
Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces


Abstract

1. Introduction
2. Electrochemical Cell Construction According to Some Fundamental Principles of Dissipative Thermodynamics
3. General Features and Classification of Suitable Electrolyte Solutions
- Oxoacids containing phosphorus (such as phosphoric or phosphonic) [8,50,70]. Experiments with etidronic acid CH3C(OH)[PO(OH)2]2 have demonstrated that this range can be also widened due to the electrolytes that have a structural relationship to phosphoric acid [71,72]. However, this group cannot so far be expanded to include acids containing other pnictogens (e.g., nitric acid [73]) because their ability to produce cellular alumina nanostructures has not been experimentally confirmed.
4. The Role of Anode Material Morphology and Microstructure
4.1. Search for a Relationship between the Nanopore Arrangement in PAA and Morphology, Microstructure, and Orientation of Al Crystallites
- The most important factor contributing to orderliness enhancement in PAA is the implementation of the two-step anodizing method (first introduced by Masuda and Fukuda [1]) and the corresponding pre-texturing of the Al surface before the main oxidation step;
- PAA formed on a rough (rolled) Al surface is mainly disordered, and mechanical polishing is able to improve the regularity only to a minor degree;
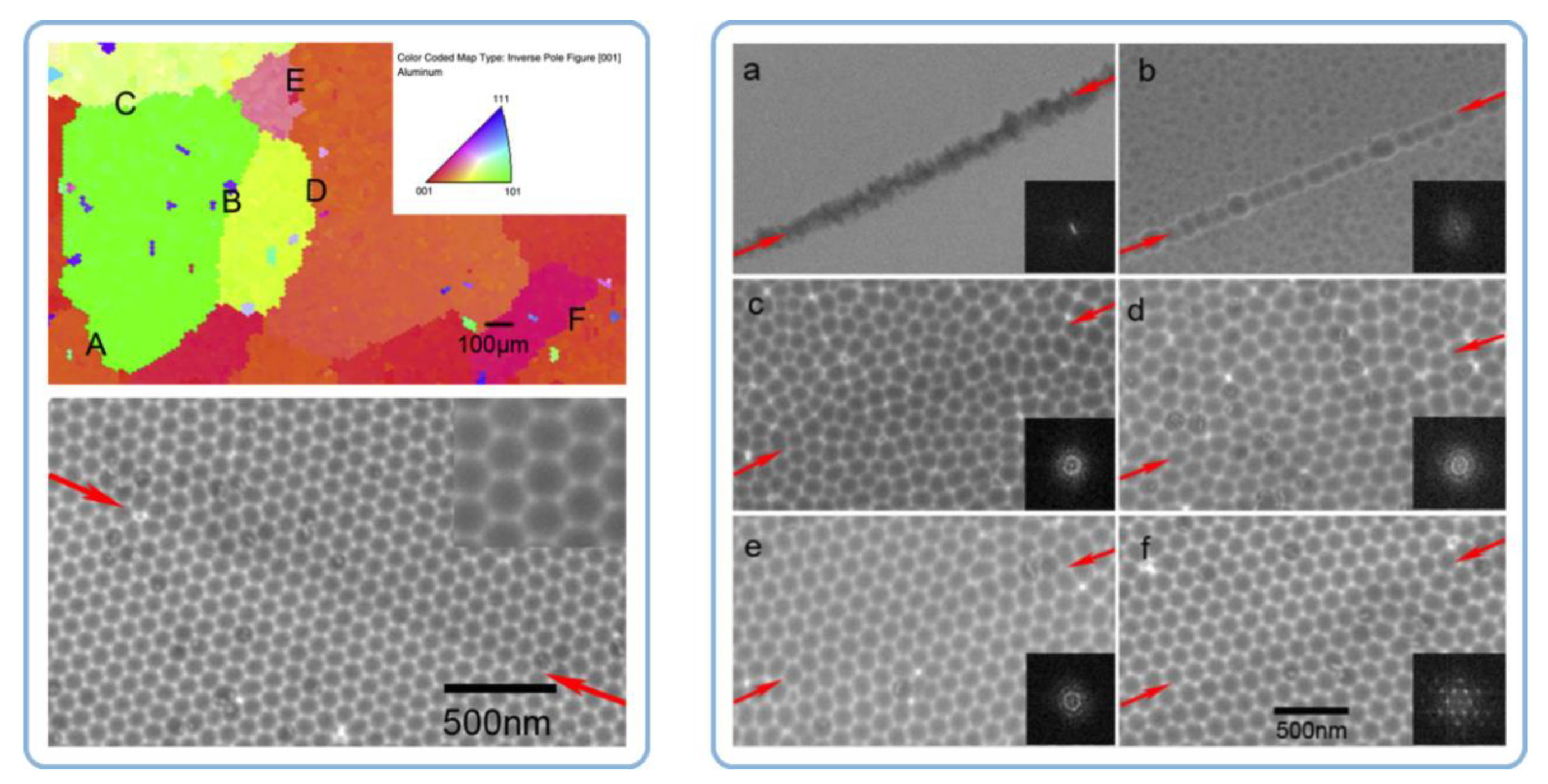
- Neither the grain size and shape nor the dislocations density in a polycrystalline substrate can significantly affect the nanopore regularity in PAA. The authors have been inclined to the view that the pore ordering is influenced by the orientation of aluminum crystals and the fraction of crystals with the cube texture component (that is, with (100) facet exposed to the surface);
- The authors have also detected a correlation between the performed annealing procedure (typically up to 500 °C, except for the magnesium-containing alloy, which was heated up to 410 °C) and the orientation effects in the FCC (face-centered cubic) aluminum phase (a large fraction of enlarged crystallites with (100) facets oriented toward the surface);
- In the case with highly pure Al substrates (99.999%), annealing and recrystallization do not improve the pore arrangement (the same good pore regularity was observed on both the annealed and untreated samples). However, the pore regularity does improve after annealing in the case with lower substrate material purity (≤99.5%). The average ordered domain size in PAA formed on both mechanically polished and unpolished 99.5% Al foils has increased from ≤200 × 200 nm to about 500 × 500 nm after their thermal treatment (however, these figures still remain below the 1 × 1 µm domain sizes observed on rough and polished 99.999% Al substrates).
