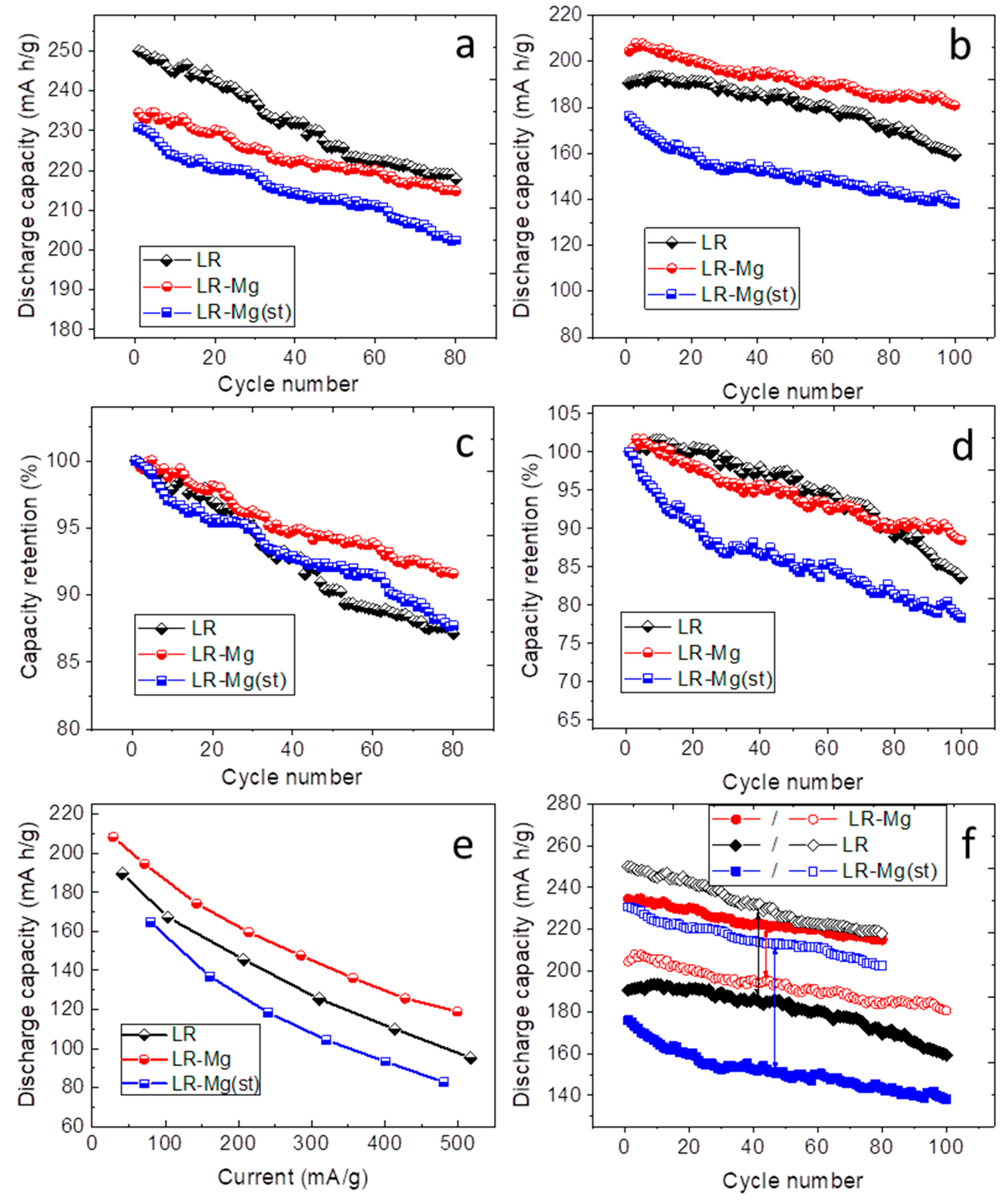
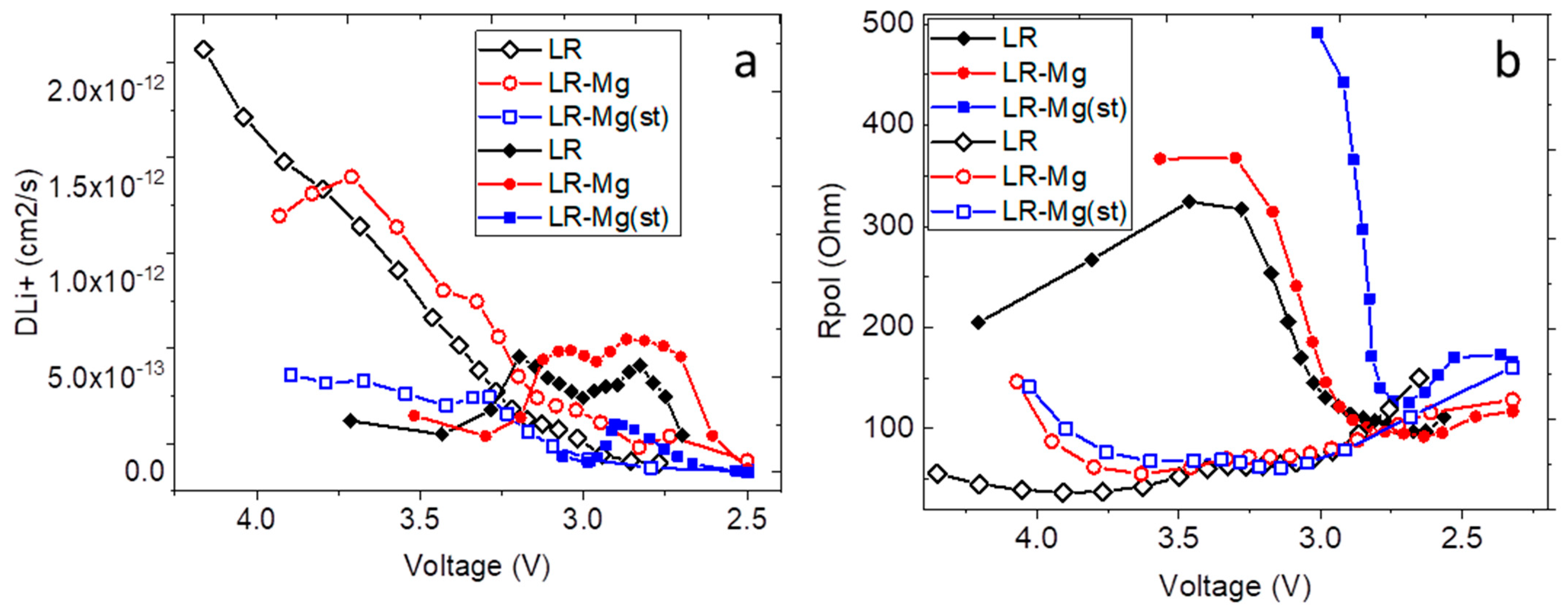
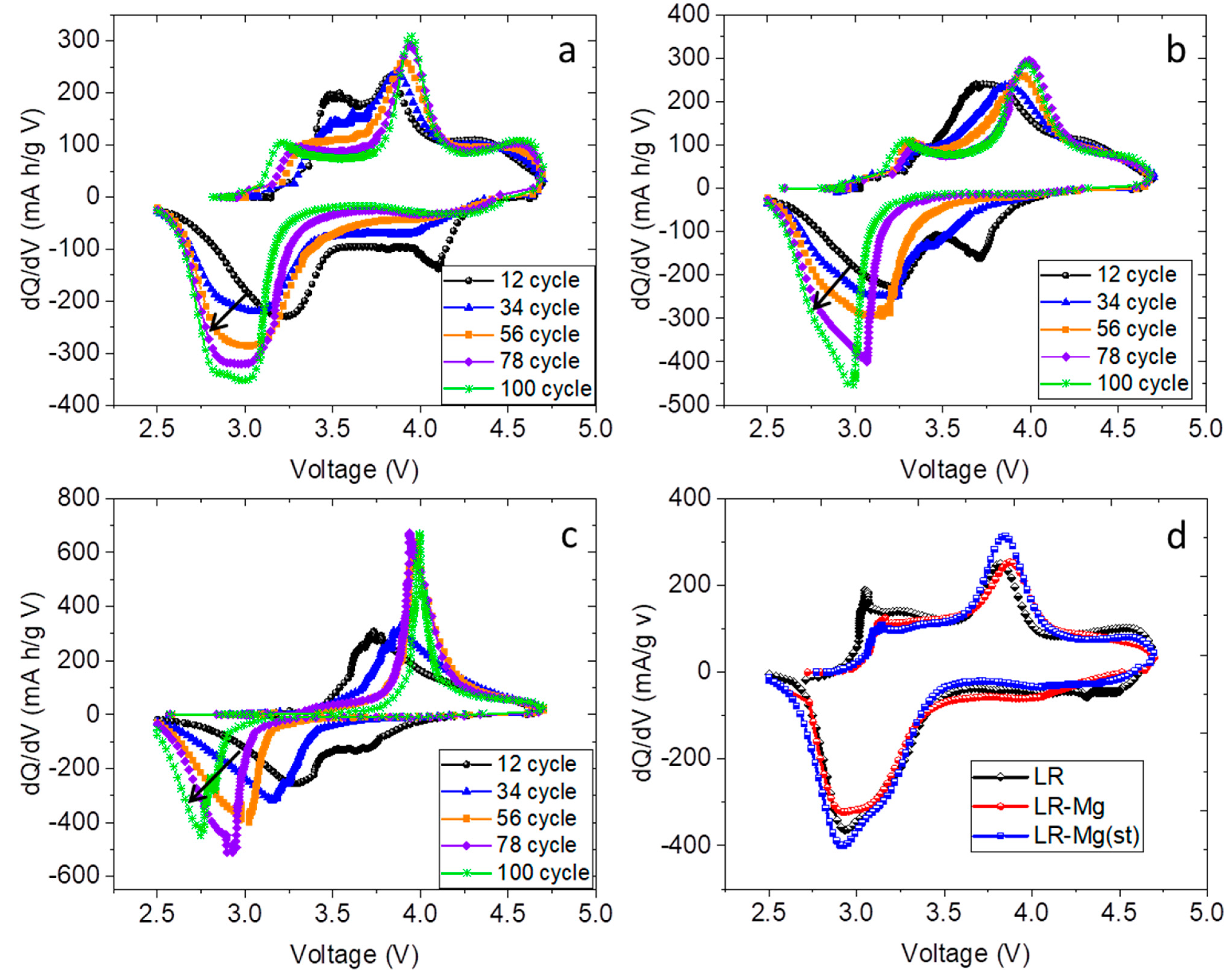
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Department of Energy. Energy Storage Grand Challenge. In Energy Storage Market Report 2020; U.S. Department of Energy: Washington, DC, USA, 2020; p. 65. [Google Scholar]
- Berg, E.J.; Villevieille, C.; Streich, D.; Trabesinger, S.; Novák, P. Rechargeable Batteries: Grasping for the Limits of Chemistry. J. Electrochem. Soc. 2015, 162, A2468–A2475. [Google Scholar] [CrossRef]
- Schipper, F.; Nayak, P.; Erickson, E.; Amalraj, S.; Srur-Lavi, O.; Penki, T.; Talianker, M.; Grinblat, J.; Sclar, H.; Breuer, O.; et al. Study of Cathode Materials for Lithium-Ion Batteries: Recent Progress and New Challenges. Inorganics 2017, 5, 32. [Google Scholar] [CrossRef]
- Johnson, C.S.; Kim, J.-S.; Lefief, C.; Li, N.; Vaughey, J.T.; Thackeray, M.M. The significance of the Li2MnO3 component in ‘composite’ xLi2MnO3·(1−x)LiMn0.5Ni0.5O2 electrodes. Electrochem. Commun. 2004, 6, 1085–1091. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Kang, S.-H.; Johnson, C.S.; Vaughey, J.T.; Benedek, R.; Hackney, S.A. Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J. Mater. Chem. 2007, 17, 3112–3125. [Google Scholar] [CrossRef]
- Koga, H.; Croguennec, L.; Ménétrier, M.; Douhil, K.; Belin, S.; Bourgeois, L.; Suard, E.; Weill, F.; Delmas, C. Reversible Oxygen Participation to the Redox Processes Revealed for Li1.20Mn0.54Co0.13Ni0.13O2. J. Electrochem. Soc. 2013, 160, 786–792. [Google Scholar] [CrossRef]
- Divakaran, A.M.; Minakshi, M.; Bahri, P.A.; Paul, S.; Kumari, P.; Divakaran, A.M.; Manjunatha, K.N. Rational design on materials for developing next generation lithium-ion secondary battery. Prog. Solid State Chem. 2021, 62, 100298. [Google Scholar] [CrossRef]
- Assat, G.; Tarascon, J.-M. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat. Energy 2018, 3, 373–386. [Google Scholar] [CrossRef]
- Xie, Y.; Saubanère, M.; Doublet, M.-L. Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries. Energy Environ. Sci. 2017, 10, 266–274. [Google Scholar] [CrossRef]
