1. Introduction
Various osmium oxide compounds exhibit attractive magnetic and electronic phenomena developed from their electron correlation effects, such as the ferromagnetic gapped state in Ba
2NiOsO
6 [
1], the singlet ground-state excitonic magnetism in Y
2OsO
7 [
2,
3,
4], the spin-driven metal to insulator transition in Pb
2CaOsO
6 [
5], and the unusual superconductivity in AOsO
6 (A = Cs, Rb, and K) [
6].
The solid-state osmium oxides are noteworthy for the following two reasons. One lies in understanding the origin of the band gap inducing of the metal to insulator transition. So far, various mechanisms for explaining the band gap opening for solid-state osmium oxide compounds have been extensively considered, such as the Mott-type mechanism, the d-level splitting pattern caused by the electron correlation effect, the Slater-type mechanism, and the orbital symmetry breaking mechanism, driven by the spin–orbit coupling (SOC) effect. As an example, the band gap opening for Cd
2Os
2O
7 and NaOsO
3 is explained by Slater-type insulators, which are associated with magnetic ordering [
7,
8,
9].
For Ba
2NaOsO
6, there has been a debate on whether it is a Mott-type insulator or a SOC effect-driven insulator. In a study by Erickson et al., the nature of the insulating phase for Ba
2NaOsO
6 is represented as a Mott-type insulator [
10,
11]. Xiang et al. suggest that the insulating behavior of Ba
2NaOsO
6 should be developed by the simultaneous effects of electron correlation and SOC [
12]. Although it is well known that the 5d-block element has considerably extended valence orbitals, resulting in weak on-site Coulomb repulsion, the magnetic insulating features of Sr
2MOsO
6 (M = Cu and Ni) are reproduced with a significantly large on-site repulsion at the Os atom site [
13]. Thus, it is of great importance to explore the origin of the band gap opening in solid-state osmium oxide compounds.
The other issue on solid-state osmium oxide is related to the various oxidation states of the Os ion. Osmium forms compounds with oxidation states, ranging from −2 to +8. As an example, the oxidation states of the Os ion in Na
2[Os(CO)], Na
2[Os
4(CO)
13], Os
3(CO)
12, OsI, OsI
2, OsBr
3, OsO
2, OsF
5, OsF
6, OsOF
5, and OsO
4 are −2, −1, 0, +1, +2, +3, +4, +5, +6, +7, and +8, respectively. A large spatial extension of Os at the 5d level is a main reason for the wide spectrum of oxidation states of the Os atom in osmium compounds. In general, the nd orbital of metal in the zero-oxidation state shows a spatial extension that increases in the order 3d < 4d < 5d, so that the widths of the d-block bands should increase in the order 3d < 4d < 5d. For this reason, 5d oxides show various valence states with the wide band widths of d-blocks. The electron correlation effects for 4d and 5d systems are weak while the effects of SOC are strong [
14].
Recently, a new double perovskite osmium oxide Ba
2CuOsO
6 was synthesized under somewhat extreme conditions (~6 GPa and ~1800 K). The crystal structure and magnetic properties of Ba
2CuOsO
6 were characterized with synchrotron X-ray diffraction, thermo-gravimetric analysis, magnetic susceptibility, isothermal magnetization, and specific heat measurements [
15]. The temperature dependence of the specific heat showed an electrically insulating behavior at all measured temperatures [
15]. They also found that the Ba
2CuOsO
6 obeys the Curie–Weiss law with the estimated Weiss temperature −13.3 K [
15]. Interestingly, a magnetic susceptibility measurement shows two T
max at ~55 K and ~70 K. This would be associated with two different magnetic ions in Ba