4.2. Applicability of the Concepts of Classical Thermodynamics to Evolution of Self-Ordered PAA Structures and Investigation of the Effect of Anodizing Time
4.3. The Pseudo-Epitaxial Growth of Self-Organized PAA Structures and the Introduction of the Concept of “In-Plane Orientational Order”
4.4. Addressing the Density of Dislocations in Al Substrates as the Key Factor Influencing the Hexagonal Pore Ordering in PAA
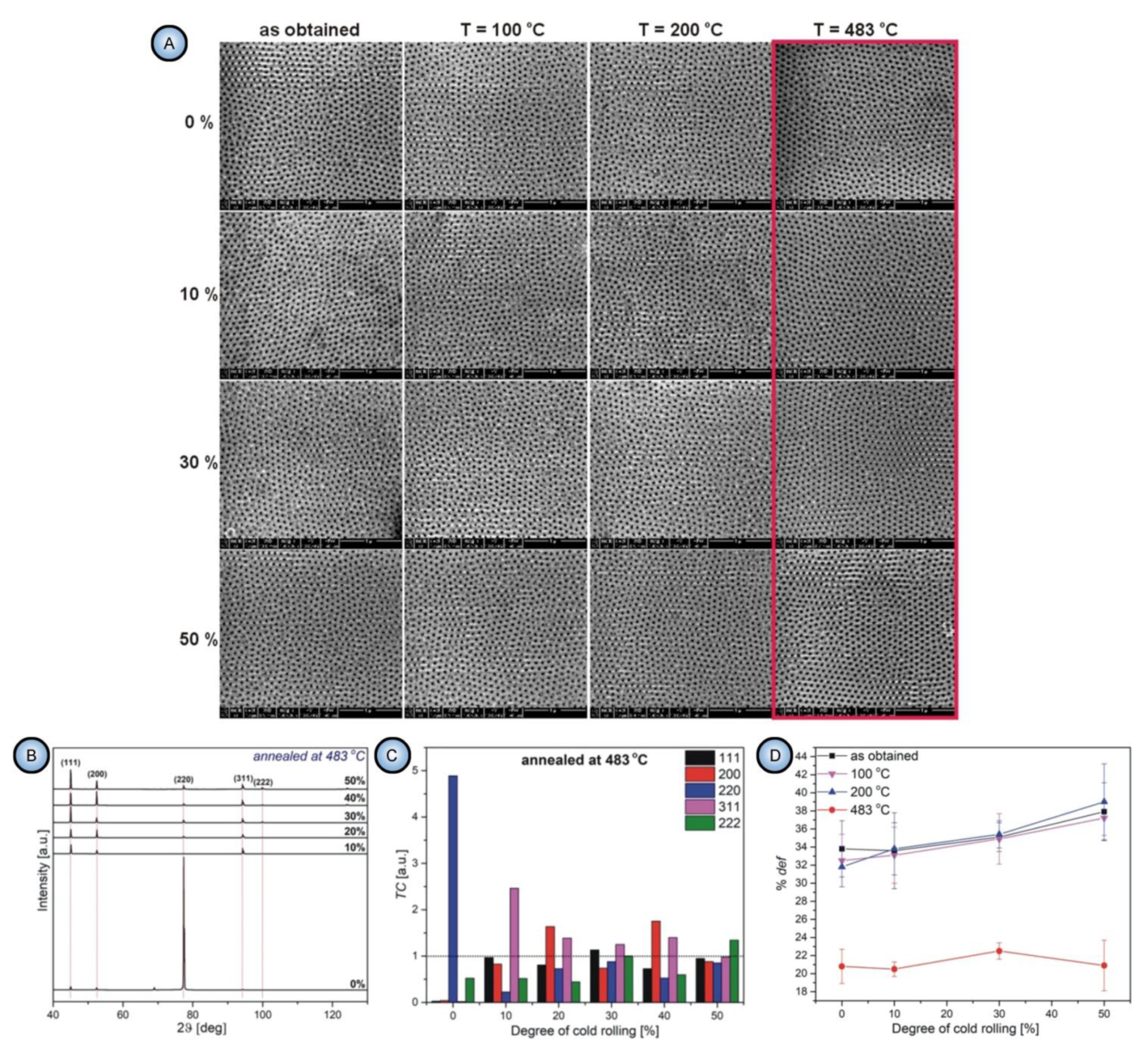
- All parameters of morphology (grain size, circularity, and elongation) and microstructure (lattice strain and surface texturing) are irrelevant to the pore formation mechanism. First, the substrates with only small percentages of mechanical deformation (0–10%) exhibited no changes in the aspect ratios of grains upon annealing at 483 °C. At the same time, they demonstrated a significant improvement of the ordered PAA structure. Second, the improvement of pore ordering upon such annealing appeared almost identical for the substrates that demonstrated a five-fold enlargement of grains and for the substrates where the grain size did not undergo such dramatic changes;
- The experimental observations from this study suggested that the density of dislocations is the only parameter of an Al substrate responsible for the pore ordering. While PAA laminas grown on polycrystalline substrates with a relatively large number of dislocations are chaotic, removing those dislocations from the substrate leads to the formation of highly ordered PAA. The dislocation density can be reduced by recrystallization upon achieving the temperature of 483 °C. Such identification of the key role of dislocations density also disagrees with the earlier results reported by Beck and Petrikowski, who found this parameter to be insignificant [95].