- Abakumov, A.M.; Fedotov, S.S.; Antipov, E.V.; Tarascon, J.-M. Solid state chemistry for developing better metal-ion batteries. Nat. Commun. 2020, 11, 4976. [Google Scholar] [CrossRef]
- Seo, D.-H.; Lee, J.; Urban, A.; Malik, R.; Kang, S.; Ceder, G. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 2016, 8, 692–697. [Google Scholar] [CrossRef]
- McCalla, E.; Abakumov, A.M.; Saubanère, M.; Foix, D.; Berg, E.J.; Rousse, G.; Doublet, M.-L.; Gonbeau, D.; Novák, P.; Van Tendeloo, G.; et al. Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 2015, 350, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qiao, Y.; Guo, S.; Xu, Z.; Zhu, H.; Zhang, X.; Yuan, Y.; He, P.; Ishida, M.; Zhou, H. Direct Visualization of the Reversible O2−/O− Redox Process in Li-Rich Cathode Materials. Adv. Mater. 2018, 30, 1705197. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Asl, S.; Lu, J.; Amine, K.; Shahbazian-Yassar, R. Oxygen Release Degradation in Li-Ion Battery Cathode Materials: Mechanisms and Mitigating Approaches. Adv. Energy Mater. 2019, 9, 1900551. [Google Scholar] [CrossRef]
- Chen, H.; Islam, M.S. Lithium Extraction Mechanism in Li-Rich Li2MnO3 Involving Oxygen Hole Formation and Dimerization. Chem. Mater. 2016, 28, 6656–6663. [Google Scholar] [CrossRef]
- Croy, J.R.; Gallagher, K.G.; Balasubramanian, M.; Long, B.R.; Thackeray, M.M. Quantifying Hysteresis and Voltage Fade in xLi 2MnO3•(1−x)LiMn0.5Ni0.5O2 Electrodes as a Function of Li2MnO3 Content. J. Electrochem. Soc. 2014, 161, A318–A325. [Google Scholar] [CrossRef]
- Dogan, F.; Long, B.R.; Croy, J.R.; Gallagher, K.G.; Iddir, H.; Russell, J.T.; Balasubramanian, M.; Key, B. Re-entrant Lithium Local Environments and Defect Driven Electrochemistry of Li- and Mn-Rich Li-Ion Battery Cathodes. J. Am. Chem. Soc. 2015, 137, 2328–2335. [Google Scholar] [CrossRef] [Green Version]
- Myung, S.-T.; Izumi, K.; Komaba, S.; Yashiro, H.; Bang, H.J.; Sun, Y.-K.; Kumagai, N. Functionality of Oxide Coating for Li[Li0.05Ni0.4Co0.15Mn0.4]O2 as Positive Electrode Materials for Lithium-Ion Secondary Batteries. J. Phys. Chem. C 2007, 111, 4061–4067. [Google Scholar] [CrossRef]