2CuOsO
6, which lead to more than two types of magnetic sublattice.
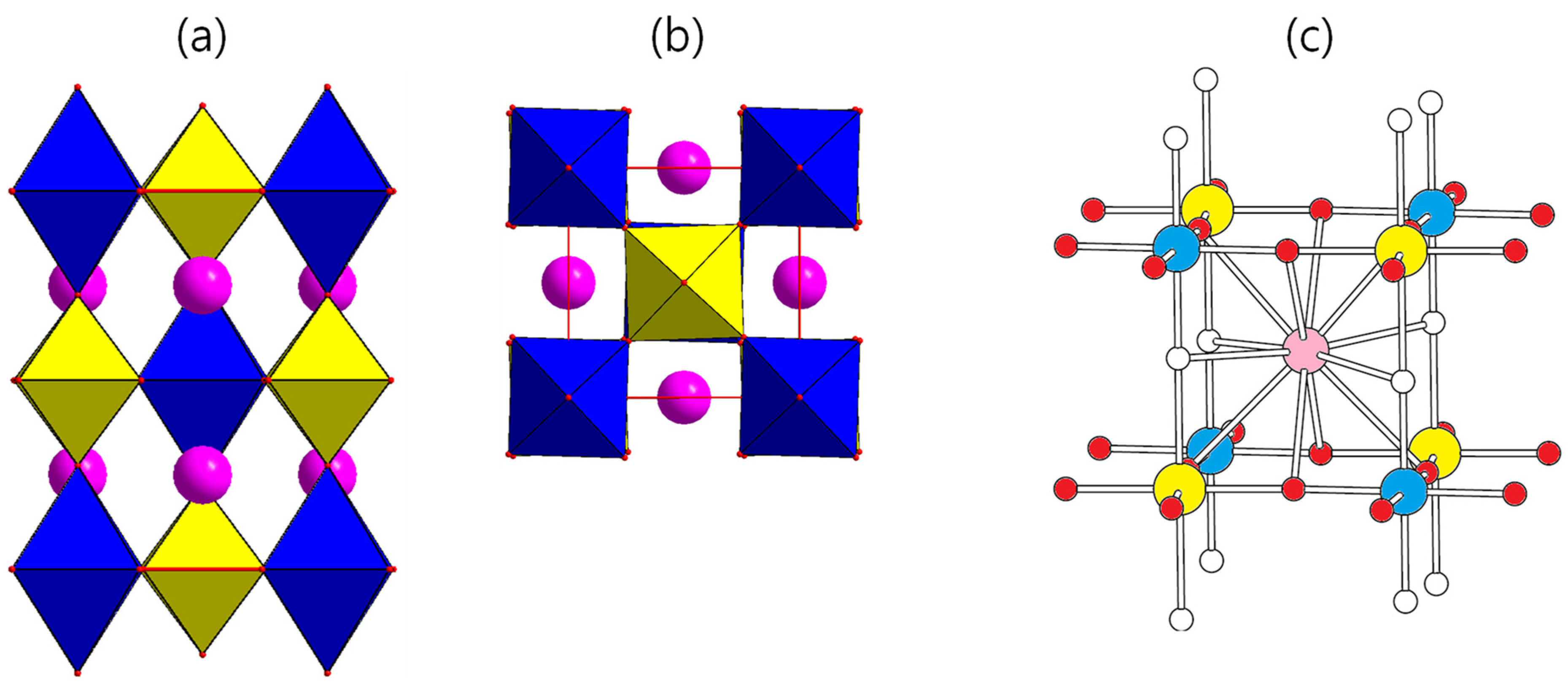
The Ba
2CuOsO
6 crystallizes in a tetragonal space group, I4/m, in which the valence states of the Os and Cu atoms are Os
6+ (d
2, S = 1) and Cu
2+ (d
9, S = 1/2), respectively. The Cu
2+ and Os
6+ ions are located on the perovskite B-site, and they form the CuO
6 and OsO
6 octahedrons, respectively. They share their corners in all crystallographic directions, such that alternating CuO
6 and OsO
6 run in all three crystallographic directions, as shown in
Figure 1. The Ba
2+ ion is located on the center of the Cu
4Os
4 cube (see
Figure 1). With the structural distortion of CuO
6 and OsO
6 octahedrons from the ideal MO
6 octahedron, the Cu-O-Os bridges in the ab plane are bent, whereas those along the c direction are linear, which should be closely related with the orbital splitting and the orbital occupancy.
One can easily predict that the axial Cu-O bond is elongated by a strong Jahn–Teller distortion, associated with the electron configuration of the Cu
2+ (d
9) ion in each CuO
6 octahedron, with two long Cu-O
ax bonds along the z
2 orbital direction (crystallographic c direction) and four short equatorial Cu-O bonds in the x
2–y
2 orbital locating plane (the crystallographic ab plane). Besides, in each OsO
6 octahedron, the (t
2g)
2 electron configuration of the Os
6+ ion exhibits a weak Jahn–Teller distortion, associated with spatial extension of 5d orbital (see
Scheme 1). As a consequence, unlike CuO
6 octahedrons, each OsO
6 octahedron is weakly compressed in the axial direction with two short Os-O
ax bonds along the c direction and four elongated Os-O
eq bonds in the ab plane.
Here, we examined the causes of several interesting and seemingly puzzling phenomena, such as the magnetic structures and the origin of the insulating phase in Ba2CuOsO6, by performing the DFT, the DFT + U, and the DFT + U + SOC electronic band structure calculations. Then, we studied the spin exchange interactions of Ba2CuOsO6 by a relative energy-mapping analysis. The magnetic properties of Ba2CuOsO6 were explained by the aspect of their orbital interactions and their spin exchange interactions. The double antiferromagnetic (AFM) anomaly at ~55 K and ~70 K of the susceptibility curve was investigated by analyzing its spin exchange interaction. Finally, we examined the reason for the insulating phase on the basis of the DFT studies and a perturbation theory analysis using as perturbation.
2. Computational Details
In our DFT calculations, we employed the frozen-core, projector-augmented wave method [
16,
17], encoded in the Vienna ab initio simulation package (VASP) [
18], and the generalized-gradient approximation of Perdew, Burke, and Ernzerhof [
19], for the exchange correlation functional, with the plane-wave cut-off energy of 450 eV and a set of 48 k-points for the irreducible Brillouin zone.
In general, the orbitals of the 5d element are much more diffuse than 3d orbitals, so the U
Os value is expected to be smaller than the U value of Cu. However, in Ba
2CuOsO
6, the oxidation state of Os ion is +6, so that 5d state of Os
6+ ion is strongly contracted. Note that the ionic size of six coordinated Cu
2+ ions (0.870 Å) is larger than that of six coordinated Os
6+ ions (0.685 Å) [
20]. Moreover, the electronegativity of six coordinated Os
6+ and Cu
2+ ions are 2.362 and 1.372, respectively [
21]. Thus, we use the larger U values on the Os
6+ ion than on the Cu
2+ ion. In our DFT calculation, we employed U value sets of U
Cu = 2, U
Os = 3 eV and U
Cu = 4, U
Os = 5 eV, respectively [
22].