4.5. A Mismatch between the Grain Boundaries in Al and the Boundaries of Coherent Domains in PAA and Attributing the Hexagonal Ordering to Repulsion between Neighboring Pores
5. Structure and Composition of PAA Laminas
5.1. Theoretical Models That Provide Insight into the Physical Properties and Composition of PAA by Considering Its Colloidal Morphology
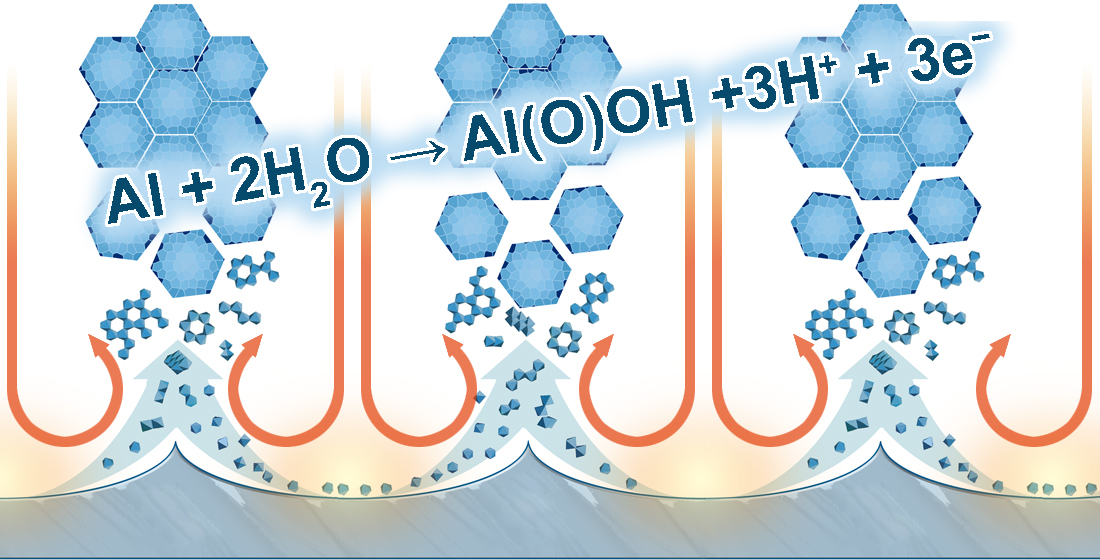
- The innermost “barrier layer” represents almost anhydrous electrolyte-contaminated alumina. Al3+ ions are ejected from the metallic substrate under the influence of the electromagnetic field and react with oxygen-containing species within this barrier layer to form new oxide;
- The electrolyte anions and OH− move toward the oxide layer, and then, they penetrate it at selected points and break it up to form an agglomeration of discrete colloidal particles;
- The outermost layer of the anodic films represents agglomerated nanocrystalline anhydrous oxide particles with gaps in between filled by a complex hydrogen-bonded system of water, electrolyte anions, OH−, and H+. The mechanisms of the oxide formation and of the appearance of an array of cylindrical pores in PAA were viewed by the authors as separate. The importance of these microscopically observable cylindrical pores in the mechanism of oxide layer formation was held to be less than in competing hypotheses;
- Oxyanions of electrolyte adsorb to the surface of colloidal alumina particles and stabilize them after separation from the barrier layer. After that, electrolyte anions unite with H2O, OH−, and H+ into one complex hydrogen-bonded system through which the ionic conduction within PAA occurs;
- The formation of PAA takes place in the vicinity of the barrier layer/porous layer interface (i.e., at a certain distance from the metal substrate surface) approached by cations and anions from the opposite directions under the influence of the electromagnetic field.
5.2. The Mechanism of Polycrystalline PAA Formation from Electrochemically Generated Colloidal Dispersion Assisted by DLVO Interaction Forces
- The reason for the rapid coagulation of hydrated alumina particles shortly after their nucleation can be their small size. In order to aggregate, colloidal particles have to overcome a potential energy barrier (a local maximum of the DLVO curve, which shows the interaction potential of two closely located spherical particles as a function of the distance between them). This barrier can be overcome due to the energy of Brownian motion whose average intensity approximates kT. This energy of a particle moving by Brownian motion should be independent of particle size, whereas the barrier to coagulation should decrease with the decrease in particle radius (this follows from the analysis of the mathematical expression for the interaction potential). This is the reason why electrocratic nano-colloids are generally difficult to stabilize [109] and also why alumina nanoparticles must be prone to rapid coagulation after performing their role in the self-organization process (probably, alumina is delivered from pore centers to the interpore walls with a moving viscous fluid not as already formed colloidal particles but in form of their precursors, i.e., intermediate polynuclear complexes resulting from the polycondensation of [Al(OH2)6]3+ ions in large concentrations; therefore, self-organization has sufficient time before the final coagulation occurs);