- Qiu, B.; Zhang, M.; Wu, L.; Wang, J.; Xia, Y.; Qian, D.; Liu, H.; Hy, S.; Chen, Y.; An, K.; et al. Gas–solid interfacial modification of oxygen activity in layered oxide cathodes for lithium-ion batteries. Nat. Commun. 2016, 7, 12108. [Google Scholar] [CrossRef]
- Zhu, Z.; Yu, D.; Yang, Y.; Su, C.; Huang, Y.; Dong, Y.; Waluyo, I.; Wang, B.; Hunt, A.; Yao, X.; et al. Gradient Li-rich oxide cathode particles immunized against oxygen release by a molten salt treatment. Nat. Energy 2019, 4, 1049–1058. [Google Scholar] [CrossRef]
- Oh, P.; Yun, J.; Park, S.; Nam, G.; Liu, M.; Cho, J. Recent Advances and Prospects of Atomic Substitution on Layered Positive Materials for Lithium-Ion Battery. Adv. Energy Mater. 2021, 11, 2003197. [Google Scholar] [CrossRef]
- Phattharasupakun, N.; Geng, C.; Johnson, M.B.; Väli, R.; Liu, A.; Liu, Y.; Sawangphruk, M.; Dahn, J.R. Impact of Al Doping and Surface Coating on the Electrochemical Performances of Li-Rich Mn-Rich Li1.11Ni0.33Mn0.56O2 Positive Electrode Material. J. Electrochem. Soc. 2020, 167, 120531. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhang, J.; Zhang, H.; Liu, X.; Wang, C.-W.; Sun, L.; Liu, X. The Synergic Effects of Zr Doping and Li 2 TiO 3 Coating on the Crystal Structure and Electrochemical Performances of Li-Rich Li1.2Ni0.2Mn0.6O2. J. Electrochem. Soc. 2019, 166, A1323–A1329. [Google Scholar] [CrossRef]
- Ning, F.; Li, B.; Song, J.; Zuo, Y.; Shang, H.; Zhao, Z.; Yu, Z.; Chu, W.; Zhang, K.; Feng, G.; et al. Inhibition of oxygen dimerization by local symmetry tuning in Li-rich layered oxides for improved stability. Nat. Commun. 2020, 11, 4973. [Google Scholar] [CrossRef] [PubMed]
- Nayak, P.K.; Erickson, E.M.; Schipper, F.; Penki, T.R.; Munichandraiah, N.; Adelhelm, P.; Sclar, H.; Amalraj, F.; Markovsky, B.; Aurbach, D. Review on Challenges and Recent Advances in the Electrochemical Performance of High Capacity Li- and Mn-Rich Cathode Materials for Li-Ion Batteries. Adv. Energy Mater. 2018, 8, 1702397. [Google Scholar] [CrossRef]
