
Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes

 , , and
, , and
Abstract
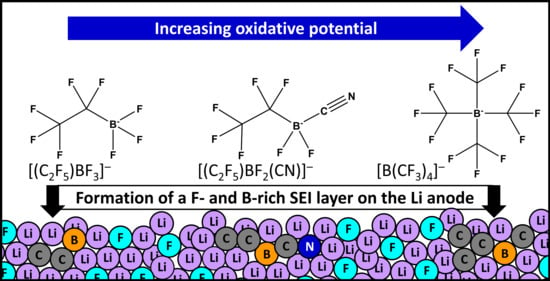
:
1. Introduction
2. Methodology
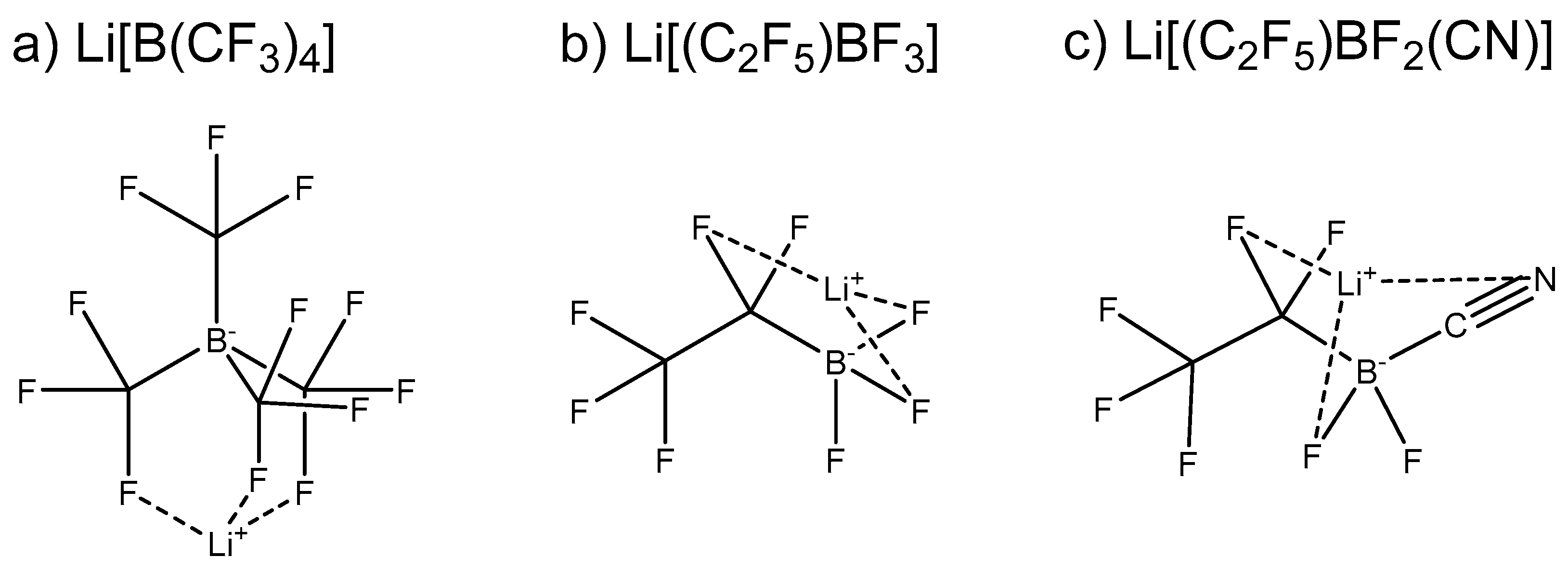
2.1. Individual Anions and Li-Salts
2.2. Li-Salt/Li(001)
3. Results and Discussion
3.1. DFT Calculations of Individual Anions and Li-Salts
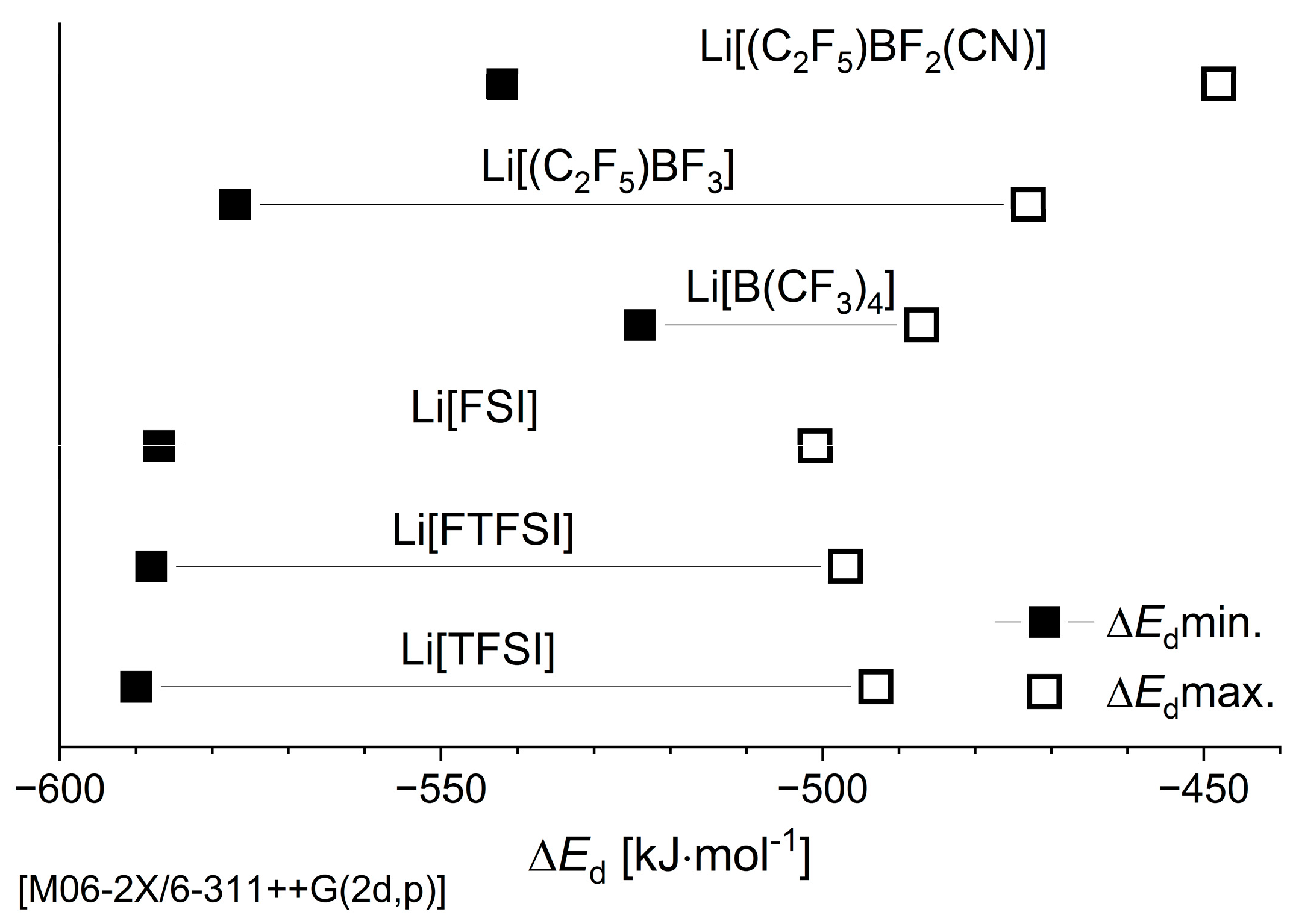
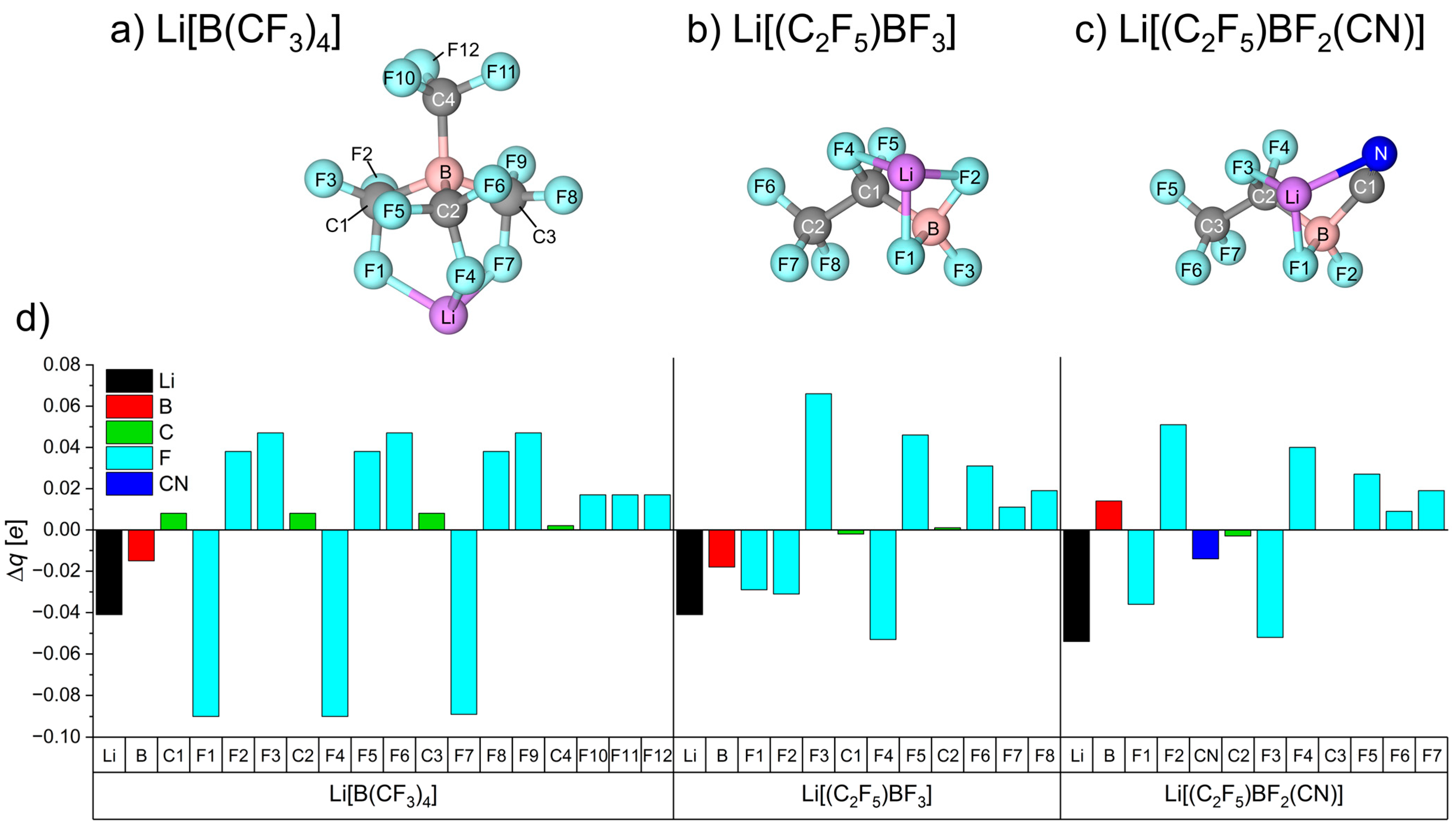
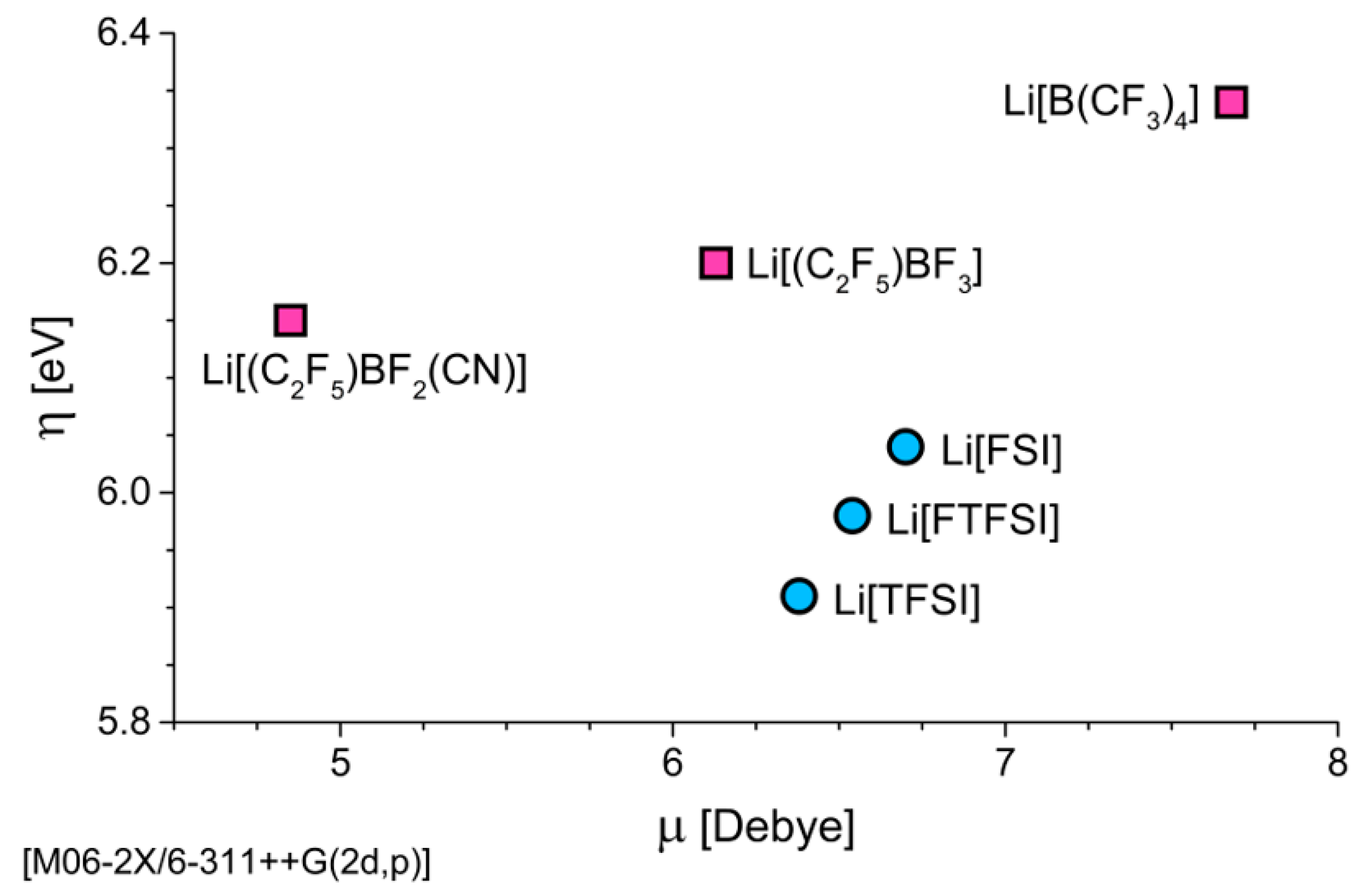
3.1.1. Electronic Dissociation Energy, Charge Distribution, and Dipole Moment for Li-Salt Systems
3.1.2. Chemical Hardness
3.1.3. Stability against Oxidation and Reduction
3.2. Li-Salt/Li(001)
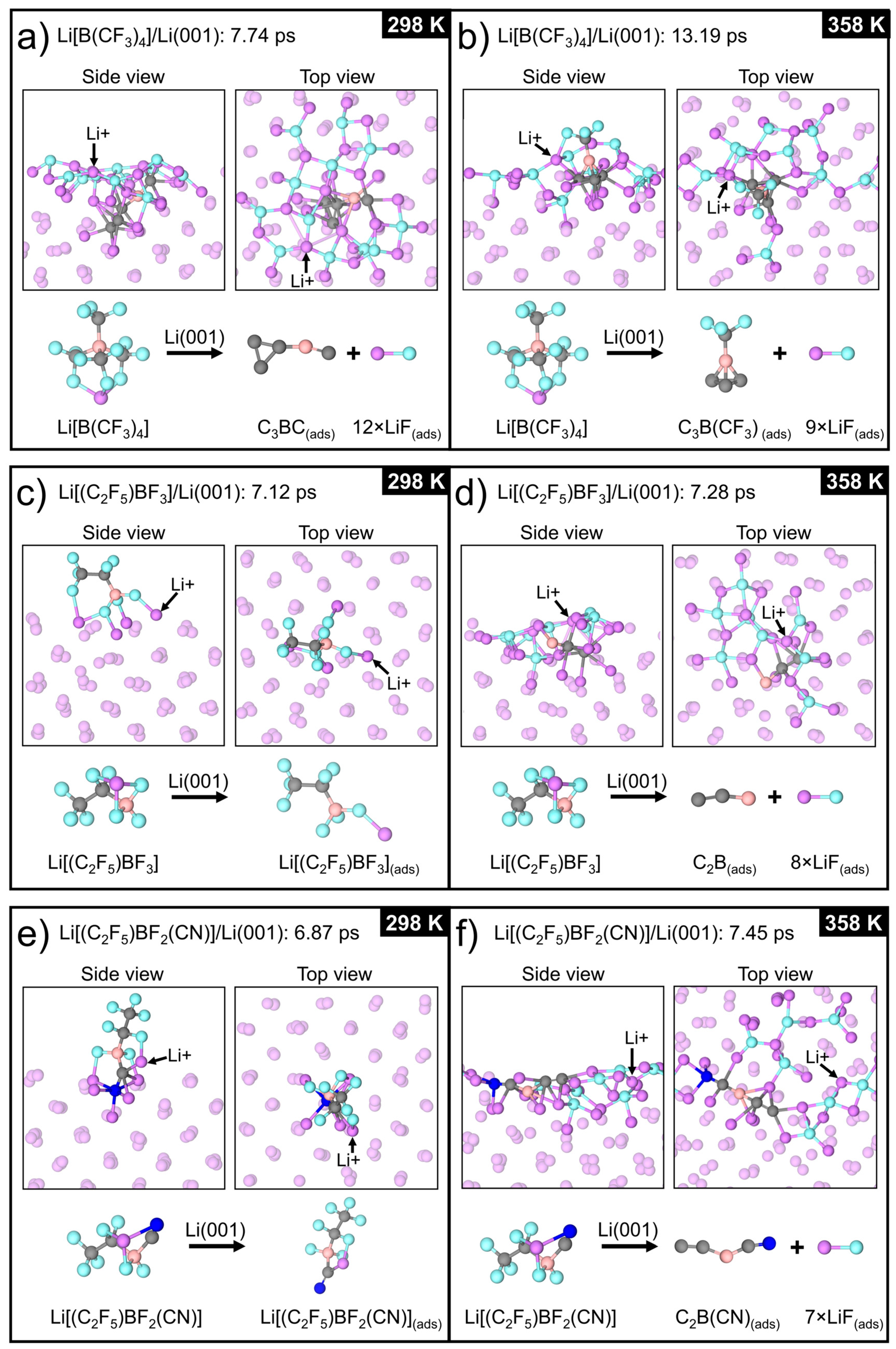
3.2.1. Li[B(CF3)4]/Li(001)
3.2.2. Li[(C2F5)BF3]/Li(001)
3.2.3. Li[(C2F5)BF2(CN)]/Li(001)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, W.; Wang, J.; Ding, F.; Chen, X.; Nasybulin, E.; Zhang, Y.; Zhang, J.-G. Lithium metal anodes for rechargeable batteries. Energy Environ. Sci. 2014, 7, 513–537. [Google Scholar] [CrossRef]
- Aurbach, D.; Zinigrad, E.; Cohen, Y.; Teller, H. A short review of failure mechanisms of lithium metal and lithiated graphite anodes in liquid electrolyte solutions. Solid State Ion. 2002, 148, 405–416. [Google Scholar] [CrossRef]
- Gireaud, L.; Grugeon, S.; Laruelle, S.; Yrieix, B.; Tarascon, J.M. Lithium metal stripping/plating mechanisms studies: A metallurgical approach. Electrochem. Commun. 2006, 8, 1639–1649. [Google Scholar] [CrossRef]
- Luo, Z.; Qiu, X.; Liu, C.; Li, S.; Wang, C.; Zou, G.; Hou, H.; Ji, X. Interfacial challenges towards stable Li metal anode. Nano Energy 2021, 79, 105507. [Google Scholar] [CrossRef]
- Hu, Z.; Li, G.; Wang, A.; Luo, J.; Liu, X. Recent Progress of Electrolyte Design for Lithium Metal Batteries. Batter. Supercaps 2020, 3, 331–335. [Google Scholar] [CrossRef]