- Increased temperature should produce increases in the repulsion between particles, which could contribute to colloid stabilization near the anode where Joule heat is produced. Since temperature is included into the corresponding mathematical expression as absolute temperature in K, this dependency under anodizing conditions is possibly not very pronounced [109]. However, numerous experiments show that the increase in anodizing temperature leads to a noticeable increase in PAA pore size, whereas the cell size remains practically unchanged and seems to be primarily voltage-dependent [53,115,116]. This effect can be attributed either to the enhanced chemical dissolution of alumina at a higher temperature, or to a higher loss of stabilized colloidal alumina particles, which leads to formation of thinner interpore walls and therefore to the increased porosity of PAA. Of course, the combination of both of those factors is also possible. In extreme cases, when a high temperature of the electrolyte solution is combined with the additional intense heat production at the anode, the etching of a hexagonal pattern on anodized aluminum surface without the deposition of any analytically detectable amount of alumina on top of it can be also observed [54];
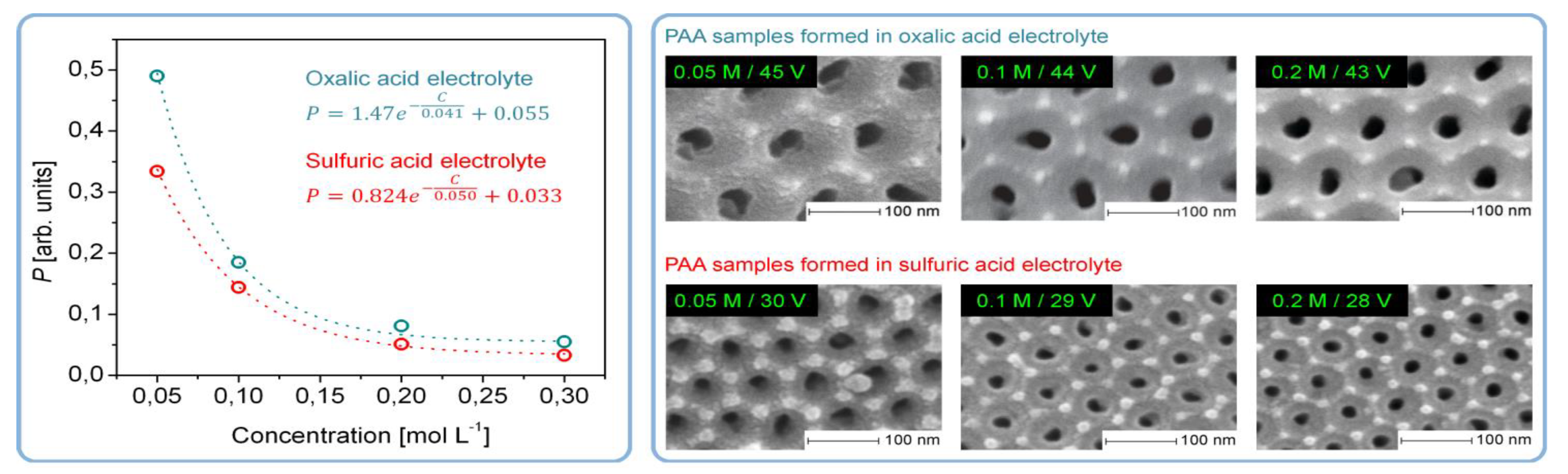
- The stability of an electrocratic dispersion very strongly depends on the indifferent electrolyte concentration and valence. The potential barrier decreases sharply with any increase in electrolyte concentration due to compression and the following collapse of the double layer [109]. The barrier goes to zero, and the colloid undergoes rapid aggregation upon reaching the so-called critical coagulation concentration (CCC) typically ranging from a few tens to a few hundreds of mM. These concentrations are very close to those used in the anodic oxidation of Al (0.3 M for the most classical electrolyte compositions), which should facilitate the aggregation of nanoparticles into polycrystalline pore walls. The dependence of the CCC on the valence of an electrolyte can be more or less pronounced depending on the specific value of the Stern potential for given particles, but for both the high and the low values of the Stern potential, the CCC either steeply or moderately decreases at higher electrolyte valences. This can be the possible explanation for why PAA synthesis is usually more successful when using moderately concentrated polyprotic acids (H2C2O4, H2SO4, H3PO4 etc.), and only negligible amounts of alumina can be deposited onto the waved surface of metallic substrates when using monoprotic electrolytes of comparable concentrations (HCOOH, [16] HIO3, HIO4 [88]);
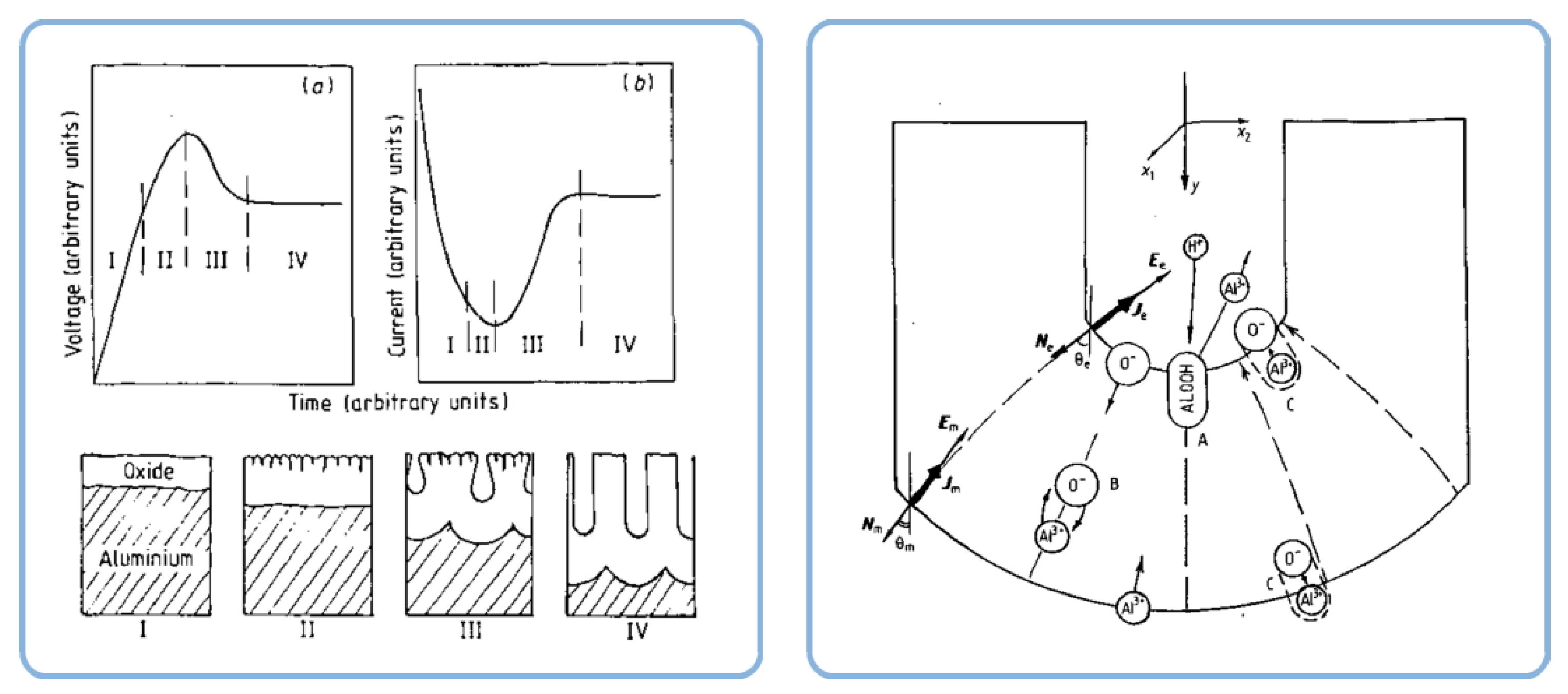
- The effect of the Stern potential on the stability of colloids is also very significant: very low potentials cannot produce a sufficient barrier to coagulation and consequently also cannot assure reasonable stability. The magnitude of the Stern potential can be controlled by varying the surface potential of a nanoparticle, which depends on the relative concentration of the potential determining ions (for oxides, the pH) [109]. Different pH regions (or strata) can also exist in close proximity to the anodized aluminum surface due to oxidation of water and generation of H+, as well as due to the attraction of negatively charged OH− ions toward the positively charged anode (these regions corresponding to low and high pH values should come into balance at a certain height above the substrate surface; see the detailed graphical explanation in Figure 23 reproduced from Ref [16] where the coagulation of colloidal alumina was rationalized on the basis of a similar principle).