The measured electrical resistivity (ρ) vs. temperature of Ba
2CuOsO
6, given in the previous study [
15], shows an insulating behavior for all temperature ranges, which means that the insulating behavior of Ba
2CuOsO
6 is an intrinsic nature, not a phenomenon coupled with magnetic ordering. Additionally, Khaliullin et al. suggest that the band gap is developed from excitonic transition from singlet to triplet in the Y
2Os
2O
7 system [
4]. The Y
2Os
2O
7 (Os
4+, d
4) belongs to the van Vleck-type Mott system, showing non-magnetic ground state, due to SOC and weak temperature dependence, such as uniform magnetic susceptibility above Neel temperature. However, magnetic susceptibility curve in Ba
2CuOsO
6 showed Curie–Weiss behavior over a broad temperature range [
15], implying that it is not a case of suppressed effective magnetic moment by SOC. Thus, we considered the ferromagnetic (FM) state of Ba
2CuOsO
6 in our DFT + U + SOC calculation, even though this system undergoes the antiferromagnetic ordering. In order to find proper U value set for reproducing the insulating state of Ba
2CuOsO
6, we perform systematic DFT + U + SOC calculations with various U value sets. However, all U sets failed to reproduce the insulating state of Ba
2CuOsO
6 except U value set of U
Cu = 4 and U
Os = 5 eV (see
Table S1 and Figure S1). Thereby, we carried out the DFT + U + SOC calculation with the U value set of U
Os = 5 and U
Cu = 4 eV for understanding the origin of the insulating behavior on Ba
2CuOsO
6.
3. Spin Exchange Interaction and Spin Lattice
In understanding the magnetic structure of Ba
2CuOsO
6, it is important to inspect its local structure and its following spin exchange paths.
Figure 2 shows eight possible spin exchange paths of Ba
2CuOsO
6. There are three spin exchanges between Cu ions, three spin exchanges between Os ions, and two Cu-Os spin exchanges. The J
1 and J
2 are the superexchange (SE) type, involving the Cu-O-Os path, while the J
3–J
8 are super-superexchange (SSE), involving M-O···M-O exchange path (M = Cu or Os). The J
1, J
3, J
4 J
7, and J
8 exchanges are ab plane interactions and the J
2, J
5, and J
6 exchanges are interactions between ab planes. The geometrical parameters associated with these paths are listed in
Table 1.
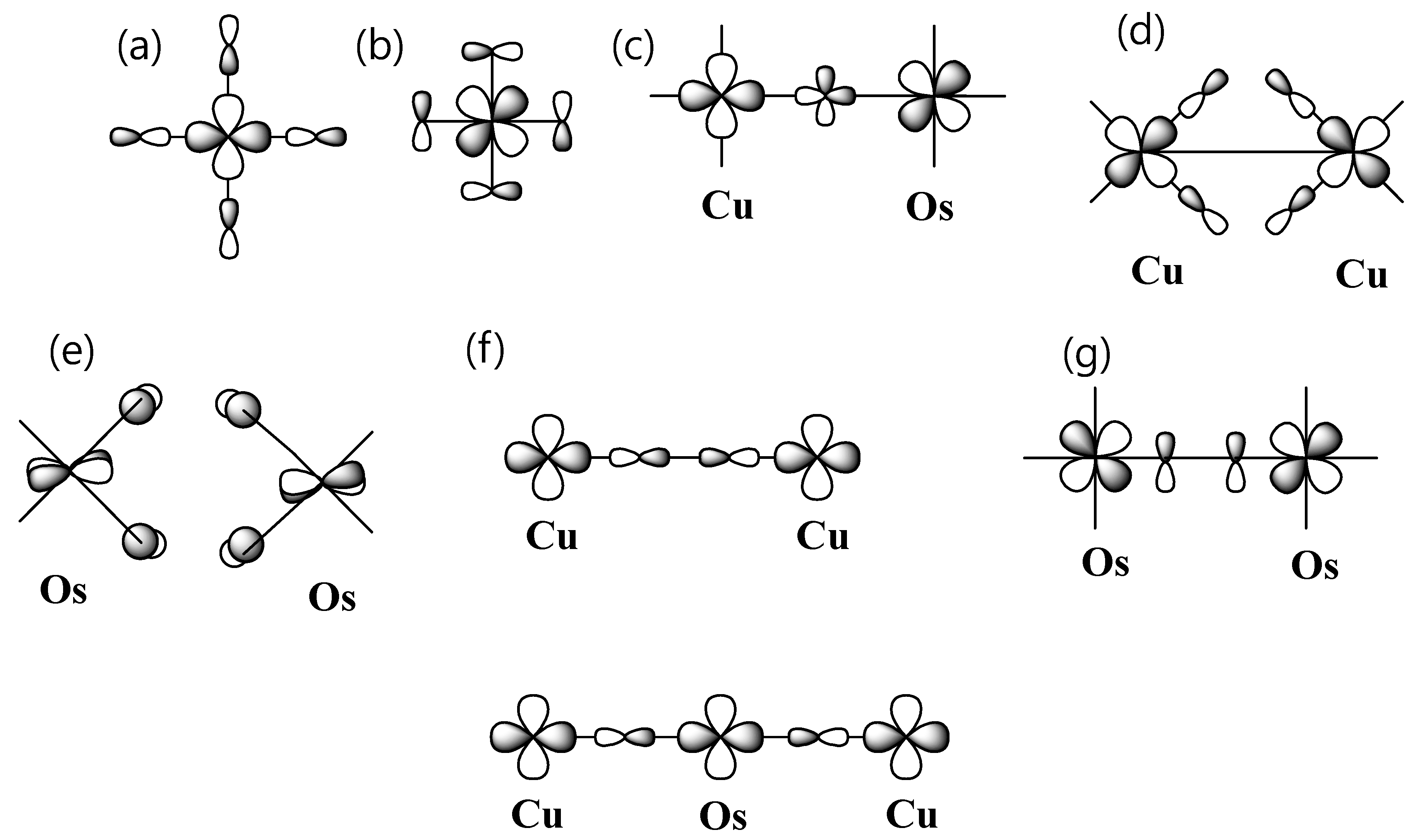
Let us examine the relationship between the spin exchange interactions J
1–J
8 and the structural parameters associated with their exchange pathways and the spin exchange interactions in a viewpoint of orbital interaction. As mentioned, the CuO
6 and OsO
6 octahedrons are distorted from regular MO
6 octahedrons. The nature of distortion should be explained by the crystal field effect, the Jahn–Teller instability, and the different magnitude of interaction between the Ba
2+ ion and the MO
6 (M = Os, and Cu) octahedron. The distortion of the CuO
6 and OsO
6 octahedrons plays an important role in the nature and the strength of the spin exchanges. The CuO
6 octahedron is axially elongated and the OsO
6 octahedron is axially shrunk by the Jahn–Teller instability. Thus, the d-orbital sequence of Cu
2+ ion is xz, yz < xy < z
2 < x
2–y
2 and the d-orbital splitting of Os
6+ ion is xy < xz,yz < x
2–y
2 < z
2. As depicted in
Figure 3a,b, the magnetic orbitals of Cu
2+ ion and Os
6+ are singly occupied x
2–y
2 orbital and two orbitals of three t
2g orbitals, respectively. The structural parameters are listed in
Table 1. The ∠Cu-O-Os angle in J
1 path is close to 180° (172.6°), so the J
1 exchange should be strong FM, because of orthogonality between Cu x
2–y
2 and Os t
2g orbitals, according to Goodenough’s role [
23]. In the J
2 exchange, the Cu-O and Os-O bonds of the Cu-O-Os linkage does not contain magnetic orbitals and the ∠Cu-O-Os angle is 180°. Thus, the Cu
2+ ion and the Os
6+ ion in the J
2 exchange do not interact. The J
2 exchange interaction should be a very weak interaction. The J
3 exchange is unsymmetrical and the ∠Cu-O···O-Cu angles are 127.6 and 142.4°, which contain magnetic orbitals, indicating possible good orbital overlap between Cu
2+ ions (see
Figure 3d). Thus, we expect that J
3 should be AFM. The geometrical structure of J
4 exchange is almost identical to that of the J