- Kong, F.; Longo, R.C.; Yeon, D.-H.; Yoon, J.; Park, J.-H.; Liang, C.; KC, S.; Zheng, Y.; Doo, S.-G.; Cho, K. Multivalent Li-Site Doping of Mn Oxides for Li-Ion Batteries. J. Phys. Chem. C 2015, 119, 21904–21912. [Google Scholar] [CrossRef]
- Liu, W.; Oh, P.; Liu, X.; Myeong, S.; Cho, W.; Cho, J. Countering Voltage Decay and Capacity Fading of Lithium-Rich Cathode Material at 60 °C by Hybrid Surface Protection Layers. Adv. Energy Mater. 2015, 5, 1500274. [Google Scholar] [CrossRef]
- Xu, H.; Deng, S.; Chen, G. Improved electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2 by Mg doping for lithium ion battery cathode material. J. Mater. Chem. A 2014, 2, 15015–15021. [Google Scholar] [CrossRef]
- Nayak, P.K.; Grinblat, J.; Levi, E.; Levi, M.; Markovsky, B.; Aurbach, D. Understanding the influence of Mg doping for the stabilization of capacity and higher discharge voltage of Li- and Mn-rich cathodes for Li-ion batteries. Phys. Chem. Chem. Phys. 2017, 19, 6142–6152. [Google Scholar] [CrossRef]
- Wang, D.; Huang, Y.; Huo, Z.; Chen, L. Synthesize and electrochemical characterization of Mg-doped Li-rich layered Li[Li0.2Ni0.2Mn0.6]O2 cathode material. Electrochim. Acta 2013, 107, 461–466. [Google Scholar] [CrossRef]
- Xiang, Y.; Li, J.; Wu, X.; Liu, Z.; Xiong, L.; He, Z.; Yin, Z. Synthesis and electrochemical characterization of Mg-doped Li-rich Mn-based cathode material. Ceram. Int. 2016, 42, 8833–8838. [Google Scholar] [CrossRef]
- Ni, J.; Zhao, Y.; Chen, J.; Gao, L.; Lu, L. Site-dependent electrochemical performance of Mg doped LiFePO4. Electrochem. Commun. 2014, 44, 4–7. [Google Scholar] [CrossRef]
- Makhonina, E.V.; Pechen, L.S.; Volkov, V.V.; Rumyantsev, A.M.; Koshtyal, Y.M.; Dmitrienko, A.O.; Politov, Y.A.; Pervov, V.S.; Eremenko, I.L. Synthesis, microstructure, and electrochemical performance of Li-rich layered oxide cathode materials for Li-ion batteries. Russ. Chem. Bull. 2019, 68, 301–312. [Google Scholar] [CrossRef]