- Albertus, P.; Babinec, S.; Litzelman, S.; Newman, A. Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries. Nat. Energy 2017, 3, 16–21. [Google Scholar] [CrossRef]
- Cheng, X.B.; Zhang, R.; Zhao, C.Z.; Zhang, Q. Toward Safe Lithium Metal Anode in Rechargeable Batteries: A Review. Chem. Rev. 2017, 117, 10403–10473. [Google Scholar] [CrossRef]
- Ghazi, Z.A.; Sun, Z.; Sun, C.; Qi, F.; An, B.; Li, F.; Cheng, H.M. Key Aspects of Lithium Metal Anodes for Lithium Metal Batteries. Small 2019, 15, 1900687. [Google Scholar] [CrossRef]
- Brennan, M.D.; Breedon, M.; Best, A.S.; Morishita, T.; Spencer, M.J.S. Surface Reactions of Ethylene Carbonate and Propylene Carbonate on the Li(001) Surface. Electrochim. Acta 2017, 243, 320–330. [Google Scholar] [CrossRef]
- Merinov, B.V.; Zybin, S.V.; Naserifar, S.; Morozov, S.; Oppenheim, J.; Goddard, W.A.; Lee, J.; Lee, J.H.; Han, H.E.; Choi, Y.C.; et al. Interface Structure in Li-Metal/[Pyr14][TFSI]-Ionic Liquid System from ab Initio Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2019, 10, 4577–4586. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.B.; Zhang, R.; Zhao, C.Z.; Wei, F.; Zhang, J.G.; Zhang, Q. A Review of Solid Electrolyte Interphases on Lithium Metal Anode. Adv. Sci. 2016, 3, 1500213. [Google Scholar] [CrossRef]
- Chen, S.; Wen, K.; Fan, J.; Bando, Y.; Golberg, D. Progress and future prospects of high-voltage and high-safety electrolytes in advanced lithium batteries: From liquid to solid electrolytes. J. Mater. Chem. A 2018, 6, 11631–11663. [Google Scholar] [CrossRef] [Green Version]
- Jang, D.H.; Oh, S.M. Electrolyte Effects on Spinel Dissolution and Cathodic Capacity Losses in 4 V Li/LixMn2O4 Rechargeable Cells. J. Electrochem. Soc. 1997, 144, 3342–3348. [Google Scholar] [CrossRef]
- Li, L.; Dai, H.; Wang, C. Electrolyte additives: Adding the stability of lithium metal anodes. Nano Select 2020, 2, 16–36. [Google Scholar] [CrossRef]