3 exchange. However, the J
4 exchange contains two magnetic orbitals, indicating multiple channel spin exchange interactions, suggesting a presence of a strong orbital overlap via the Os-O···O-Os linkage (
Figure 2a). The J
4 spin exchange should have strong AFM because the two magnetic orbitals of the t
2g orbitals can overlap across their O···O contacts, as shown in
Figure 3e. The J
5 and J
6 exchanges are the interactions between the adjacent ab layers, and the O···O contact distances of the SSE path J
5 and J
6 are 4.264 Å. The M-O bonds of the M-O···O-M linkage are not located in the magnetic orbital plane. One can predict that the magnetic interaction of J
5 and J
6 exchange path is negligibly weak.
The ∠M-O···O-M angles of the SE paths J
7 and J
8 are close to 180° (172.6°) and the M-O bond of M-O···M-O linkage contains a magnetic orbital. Thus, the spin exchange interaction J
7 and J
8 should have strong AFM. However, the magnitude of the J
7 spin exchange is much stronger than that of J
8. It is due to the fact that the orbital interaction in the J
7 exchange is a π-type orbital interaction, while the orbital interaction in J
8 is a σ-type orbital interaction, as shown in
Figure 3f,g.
We extracted the spin exchange interactions using the DFT + U + SOC calculation to elucidate our analysis of the spin exchange interactions in terms of the local geometrical structure and the orbital interaction analysis. To extract the values for the J
1–J
8 exchanges, we carried out a total energy calculation of the nine ordered spin configurations of Ba
2CuOsO
6, shown in
Figure 4. To obtain the values of J
1–J
8, we determined the relative energies of the abovementioned cases obtained from the DFT + U + SOC calculations with U
Cu = 2, U
Os = 3 eV and U
Cu = 4, U
Os = 5 eV. The relative energies of various AFM cases obtained are summarized in
Figure 4. In terms of the spin Hamiltonian
, where J
ij = J
1–J
8, the total spin exchange energies of these states per formula units (FUs) are expressed as follows:
by using the energy expressions obtained for spin dimers with M and N unpaired spins per spin site (here, M(Cu
2+) = 1 and N(Os
6+) = 2) [
24]. The values of n
1−n
8 for the eight ordered spin states, FM, and AF1–AF8, are described in
Table S2.
Thus, by mapping the relative energies of the nine spin arrangements presented in
Figure 2 onto corresponding energies expected from Equation (1), we obtain J
1–J
8, as summarized in
Table 2.
The determined spin exchange interactions, based on the DFT + U + SOC calculation (U = 4 and 5 eV for Cu and Os), show that the relative strength of the spin exchange interactions associated with their strongest interaction, J
7, are |J
1| ≈ 0.385 |J
7|, |J
2| ≈ 0.005 |J
7|, |J
3| ≈ 0.283 |J
7|, |J
4| ≈ 0.850 |J
7|, |J
5| ≈ 0.015 |J
7|, |J
6| ≈ 0.025 |J
7|, and |J
8| ≈ 0.339|J
7|, respectively. This suggests that the magnetic property of Ba
2CuOsO
6 is mainly governed by J
1, J
3, J
4, J
7, and J
8 exchanges, in which all of them are AFM except J
1. The J
1 exchange is an FM interaction, as expected in the previous section. The J
4 and J
7 spin exchanges are much stronger than others. As a consequence, the magnetic property of the Ba
2CuOsO
6 system mainly comes from the J
4 and J
7 spin exchange interactions, which would deduce the existence of magnetic sublattice. In addition, the observed phenomena of two T
max at ~55 K and ~70 K in susceptibility measurement would support such a possibility of magnetic sublattice [
15]. The presence of two different magnetic ions in Ba
2CuOsO
6 system should be related with the double AFM-like anomaly at ~55 K and ~70 K. We roughly obtained Neel temperature T
N using a mean field approximation with the extracted spin exchange interactions, based on the DFT + U + SOC calculation (U
Cu = 4 and U
Os = 5 eV). The calculated Neel temperatures for the Cu
2+ ion and Os
6+ ion sublattices are 256 K and 297 K, respectively. The expected T
N for the Cu and Os sublattices is largely overestimated. The overestimation of the calculated T
N is comprehensible because it is well known that the DFT calculations generally overestimate the magnitude of spin exchange interactions by a factor of, approximately, up to four [
25,
26,
27,
28].