- Weppner, W.; Huggins, R.A. Determination of the Kinetic Parameters of Mixed-Conducting Electrodes and Application to the System Li3Sb. J. Electrochem. Soc. 1977, 124, 1569–1578. [Google Scholar] [CrossRef]
- Wen, C.J.; Boukamp, B.A.; Huggins, R.A.; Weppner, W. Thermodynamic and Mass Transport Properties of “LiAl”. J. Electrochem. Soc. 1979, 126, 2258–2266. [Google Scholar] [CrossRef]
- Pechen, L.; Makhonina, E.; Volkov, V.; Rumyantsev, A.; Koshtyal, Y.; Politov, Y.; Pervov, V.; Eremenko, I. Investigation of Capacity Fade and Voltage Decay in Li-Rich Cathode Materials with Different Phase Composition. In Proceedings of the 11th International Conference on Nanomaterials-Research & Application, Hotel Voronez I, Brno, Czech Republic, 16–18 October 2019; pp. 144–149. [Google Scholar]
- Koga, H.; Croguennec, L.; Ménétrier, M.; Mannessiez, P.; Weill, F.; Delmas, C. Different oxygen redox participation for bulk and surface: A possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. J. Power Sources 2013, 236, 250–258. [Google Scholar] [CrossRef]
- Makhonina, E.V.; Maslennikova, L.S.; Volkov, V.V.; Medvedeva, A.E.; Rumyantsev, A.M.; Koshtyal, Y.M.; Maximov, M.Y.; Pervov, V.S.; Eremenko, I.L. Li-rich and Ni-rich transition metal oxides: Coating and core-shell structures. Appl. Surf. Sci. 2019, 474, 25–33. [Google Scholar] [CrossRef]
- Muhammad, S.; Kim, H.; Kim, Y.; Kim, D.; Song, J.H.; Yoon, J.; Park, J.-H.; Ahn, S.-J.; Kang, S.-H.; Thackeray, M.M.; et al. Evidence of reversible oxygen participation in anomalously high capacity Li- and Mn-rich cathodes for Li-ion batteries. Nano Energy 2016, 21, 172–184. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.R.; Paterson, A.J.; Dupré, N.; Grey, C.P.; Bruce, P.G. Structural Evolution of Layered LixMnyO2: Combined Neutron, NMR, and Electrochemical Study. Chem. Mater. 2007, 19, 1016–1023. [Google Scholar] [CrossRef]