- Wu, F.; Yuan, Y.-X.; Cheng, X.-B.; Bai, Y.; Li, Y.; Wu, C.; Zhang, Q. Perspectives for restraining harsh lithium dendrite growth: Towards robust lithium metal anodes. Energy Storage Mater. 2018, 15, 148–170. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, J.-G.; Xu, W. Advancing Lithium Metal Batteries. Joule 2018, 2, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüther, T.; Bhatt, A.I.; Best, A.S.; Harris, K.R.; Hollenkamp, A.F. Electrolytes for Lithium (Sodium) Batteries Based on Ionic Liquids: Highlighting the Key Role Played by the Anion. Batter. Supercaps 2020, 3, 793–827. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Z.; Sun, X.G.; Hu, Y.S.; Xing, H.; Dai, S. Ionic liquids and derived materials for lithium and sodium batteries. Chem. Soc. Rev. 2018, 47, 2020–2064. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, K.; Wang, X.; Zhong, Y.; Liu, M.; Liu, X.; Xu, K.; Fan, W.; Yu, L.; Li, W. Lithium Bis(oxalate)borate Reinforces the Interphase on Li-Metal Anodes. ACS Appl. Mater. Interfaces 2019, 11, 20854–20863. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, S.; Lee, H.; Zhang, L.; Engelhard, M.H.; Li, Q.; Jiao, S.; Liu, J.; Xu, W.; Zhang, J.-G. A Localized High-Concentration Electrolyte with Optimized Solvents and Lithium Difluoro(oxalate)borate Additive for Stable Lithium Metal Batteries. ACS Energy Lett. 2018, 3, 2059–2067. [Google Scholar] [CrossRef]
- Schedlbauer, T.; Krüger, S.; Schmitz, R.; Schmitz, R.W.; Schreiner, C.; Gores, H.J.; Passerini, S.; Winter, M. Lithium difluoro(oxalato)borate: A promising salt for lithium metal based secondary batteries? Electrochim. Acta 2013, 92, 102–107. [Google Scholar] [CrossRef]
- Qiao, L.; Cui, Z.; Chen, B.; Xu, G.; Zhang, Z.; Ma, J.; Du, H.; Liu, X.; Huang, S.; Tang, K.; et al. A promising bulky anion based lithium borate salt for lithium metal batteries. Chem. Sci. 2018, 9, 3451–3458. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Chen, L.; Borodin, O.; Ji, X.; Chen, J.; Hou, S.; Deng, T.; Zheng, J.; Yang, C.; Liou, S.-C.; et al. Non-flammable electrolyte enables Li-metal batteries with aggressive cathode chemistries. Nat. Nanotechnol. 2018, 13, 715–722. [Google Scholar] [CrossRef]