Although there is a possibility of spin frustration occurred by (J
1,J
1,J
7), (J
1,J
1,J
8), (J
1,J
1,J
3), (J
1,J
1,J
4), (J
3,J
3,J
7), and (J
4,J
4,J
8) triangles, it does not occur due to the fact that the strong J
4 and J
7 exchanges are forced to avoid spin frustration. This is in good agreement with the experimental result, in which the spin frustration factor (f = |θ|/|T
N|) is just ~0.24, and there is no divergence between the zero-field and field susceptibility curves in Ba
2CuOsO
6 indicating the absence of spin frustration [
15]. For a spin frustrated magnetic system, it is generally expected that the ratio, f = |θ|/|T
N|, is greater than 6 [
29,
30,
31]. The spin frustration factor f-value for Ba
2CuOsO
6 system is much lower than this critical value. Thus, the low f-value suggests that the spin frustration is very weak or does not occur in Ba
2CuOsO
6, despite its similar system (Sr
2CuOsO
6, Sr
2CuIrO
6) [
32,
33] showing strong spin frustration.
In summary, the overall magnetic property of the Ba2CuOsO6 system should be explained by AFM, and it does not show spin frustration.
4. Describing the Magnetic Insulating Behavior of Ba2CuOsO6
As mentioned in the introduction section, the Ba
2CuOsO
6 system is a magnetic insulator. Feng et al. [
15] measured the temperature dependence of resistivity ρ of polycrystalline Ba
2CuOsO
6, in which it showed an insulating behavior at all measured temperature ranges. This reveals that the insulating behavior of Ba
2CuOsO
6 is an intrinsic nature, not a phenomenon coupled with magnetic ordering.
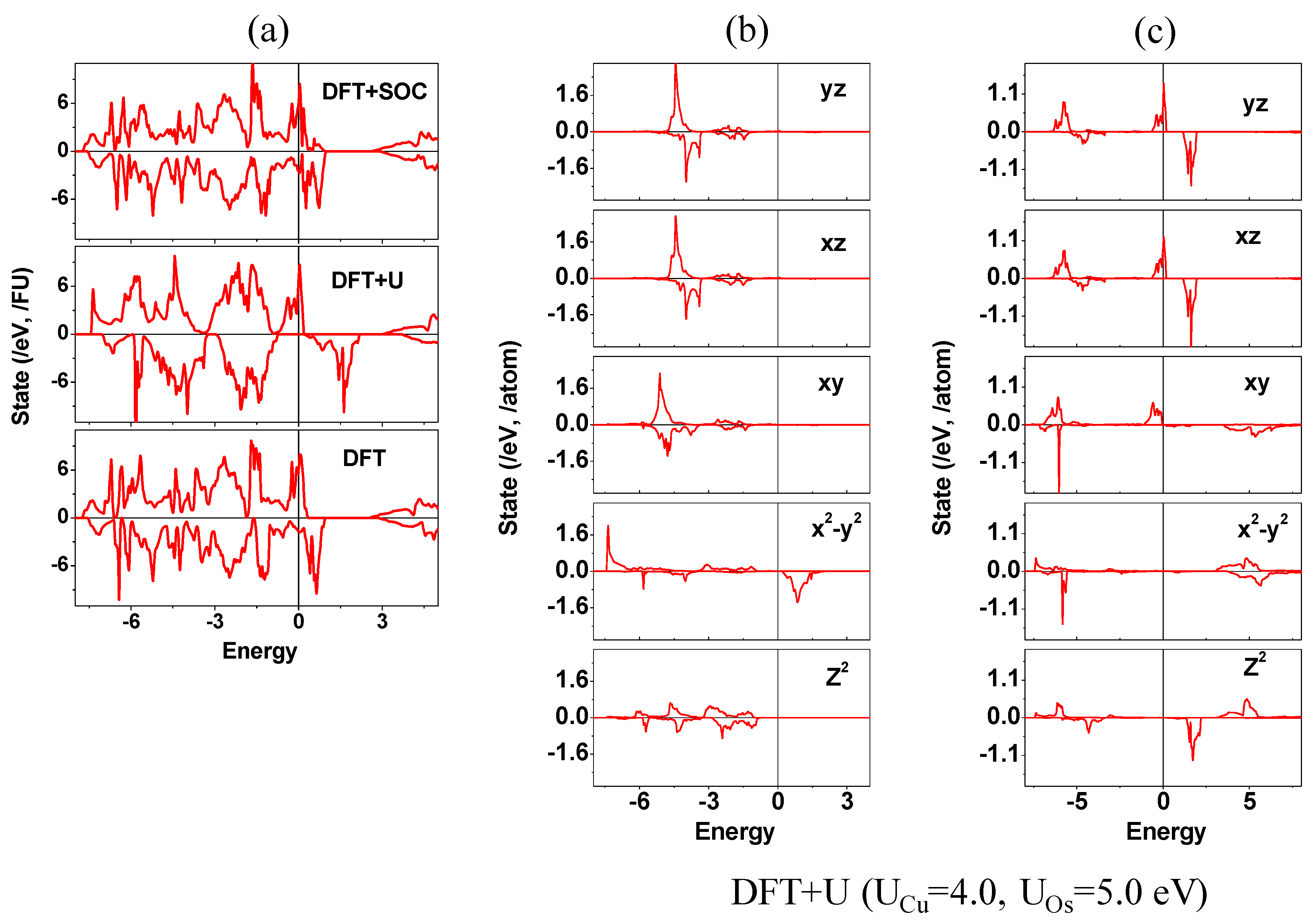
Although they mentioned the origin of the band gap in terms of a theoretical approach, they did not show evidence as well as a discussion for opening the band gap. Here, we discuss the origin of the insulating state of Ba
2CuOsO
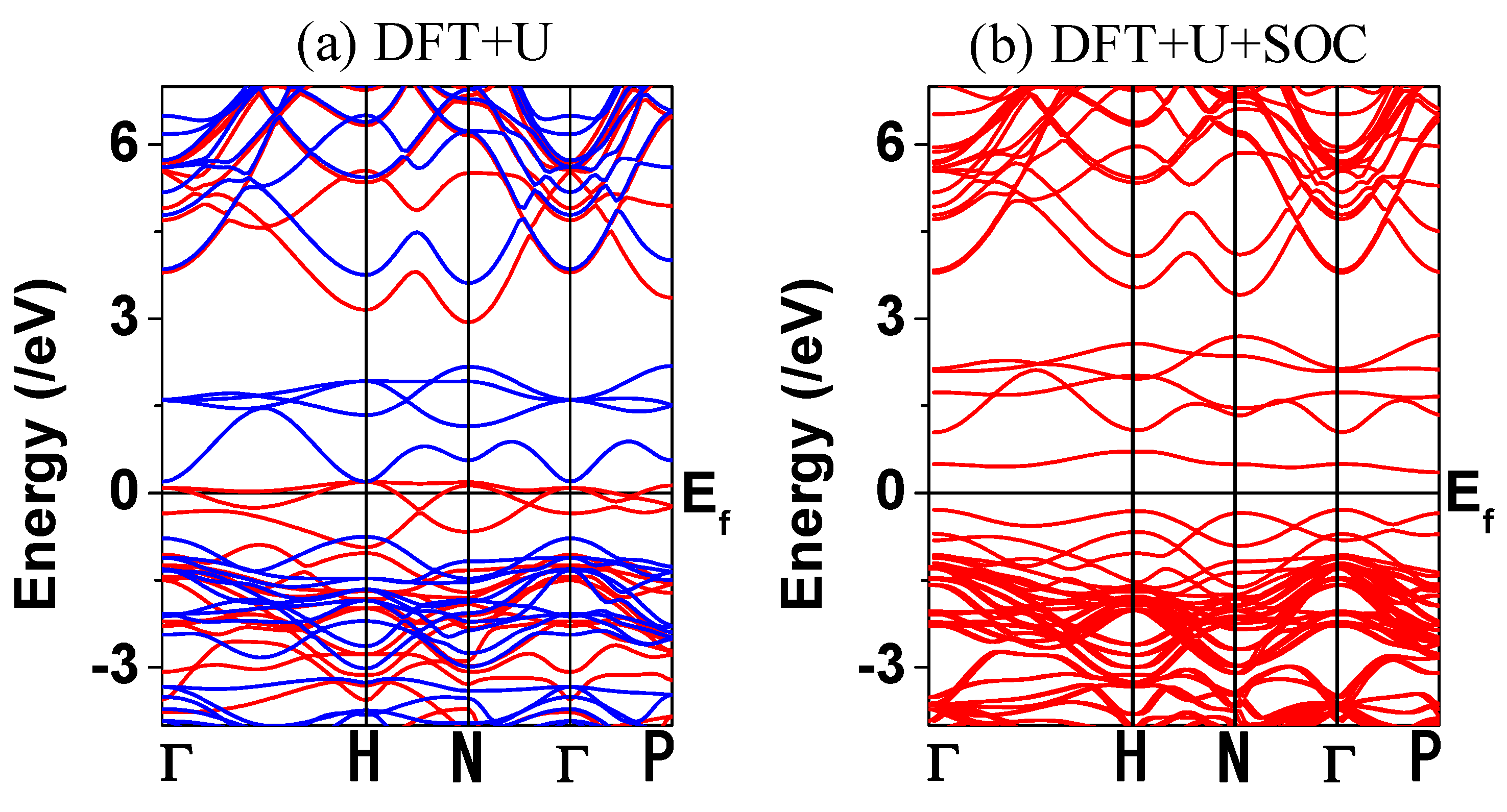
6. The calculated electronic structures for Ba
2CuOsO
6 are presented in
Figure 5 and
Figure 6. The main distribution near the Fermi level comes from the Os
6+ ion rather than the Cu
2+ ion, which connotes that the Os
6+ 5d states are mainly concerned with the insulating behavior of Ba
2CuOsO
6 (see
Figure 5b,c).