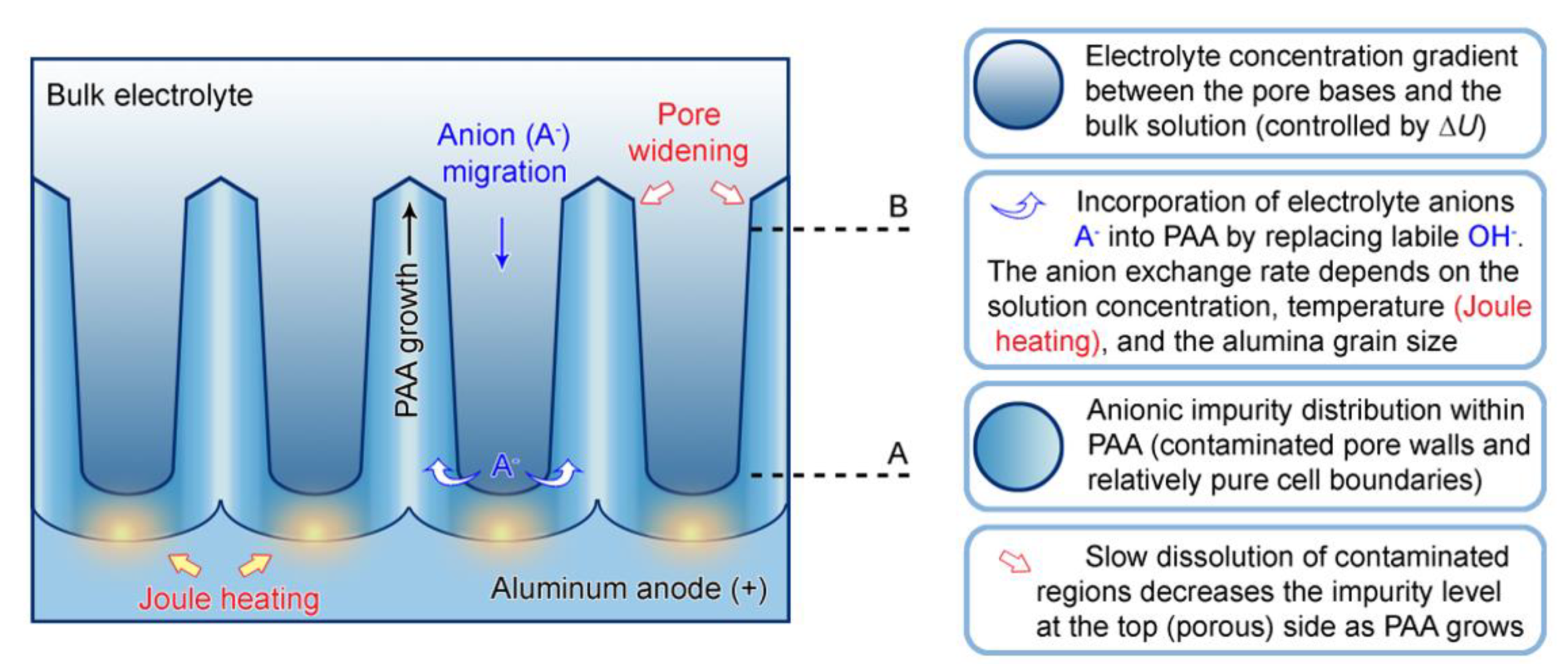
5.3. The Incorporation of Electrolyte Impurity into PAA through a Process of Anion Exchange
- A period of time, during which a particular segment of a growing cylindrical nanochannel stays in contact with the electrolyte solution, and the corresponding exchange of ions between the liquid and the solid phase takes place;
- The concentration of electrolyte in the aqueous phase within PAA nanochannels (because the difference in concentrations is the driving force of counterion diffusion toward the sites within the solid phase where ion exchange occurs). It should be noted that the overall concentration of an electrolyte in the bath and the local concentration within PAA nanochannels are not the same. Due to the migration of anions to the anode under the electromagnetic force, a concentration gradient is established (electrolyte species are accumulated near the anode surface but are not consumed in RedOx reactions). Some early investigations pointed to the electrolyte concentration at the pore base three times higher than the bulk electrolyte concentration during anodizing [120]. Thus, higher anionic impurity content can be expected in PAA if a larger potential difference is applied between the electrodes. This has been experimentally confirmed by the compositional analysis of PAA laminas formed under HA conditions, which demonstrated an increase in the impurity level by 88% in comparison with PAA layers prepared under MA conditions [122]);
- Since the rate of grain boundary diffusion depends on temperature, higher impurity contents can be expected if anodizing is performed either in solutions at high temperatures or under high voltages and current densities when Joule heating of the anode becomes important (this additionally explains why PAA samples synthesized under HA conditions have higher contamination levels);
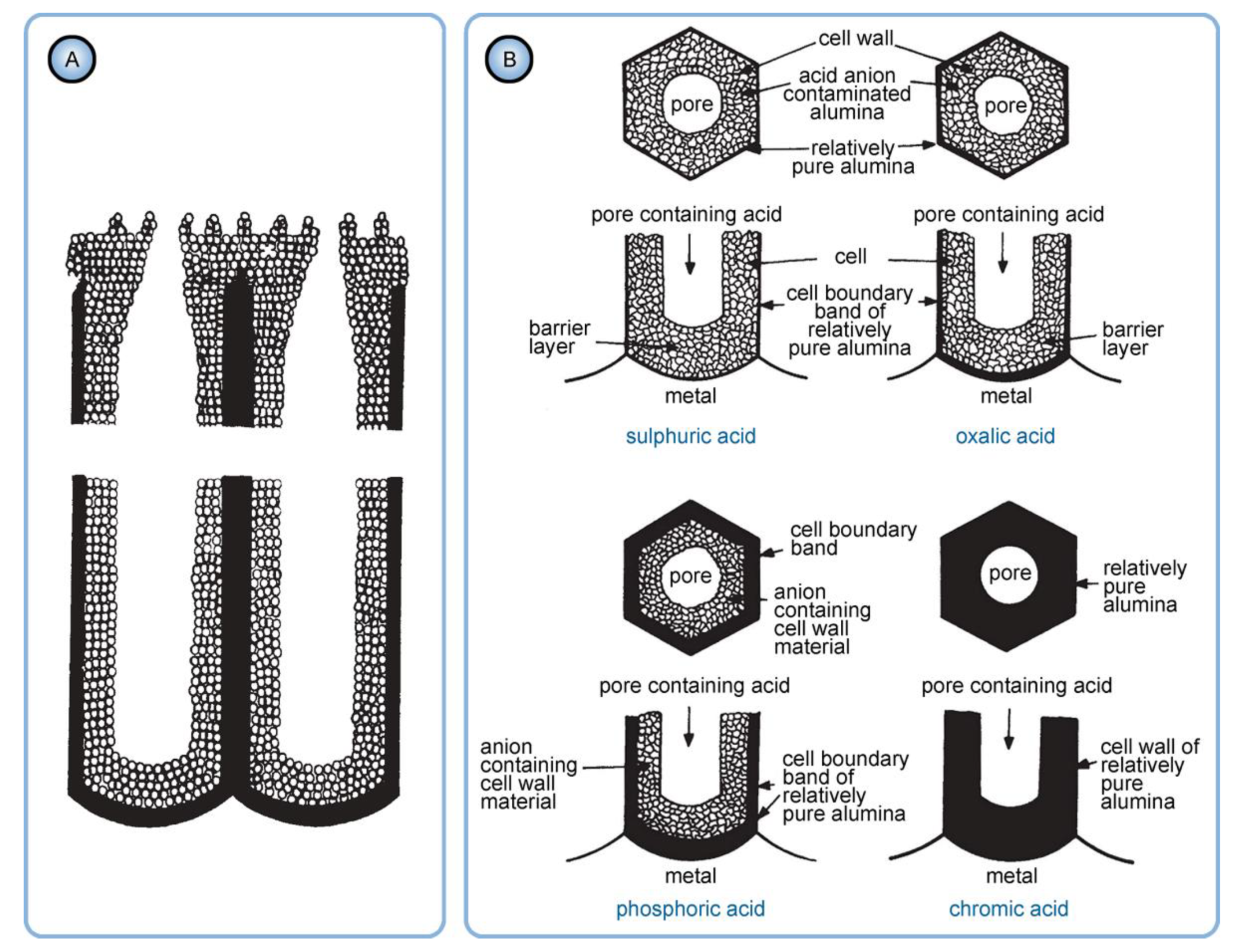
- As mentioned above, hydrated alumina is noticeably soluble in acidic environment (at pH < 4). It is well-known that PAA cells consist of two layers: the relatively pure cell boundaries and the electrolyte-contaminated pore walls (i.e., polycrystalline regions directly around the pores where adsorbed counterions are most prominently contained) [56]. As the tubular nanochannels grow and propagate further in the direction of the metallic substrate, the earlier formed sections of contaminated walls can be again moderately dissolved in the aggressive electrolyte medium (because of such chemical dissolution, the pore openings in PAA acquire the well-known “trumpet shape” [13]). Slow etching of the electrolyte-contaminated region results in the increase in the fraction of relatively pure alumina that remains protected from a direct contact with the etchant. Thus, chemical dissolution is the only involved competing process which increases the purity of a PAA nanochannel segment upon a longer exposure to the aqueous electrolyte solution (see Figure 16).