- Suo, L.; Xue, W.; Gobet, M.; Greenbaum, S.G.; Wang, C.; Chen, Y.; Yang, W.; Li, Y.; Li, J. Fluorine-donating electrolytes enable highly reversible 5-V-class Li metal batteries. Proc. Natl. Acad. Sci. USA 2018, 115, 1156–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Fan, X.; Cao, L.; Chen, J.; Hou, S.; Ji, X.; Chen, L.; Li, S.; Zhou, X.; Hu, E.; et al. Designing In-Situ-Formed Interphases Enables Highly Reversible Cobalt-Free LiNiO2 Cathode for Li-ion and Li-metal Batteries. Joule 2019, 3, 2550–2564. [Google Scholar] [CrossRef]
- Li, T.; Zhang, X.-Q.; Shi, P.; Zhang, Q. Fluorinated Solid-Electrolyte Interphase in High-Voltage Lithium Metal Batteries. Joule 2019, 3, 2647–2661. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Cheng, X.-B.; Chen, X.; Yan, C.; Zhang, Q. Fluoroethylene Carbonate Additives to Render Uniform Li Deposits in Lithium Metal Batteries. Adv. Funct. Mater. 2017, 27, 1605989. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, F.; Bai, Y.; Li, Y.; Chen, G.; Wang, Z.; Wu, C. Regulating Li deposition by constructing LiF-rich host for dendrite-free lithium metal anode. Energy Storage Mater. 2019, 16, 411–418. [Google Scholar] [CrossRef]
- Fan, X.; Chen, L.; Ji, X.; Deng, T.; Hou, S.; Chen, J.; Zheng, J.; Wang, F.; Jiang, J.; Xu, K.; et al. Highly Fluorinated Interphases Enable High-Voltage Li-Metal Batteries. Chem 2018, 4, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, P.; Wieczorek, W.; Johansson, P. New boron based salts for lithium-ion batteries using conjugated ligands. Phys. Chem. Chem. Phys. 2016, 18, 16274–16280. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, E.; Henkel, G.; Willner, H.; Pawelke, G.; Bürger, H. Synthesis and Properties of the Tetrakis(trifluoromethyl)borate Anion, [B(CF3)4]−: Structure Determination of Cs[B(CF3)4] by Single-Crystal X-ray Diffraction. Chem. Eur. J. 2001, 7, 4696–4705. [Google Scholar] [CrossRef]