- Levasseur, S.; Ménétrier, M.; Delmas, C. On the Dual Effect of Mg Doping in LiCoO2 and Li1+δCoO2: Structural, Electronic Properties, and 7Li MAS NMR Studies. Chem. Mater. 2002, 14, 3584–3590. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, Z.; Zheng, X.; Guo, H.; Li, X.; Jing, Q.; Yang, Z. Effect of Mg doping on the structural and electrochemical performance of LiNi0.6Co0.2Mn0.2O2 cathode materials. Electrochim. Acta 2015, 182, 795–802. [Google Scholar] [CrossRef]
- Venkatraman, S.; Manthiram, A. Comparison of the phase relationships of chemically delithiated layered LiCoMO (M=Al and Mg) oxides. Solid State Ionics 2005, 176. [Google Scholar] [CrossRef]
- Boulineau, A.; Simonin, L.; Colin, J.-F.; Bourbon, C.; Patoux, S. First Evidence of Manganese–Nickel Segregation and Densification upon Cycling in Li-Rich Layered Oxides for Lithium Batteries. Nano Lett. 2013, 13, 3857–3863. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lyu, Y.; Gu, L.; Wu, H.; Bak, S.-M.; Zhou, Y.; Amine, K.; Ehrlich, S.N.; Li, H.; Nam, K.-W.; et al. Understanding the Rate Capability of High-Energy-Density Li-Rich Layered Li1.2Ni0.15Co0.1Mn0.55O2 Cathode Materials. Adv. Energy Mater. 2014, 4, 1300950. [Google Scholar] [CrossRef]
- Rozier, P.; Tarascon, J.M. Review—Li-Rich Layered Oxide Cathodes for Next-Generation Li-Ion Batteries: Chances and Challenges. J. Electrochem. Soc. 2015, 162, A2490–A2499. [Google Scholar] [CrossRef]
- Xu, B.; Fell, C.R.; Chi, M.; Meng, Y.S. Identifying surface structural changes in layered Li-excess nickel manganese oxides in high voltage lithium ion batteries: A joint experimental and theoretical study. Energy Environ. Sci. 2011, 4, 2223. [Google Scholar] [CrossRef]
- Croy, J.R.; Kim, D.; Balasubramanian, M.; Gallagher, K.; Kang, S.-H.; Thackeray, M.M. Countering the Voltage Decay in High Capacity xLi2 MnO3•(1–x)LiMO2 Electrodes (M = Mn, Ni, Co) for Li+ -Ion Batteries. J. Electrochem. Soc. 2012, 159, A781–A790. [Google Scholar] [CrossRef]
- Gu, M.; Belharouak, I.; Zheng, J.; Wu, H.; Xiao, J.; Genc, A.; Amine, K.; Thevuthasan, S.; Baer, D.R.; Zhang, J.-G.; et al. Formation of the Spinel Phase in the Layered Composite Cathode Used in Li-Ion Batteries. ACS Nano 2013, 7, 760–767. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Monoclinic Phase Portion, % | a, Å | c, Å | V, Å3 | c/a | Rwp (%) | Goodness of Fit, S |