6. Theoretical Models Explaining PAA Formation
6.1. The PAA Growth Models Based on Ionic Migration and Diffusion through a Solid “Barrier Layer” and Electric-Field-Assisted Oxide Dissolution
- In addition to the movement of Al3+ ions toward the oxide/electrolyte interface at the pore base, some oxygen must also pass through the film in the opposite direction and neutralize Al3+ ions near the metal/oxide interface, increasing the separation between pore bottoms and etched concavities on the metal (Figure 18, left);
- The acid must dissolve the oxide at the pore base, keeping the compact (“barrier”) layer thickness constant;
- Both of the foregoing conditions are satisfied if the following reaction between protons from the electrolyte and O2− ions from the oxide lattice (Figure 18, right) takes place at the pore bottom: O2− + H+ ↔ OH−. First, hydroxyls are more mobile than O2− under the influence of the field and thus can move more easily toward the metal and deliver oxygen more efficiently. Second, Al3+ cations can be ejected from the oxide surface into the electrolyte solution upon such removal of anions from the lattice, which agrees with the experimental data;
- For a certain compact layer thickness, the rate of Al3+ transfer (which reacts with O2− at the oxide/electrolyte interface to form new oxide) will be equal to the rate of OH− transfer (whose loss is compensated by the ejection of Al3+ from the lattice into the electrolyte solution). That is, the increase in film thickness will come into balance with the decrease;
- The model by Hoar and Mott also implied a self-adjustment mechanism for the preservation of the porous structure: if the curvature of a pore bottom becomes too great (i.e., a flattening process starts), the field, the oxide dissolution rate, and the ion transfer rates will increase at this location and promote pore deepening.
- Diffusion of Al3+ from metal and O2−/OH− from the side of electrolyte solution toward the metal/oxide interface where new oxide is formed;
- Ejection of hydrated Al3+ ions to the electrolyte solution.
- A scalloped native barrier-type oxide grows over the also slightly scalloped Al surface left by the electropolishing procedure;
- In the beginning of the anodizing, pores initiate at cracks and surface imperfections, leaving an electric field concentrated below the regions where the oxide film is thinner, hence the local dissolution of oxide (Figure 19b);
- The pore bottom deepening leads to the formation of a major pore at the expense of the former shallow pores (Figure 19c,d).