- Landmann, J.; Sprenger, J.A.P.; Hennig, P.T.; Bertermann, R.; Grune, M.; Wurthner, F.; Ignat’ev, N.V.; Finze, M. Perfluoroalkyltricyanoborate and Perfluoroalkylcyanofluoroborate Anions: Building Blocks for Low-Viscosity Ionic Liquids. Chem. Eur. J. 2018, 24, 608–623. [Google Scholar] [CrossRef]
- Zhou, Z.B.; Matsumoto, H.; Tatsumi, K. Cyclic quaternary ammonium ionic liquids with perfluoroalkyltrifluoroborates: Synthesis, characterization, and properties. Chem. Eur. J. 2006, 12, 2196–2212. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Takeda, M.; Fujii, T.; Ue, M. Li[C2F5BF3] as an Electrolyte Salt for 4 V Class Lithium-Ion Cells. J. Electrochem. Soc. 2005, 152. [Google Scholar] [CrossRef]
- Zhang, H.; Oteo, U.; Judez, X.; Eshetu, G.G.; Martinez-Ibañez, M.; Carrasco, J.; Li, C.; Armand, M. Designer Anion Enabling Solid-State Lithium-Sulfur Batteries. Joule 2019, 3, 1689–1702. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16 Rev. C.01; Semichem Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, P.; Wieczorek, W.; Johansson, P. SEI-forming electrolyte additives for lithium-ion batteries: Development and benchmarking of computational approaches. J. Mol. Model. 2017, 23, 6. [Google Scholar] [CrossRef] [Green Version]
- Osborne, D.A.; Breedon, M.; Rüther, T.; Spencer, M.J.S. Towards Higher Voltage Electrolytes: Lithium Salt Design Through in silico Screening. J. Phys. Chem. C. Unpublished work, submitted.
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Pearson, R.G. Absolute Electronegativity and Hardness: Applications to Organic Chemistry. J. Org. Chem. 1989, 54, 1423–1430. [Google Scholar] [CrossRef]