|---|---|---|---|---|---|---|---|
| LR | 33(3) | 2.84678(6) | 14.2180(5) | 99.788(5) | 4.9944 | 2.81 | 4.12 |
| LR-Mg | 32(1) | 2.84911(4) | 14.2266(5) | 100.011(4) | 4.9933 | 0.91 | 2.87 |
| LR-Mg(st) | 31(4) | 2.85207(7) | 14.2379(4) | 100.299(5) | 4.9921 | 2.20 | 4.23 |
| Sample | C1s, Peak 1 ± 0.1 eV | Mn2p3 ± 0.3 eV | Co2p3 ± 0.2 eV | Ni2p3 ± 0.2 eV | Mg1s ± 0.2 eV | O1s Peak 1 ± 0.2 эB | O1s Peak 2 ± 0.2 эB |
|---|---|---|---|---|---|---|---|
| LR | 284.9 | 642.0 | 779.9/1.3 | 854.4/1.6 | - | 529.3 | 531.0 |
| LR-Mg(st) | 284.9 | 642.0 | 779.9/1.3 | 854.5/1.7 | 1302.9/1.2 | 529.2 | 531.3 |
| LR-Mg | 284.9 | 642.0 | 779.9/1.3 | 854.8/1.9 | 1302.9/1.5 | 529.1 | 531.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makhonina, E.; Pechen, L.; Medvedeva, A.; Politov, Y.; Rumyantsev, A.; Koshtyal, Y.; Volkov, V.; Goloveshkin, A.; Eremenko, I. Effects of Mg Doping at Different Positions in Li-Rich Mn-Based Cathode Material on Electrochemical Performance. Nanomaterials 2022, 12, 156. https://doi.org/10.3390/nano12010156
Makhonina E, Pechen L, Medvedeva A, Politov Y, Rumyantsev A, Koshtyal Y, Volkov V, Goloveshkin A, Eremenko I. Effects of Mg Doping at Different Positions in Li-Rich Mn-Based Cathode Material on Electrochemical Performance. Nanomaterials. 2022; 12(1):156. https://doi.org/10.3390/nano12010156
Chicago/Turabian StyleMakhonina, Elena, Lidia Pechen, Anna Medvedeva, Yury Politov, Aleksander Rumyantsev, Yury Koshtyal, Vyacheslav Volkov, Alexander Goloveshkin, and Igor Eremenko. 2022. "Effects of Mg Doping at Different Positions in Li-Rich Mn-Based Cathode Material on Electrochemical Performance" Nanomaterials 12, no. 1: 156. https://doi.org/10.3390/nano12010156
APA StyleMakhonina, E., Pechen, L., Medvedeva, A., Politov, Y., Rumyantsev, A., Koshtyal, Y., Volkov, V., Goloveshkin, A., & Eremenko, I. (2022). Effects of Mg Doping at Different Positions in Li-Rich Mn-Based Cathode Material on Electrochemical Performance. Nanomaterials, 12(1), 156. https://doi.org/10.3390/nano12010156