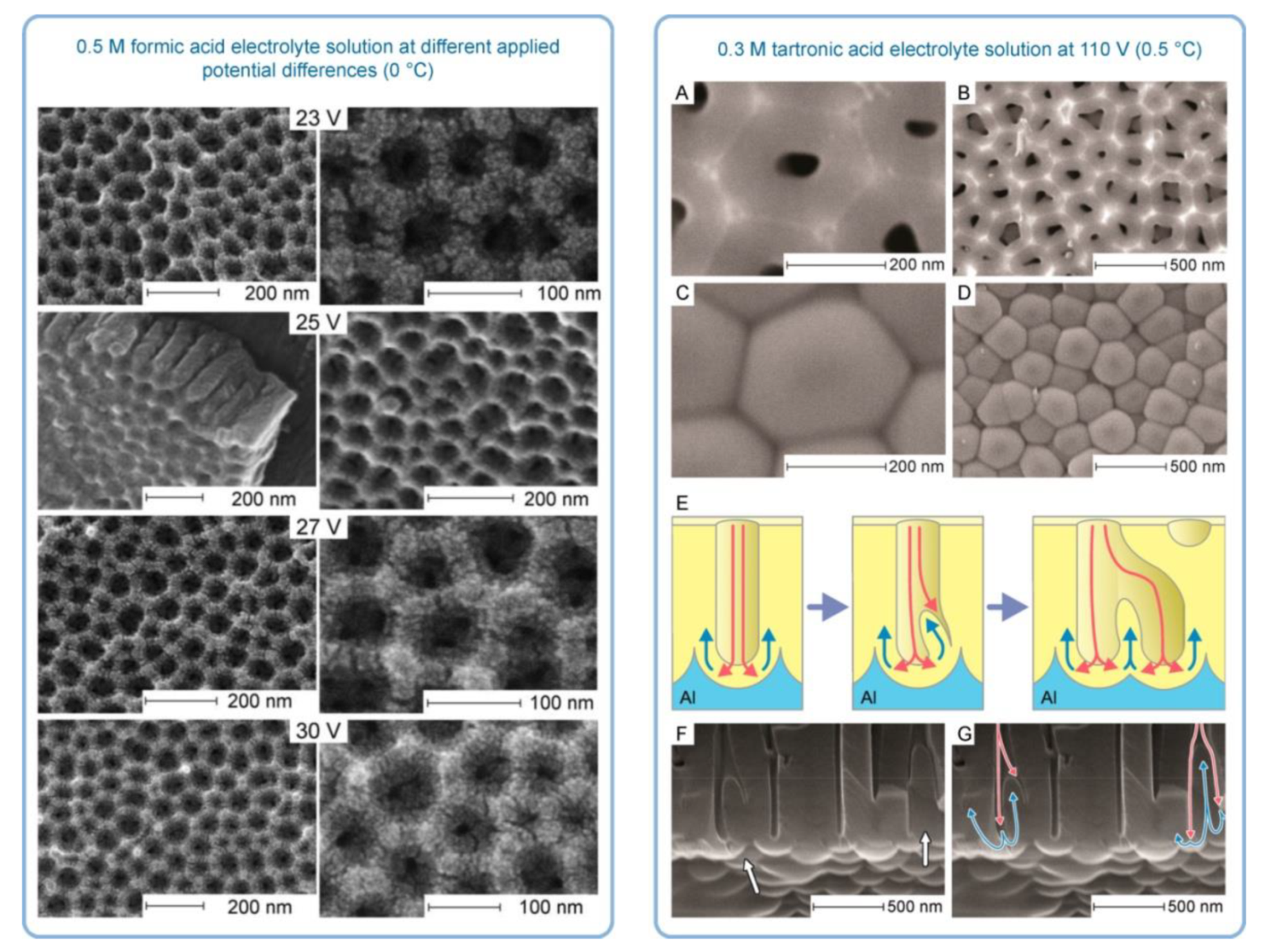
6.2. The Theories Explaining PAA Formation by Electrohydrodynamic Convective Flows
- The gradual evolution of hydrated alumina composition and structure (as it is transported from the pore middle to the pore walls) from complex polymeric cations to an electrocratic nano-colloid which is prone to rapid coagulation;
- The temperature gradient near the anode surface (due to Joule heating), which can cause variation in repulsion between colloidal alumina particles;
- The gradient in stabilizing electrolyte ions concentration (acid residues are attracted to the anode by Coulomb forces but are not consumed in RedOx reactions);
- A pH gradient due to generated H+ in close proximity of the anode surface (see chemical Equation (1)), on the one hand, and attracted OH− from the direction of the bulk electrolyte, on the other hand (the concept proposed in the original paper where this theory was first reported; see Figure 23);
7. New Directions for Theoretical Investigation and Flexible Design of PAA Structures Offered by the Electroconvective Model
7.1. First Attempts of Mathematical Investigation: The Heinschke–Schneider Model
- In the beginning of their theorizing, the authors state that: “The electrolytic system consists of two equal-sized rectangular aluminum electrodes (area Ar). These are localized in an acidic electrolyte with a defined constant temperature T. Thus, the system is isolated against heat exchange with the environment.” Thus, this model contains an apparent confusion between isothermal and adiabatic conditions. Of course, nothing forbids heat exchange between an open system kept at a constant temperature and its surroundings (this is how, for example, different substances melt or evaporate), and the change of entropy in such cases equals to Q/T, where Q is the amount of heat (either gained or lost) and T is thermodynamic temperature. Moreover, since the Heinschke–Schneider model relies on Glansdorff–Prigogine theory, the words of Prigogine himself are relevant here: “The entropy produced by the irreversible heat flow leads to an ordering process, which would be impossible if taken independently from the heat flow.” [47]. Despite this known principle, Heinschke and Schneider wrote in their work: “The entropy production is characterized by the intrinsic entropy changes dinS/dt and the entropy gain by heat exchange with the surrounding environment denS/dt. As the system is isolated with a bath that is held at constant temperature, denS/dt is equal to 0.” [150]. By setting this condition, the authors have eliminated from their modeled system the critical factor that enables the formation of dissipative structures (i.e., the self-organization) as such.