- Clarke-Hannaford, J.; Breedon, M.; Best, A.S.; Spencer, M.J.S. The interaction of ethylammonium tetrafluoroborate [EtNH3(+)][BF4(-)] ionic liquid on the Li(001) surface: Towards understanding early SEI formation on Li metal. Phys. Chem. Chem. Phys. 2019, 21, 10028–10037. [Google Scholar] [CrossRef]
- Budi, A.; Basile, A.; Opletal, G.; Hollenkamp, A.F.; Best, A.S.; Rees, R.J.; Bhatt, A.I.; O’Mullane, A.P.; Russo, S.P. Study of the Initial Stage of Solid Electrolyte Interphase Formation upon Chemical Reaction of Lithium Metal and N-Methyl-N-Propyl-Pyrrolidinium-Bis(Fluorosulfonyl)Imide. J. Phys. Chem. C 2012, 116, 19789–19797. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Strauss, S.H. The search for larger and more weakly coordinating anions. Chem. Rev. 1993, 93, 927–942. [Google Scholar] [CrossRef]
- Rupp, A.B.; Krossing, I. Ionic Liquids with Weakly Coordinating [MIII(ORF)4]− Anions. Acc. Chem. Res. 2015, 48, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P. Electronic structure calculations on lithium battery electrolyte salts. Phys. Chem. Chem. Phys. 2007, 9, 1493–1498. [Google Scholar] [CrossRef]
- Younesi, R.; Veith, G.M.; Johansson, P.; Edström, K.; Vegge, T. Lithium salts for advanced lithium batteries: Li–metal, Li–O2, and Li–S. Energy Environ. Sci. 2015, 8, 1905–1922. [Google Scholar] [CrossRef] [Green Version]
- Jónsson, E.; Armand, M.; Johansson, P. Novel pseudo-delocalized anions for lithium battery electrolytes. Phys. Chem. Chem. Phys. 2012, 14, 6021–6025. [Google Scholar] [CrossRef]
- Barthel, J.; Gores, H.J.; Neueder, R.; Schmid, A. Electrolyte solutions for technology–new aspects and approaches. Pure Appl. Chem. 1999, 71, 1705–1715. [Google Scholar] [CrossRef]
- Gores, H.J.; Barthel, J.; Zugmann, S.; Moosbauer, D.; Amereller, M.; Hartl, R.; Maurer, A. Liquid Nonaqueous Electrolytes. In Handbook of Battery Materials, 2nd ed.; Daniel, C., Besenhard, J.O., Eds.; Wiley-VCH Verlag & Co.: Weinheim, Germany, 2011; pp. 525–626. [Google Scholar]