For Sr
2CuOsO
6, it should be required to use larger Hubbard value on the Os atom than usual in realizing the magnetic insulating state [
13]. Each OsO
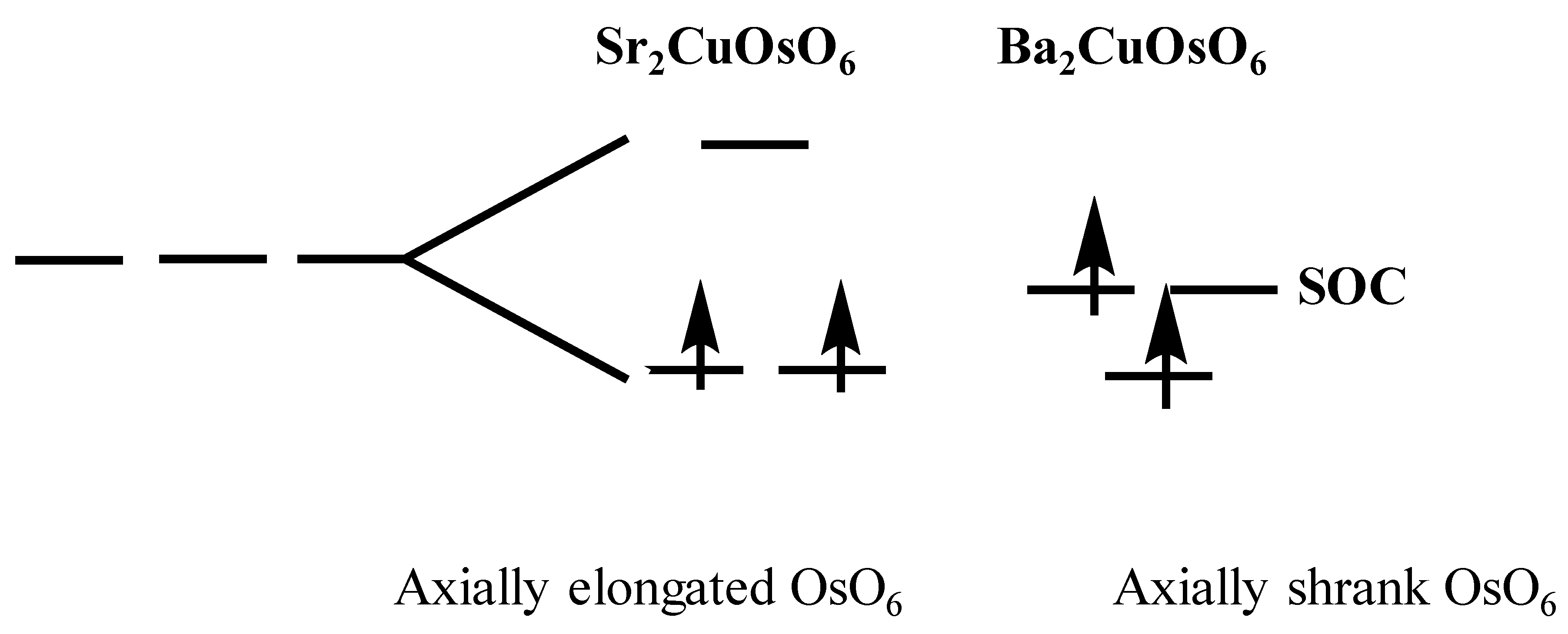
6 octahedron with axially elongated OsO
6 octahedron (i.e., Os-O
ax = 1.928 (×2) Å, Os-O
eq = 1.888 (×4) Å) appears slightly distorted, compared with the CuO
6 octahedron, caused by a weak Jahn–Teller instability. Therefore, the Os 5d state of axially elongated OsO
6 octahedron is split into four states (xz,yz < xy < z
2 < x
2–y
2) by the Jahn–Teller distortion. Two t
2g electrons in the Os
6+ ion are now occupied to the degeneracy-lifted xz and yz states (see
Scheme 1). The band gap is then created by the energy difference between occupied (xz, yz) and unoccupied states (xy). The large Hubbard U value, which enhances the electron correlation effect, is enough to describe the insulating behavior of Sr
2CuOsO
6. On the other hand, each OsO
6 of Ba
2CuOsO
6 has an axially shrunk octahedron, indicating the different types of Os 5d state splitting with Sr
2CuOsO
6. The Os 5d state of Ba
2CuOsO
6 is split into four states (xy < xz,yz < x
2–y
2 < z
2). However, the Os 5d state splitting of Ba
2CuOsO
6 is very weak compared to that of Sr
2CuOsO
6, as shown in the
Scheme 1 and
Figure 5c. Split Os 5d state of Ba
2CuOsO
6 is presented in
Figure 5c, in terms of the projected DOS, which shows that the t
2g level of the Os 5d state is very weakly separated into the two states. In the (t
2g)
2 electron of the Os
6+, one electron is occupied in the lowest xy state, and the remaining electron is occupied in doubly degenerated xz and yz states, as depicted in
Scheme 1. Thus, one can predict that the simple DFT and DFT + U approaches are insufficient to describe the magnetic insulating behavior of Ba
2CuOsO
6. The crucial key to describing the insulating behavior of Ba
2CuOsO
6 is in splitting the degenerate xz and yz states of the Os
6+ ion. The SOC effect splits the t
2g state into three substates by the orbital symmetry breaking. Thus, one can speculate that the SOC effect should play an important role in describing the insulating behavior of Ba
2CuOsO
6, thereby it is necessary to consider the SOC effect.
Moreover, since the Os atom belongs to a heavy element group, the SOC effect should be expected to have a dramatically strong effect on the electronic structure of Ba2CuOsO6.
The spin–orbital part of the Hamiltonian in the Os sphere is then given by the following:
where the SOC constant
λ > 0 for the Os
6+ (d
2) ion, with less than half-filled t
2g levels. With
θ and
φ as the azimuthal and polar angles of the magnetization in the rectangular crystal coordinate system, respectively, the
and
terms are rewritten [
34,
35] as follows:
Since the spin-up and spin-down t
2g states are separated by the exchange splitting in the first order approximation, there is no need to consider interactions between different spin-up and -down states in SOC. Thus, one simply needs to consider only spin-up parts of t
2g states in using the degenerate perturbation theory, which requires calculation of the matrix elements
(
i,
j =
xy,
yz,
xz) [
6]
. For that reason, only the
Sz operator term, as in the first line of Equation (3), brings about non-zero matrix elements. In evaluating these matrix elements, it is convenient to rewrite the angular parts of the
xy,
yz, and
xz orbitals in terms of the spherical harmonics [
12], as follows:
Using these functions, the matrix representation elements
(
i,
j =
xy,
yz,
xz) are found as follows:
By the diagonalization of Equation (5), we obtain eigenvalues of the three spin–orbit coupled states,
,
, and
. The associated eigenfunctions
,
, and
are given [
12,
34,
35] by the following:
The above analysis indicates that the SOC effect splits the t2g states into three substates by orbital symmetry breaking. For the Os6+ (d2) ion, two electrons of t2g state should occupy and. Therefore, the band gap around the Fermi level should be developed in between the occupied and the unoccupied state.