- Matsumoto, H.; Sakaebe, H.; Tatsumi, K. Li/LiCoO2 Cell Performance Using Ionic Liquids Composed of N,N-Diethyl-N-methyl-N-(2-methoxyethyl)ammonium—Effect of Anionic Structure. ECS Trans. 2009, 16, 59–66. [Google Scholar] [CrossRef]
- Zhou, Z.B.; Matsumoto, H.; Tatsumi, K. Low-melting, low-viscous, hydrophobic ionic liquids: Aliphatic quaternary ammonium salts with perfluoroalkyltrifluoroborates. Chem. Eur. J. 2005, 11, 752–766. [Google Scholar] [CrossRef]
- Ue, M.; Fujii, T.; Zhou, Z.-B.; Takeda, M.; Kinoshita, S. Electrochemical properties of Li[CnF2n+1BF3] as electrolyte salts for lithium-ion cells. Solid State Ion. 2006, 177, 323–331. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, Y.S.; Lee, H.; Han, Y.-K. Low Li+ binding affinity: An important characteristic for additives to form solid electrolyte interphases in Li-ion batteries. J. Power Sources 2011, 196, 5109–5114. [Google Scholar] [CrossRef]
- Halls, M.D.; Tasaki, K. High-throughput quantum chemistry and virtual screening for lithium ion battery electrolyte additives. J. Power Sources 2010, 195, 1472–1478. [Google Scholar] [CrossRef]
- Yildirim, H.; Haskins, J.B.; Bauschlicher, C.W.; Lawson, J.W. Decomposition of Ionic Liquids at Lithium Interfaces. 1. Ab Initio Molecular Dynamics Simulations. J. Phys. Chem. C 2017, 121, 28214–28234. [Google Scholar] [CrossRef]
- Kaneko, T.; Sodeyama, K. First-principles molecular dynamics study for S−O bond dissociation of sulfolane on Li-metal negative electrode. Chem. Phys. Lett. 2021, 762, 138199. [Google Scholar] [CrossRef]
- Clarke-Hannaford, J.; Breedon, M.; Rüther, T.; Spencer, M.J.S. Stability of Boronium Cation-Based Ionic Liquid Electrolytes on the Li Metal Anode Surface. ACS Appl. Energy Mater. 2020, 3, 5497–5509. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Marin, T.W.; Zhu, Y.; Abraham, D.P. Why Bis(fluorosulfonyl)imide Is a “Magic Anion” for Electrochemistry. J. Phys. Chem. C 2014, 118, 19661–19671. [Google Scholar] [CrossRef]