To elucidate the above discussion, we carried out calculations to reproduce the insulating state of Ba
2CuOsO
6 by employing different theoretical methods, namely, simple DFT, DFT + U (U
Cu = 4, U
Os = 5 eV), and DFT + SOC methods, but all of them failed to reproduce the insulating behavior for Ba
2CuOsO
6 (see
Figure 5a). Metallic electronic structures obtained from the DFT and the DFT + U calculations are already expected because the exchange splitting is not enough condition to open the band gap in Ba
2CuOsO
6. However, the electronic structure obtained from the DFT + SOC still failed to reproduce the insulating behavior for Ba
2CuOsO
6 (See
Figure 5a). Presented in
Figure 3a, the spin-up and spin-down bands are overlapped, which leads to a metallic state for Ba
2CuOsO
6. This means that not only the breaking of orbital symmetry by adapting the SOC effect, but also the separation of energy between the filled level (spin-up) and the empty level (spin-down) by increasing exchange splitting, should be required to describe the insulating state of Ba
2CuOsO
6. On-site repulsion, which properly describes the electron correlation effect, gives help to enhance the exchange splitting; thereby, the energy separation between the filled and the unfilled states within each spin channel is increased. Thus, the on-site repulsion should be also a crucial key to be considered in explaining the insulating state of Ba
2CuOsO
6.
To gain insight into this analysis, we carried out the band structure calculation, considering the electron correlation and the SOC effect simultaneously. The band structure calculated for the FM state of Ba
2CuOsO
6 using the DFT + U + SOC (U
eff = 4 and 5 eV on Cu and Os) is shown in
Figure 6b. Indeed, the calculated electronic structure clearly shows an insulating band gap. In consequence, the interplay between the electron correlation and the SOC effect plays an essential role in opening a band gap for Ba
2CuOsO
6.
Meanwhile, there remains one question about the origin of the band gap opening in Ba
2CuOsO
6. The insulating behavior of Ba
2CuOsO
6 is intimately linked with the structural distortion of the OsO
6 octahedron. We remind that the OsO
6 octahedron for Ba
2CuOsO
6 is an axially compressed octahedral shape, while the OsO
6 octahedron of Sr
2CuOsO
6 is an axially elongated octahedral shape. This implies that they undergo different types of structural distortion by the Jahn–Teller instability, which leads to different orbital splitting at the Os
6+ t
2g level. Thus, they show different chemical and physical properties, especially in explaining the mechanism of band gap opening. The only difference between Sr
2CuOsO
6 and Ba
2CuOsO
6 is an ionic size of the A-site ion, namely, the ionic size of the Ba
2+ ion is larger than that of the Sr
2+ ion. In A
2CuOsO
6 (A = Sr and Ba), the A-site ion is located in the center of Cu
4Os
4 distorted cubes and distorted (O
ax)
4 squares, presented in
Figure 1c. It is connected with 8 O
eq and 4 O
ax atoms to form a 12-coordinate AO
12 (see
Figure 1c). In A
2CuOsO
6, a change in the A-O
eq distance affects a, b, and c lattice parameters symmetrically; whereas, a change of A-O
ax distance affects the lattice parameter a (=b). Assuming that the increase of the A-O
ax distance leads to structural distortion forming axially shrunk type-OsO
6 octahedrons, by increasing the Os-O
eq distance in the ab plane, while the decrease of the A-O
ax distance causes structural distortion to have axially elongated OsO
6 octahedrons by decreasing the Os-O
eq distance. The A-O
ax distance should be mainly dominated by the ionic size of the A-site ion. Since the ionic size of the Ba
2+ ion is larger than that of the Sr
2+ ion, the Ba-O
ax distance is longer than the Sr-O
ax distance, which gives rise to a much longer Os-O
eq distance on Ba
2CuOsO
6, associated with the expansion of its lattice parameter a (=b). On the other hand, the effect of ionic size of the A-site on the change of the Os-O
ax distance is relatively insignificant. Indeed, the Os-O
eq distances in Ba
2CuOsO
6 and Sr
2CuOsO
6 are 1.960 and 1.888 Å, respectively, while the Os-O
ax distances in Ba
2CuOsO
6 and Sr
2CuOsO
6 are 1.928 Å and 1.946 Å, respectively [
15,
33]. Hence, the ionic size of the A-site ion should play an important role in determining the shape of the OsO
6 octahedron in A
2CuOsO
6.
To verify the importance of the ionic size effect, we examine the aforementioned questions with the DFT + U + SOC (U
Cu = 4, U
Os = 5 eV) calculation for Ba
2CuOsO
6. We imagine the hypothetical compounds of A
2CuOsO
6 (A = Sr, Ca) by replacing the Ba atoms of Ba
2CuOsO
6 with other alkali earth atoms—Sr and Ca. A
2CuOsO
6 (A = Ba, Sr, and Ca) are fully optimized with the DFT + U + SOC (U
Cu = 4, U
Os = 5 eV) calculation. The optimized atomic positions and cell parameters are presented in the
Supplementary Materials, Table S3. Results show that the lattice parameters decrease gradually with the decreasing ionic size of the A-site ion, but the decreasing of the a lattice parameter is greater than that of the c lattice parameter, which means that each Os-O
eq distance is increased as the A-site ionic size increases. Therefore, the cooperative effect of the A-site ionic size and the Jahn–Teller distortion, is responsible not only for the axially compressed OsO
6 octahedrons in Ba
2CuOsO
6 but also for the axial elongation of OsO
6 octahedrons in Sr
2CuOsO
6; this is closely related to the description of the insulating behavior of A
2CuOsO
6 (A = Ba, Sr, and Ca).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}