- Zhi, H.; Xing, L.; Zheng, X.; Xu, K.; Li, W. Understanding How Nitriles Stabilize Electrolyte/Electrode Interface at High Voltage. J. Phys. Chem. Lett. 2017, 8, 6048–6052. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System [cation][anion] | Temp. (K) | Time (ps) | Bond Broken | Reference |
|---|---|---|---|---|
| Li[B(CF3)4] | 298 | 0.95 | C–F | This study |
| 358 | 2.01 | C–F | ||
| Li[(C2F5)BF3] | 298 | 3.85 | C–F | This study |
| 358 | 1.79 | C–F | ||
| 2.35 | B–F | |||
| Li[(C2F5)BF2(CN)] | 298 | 0.42 | C–F | This study |
| 0.60 | B–F | |||
| 358 | 0.37 | C–F | ||
| 2.70 | B–F | |||
| [NNBH2][TFSI] | 298 | 0.50 | C–F | [67] |
| 358 | 2.25 | N–S | ||
| [Pyr14][TFSI] | 298 | ∼0.08 | S–C | [65] |
| N–S | ||||
| [Pyr13][FSI] | 298 | 0.05 | S–F | [44] |
| Sulfolane-Li[BF4] | 300 | 65 | – | [66] |
| 450 | 65 | – | ||
| [EMIm][BF4] | 289 | 35 | – | [65] |
| 2500 | N/A | B–F |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke-Hannaford, J.; Breedon, M.; Rüther, T.; Spencer, M.J.S. Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes. Nanomaterials 2021, 11, 2391. https://doi.org/10.3390/nano11092391
Clarke-Hannaford J, Breedon M, Rüther T, Spencer MJS. Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes. Nanomaterials. 2021; 11(9):2391. https://doi.org/10.3390/nano11092391
Chicago/Turabian StyleClarke-Hannaford, Jonathan, Michael Breedon, Thomas Rüther, and Michelle J. S. Spencer. 2021. "Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes" Nanomaterials 11, no. 9: 2391. https://doi.org/10.3390/nano11092391
APA StyleClarke-Hannaford, J., Breedon, M., Rüther, T., & Spencer, M. J. S. (2021). Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes. Nanomaterials, 11(9), 2391. https://doi.org/10.3390/nano11092391