- In the introductory section of this review, the importance of the properly operating feedback loops as the necessary kinetic condition for spontaneous self-organization was discussed, and temporal oscillations were mentioned as a typical manifestation of the existence of such feedback loops [6]. The work by Lee et al. was also cited as an example of spontaneous electrical current oscillations observed during PAA synthesis under a potentiostatic regime (see Figure 2C,D) [12]. However, under some anodizing conditions, the amplitude of such oscillations and fluctuations can be decreased with time, so that they are no more easily recognizable on the I-t curves due to the limited sensitivity of standard laboratory equipment [91,92]. Such conditions are typically “mild”, but exceptions are also possible. In general, the oscillations are the result of the forces acting on ions, and the chances to observe them using the same electrolyte should increase with increasing applied voltage [66,72]. One of the cornerstones of the Heinschke–Schneider model is the existence of a critical moment in time tc after which a stationary state is established and the electrical current (denoted in the work as iq(tc)) becomes constant in time. Thus, although the authors claim the general nature of their assumptions, their work in fact considers just one particular type of PAA formation processes in which temporal oscillations and fluctuations of the electrical current are not readily observable due to some reasons. Under experimental conditions similar to those reported by Lee et al. [12], when the amplitude of spontaneous current oscillations can even increase with time instead of decreasing, and self-organized PAA structures are still formed (actually, they even demonstrate a higher complexity in three dimensions), it would be absolutely impossible to determine the location of the point tc dividing the whole process into the nonstationary and stationary phases in the interpretation of Heinschke and Schneider. Of course, the position of tc could be simply chosen arbitrarily in such cases. In this connection, it would be again appropriate to cite here the words of Prigogine whose “general evolution criterion” was also used in the Heinschke–Schneider model: “Contrary to what happens at equilibrium, or near equilibrium, systems far from equilibrium do not conform to any minimum principle that is valid for functions of free energy or entropy production. As a consequence, there is no guarantee that fluctuations are damped. We can only achieve a formulation of sufficient conditions for stability, which we call the “general evolution criterion”. [47]. Thus, the flaw of the Heinschke–Schneider model seems to be fundamental because it relies on the very specific parameter tc, which in fact does not exist in a generic experimental situation, and it also does not formulate the sufficient conditions for stable PAA formation in a broader theoretical sense. Since Pashchanka theory served as a starting point for the authors’ theorizing, the reason for such a too-idealized representation of a typical I-t (or j-t) curve is not entirely clear. It is worth remarking here that it was mentioned in the original theoretical paper in 2011 that: “The competition of two driving forces (electromagnetic attraction against the diffusion) creates concentration fluctuations near the anode which lead to fluctuations of current with time… This results in damped oscillations of the I–t curve until the equilibrium is established. Only one local maximum in this oscillation has been observed for a long time (presumably due to the restricted sensitivity of measuring equipment). However, damped oscillations are already reported …” [16]. Thus, the oscillatory behavior of I-t curves (amply documented by now under different experimental conditions) cannot be considered as non-relevant, but they could be envisaged using the originally proposed convection-based mechanism. In light of contemporary evidence (including the advances made since 2011), the using of constant electric current as the basis for a new model does not seem well founded and can be even considered a step backward. It should also be taken into consideration that the shapes of anodizing curves recorded in some experimental situations are very different from the example demonstrated in the Heinschke–Schneider model (irrespective of whether or not oscillations and fluctuations have been detected; see Figure 30). For instance, current–time transients for HA using 0.3 M oxalic acid solution show a continuous decrease in current density with time, i.e., defining the position of a critical moment tc for such curves is evidently impossible (Figure 30E) [3].
- The Heinschke–Schneider model considers only two half-reactions within the entire system: reduction of protons at the cathode expressed by Equation (5) and oxidation of aluminum at the anode:Al → Al3+ + 3e−.The authors write that such two reactions “ensure the electron transfer between the electrodes” and that “no other reactions related to the described irreversible process are considered.” [150]. It is not clear why the reaction of water oxidation, which is essential to the PAA formation process and whose contribution to the ionic current between two electrodes is much more significant than that one from Al3+ ions, was not included into this model. Water oxidation is incorporated into Equation (1), and it can be separately written as:2H2O → 4e− + 4H+ + O2.Disregarding the water oxidation process further leads to the next incorrect assumption in the Heinschke–Schneider model: near the anode (denoted as the position x0 in their one-dimensional coordinate system), the concentration of H+ is minimized because of repulsion forces.
- The authors of the model considered the criterion P to be directly related to the entropy production and also “associated with the charge resulting from the Al oxidation reaction.” They made the following assumptions: “the majority of ions flowing toward the bulk are assumed to be Al3+ ions and anions. Here, only the former ones represent a local excess charge… Thus, only Al3+ ions are mentioned further for the determination of ion flow toward the bulk and the entropy production related thereof.” At this point, it is useful to clarify that hydrated ions ejected into an aqueous solution can contribute to the entropy production not because they are formed or because they are charged but because they are moving. The entropy of formation of hydrated Al3+(aq) ions is negative (relative to hydrogen ions): ∆forS° = −321.7 J∙K−1∙mol−1 [151]. However, from general considerations, the motion of hydrated ions through the solution should increase the entropy due to frictional forces and viscous dissipation (the production of entropy in this case equals to the Rayleigh dissipation function R divided by absolute temperature T). From this standpoint, the motion of hydrated electrolyte anions (acid residues) should make a much larger contribution to the production of entropy, whereas the effect of small concentrations of Al3+ ions should be only minor. It should be mentioned here that the formation of hexagonal patterns due to charge carrier currents coupled with fluid flows can be also observed in purely physical systems without any chemical reactions. Thus, the Al oxidation reaction can be only of secondary importance for the emergence of self-organized flows in this case.
- Heinschke and Schneider are using idealized models of a primitive PAA cell (two coaxial cylinders of diameters corresponding to the pore and cell sizes; see Figure 31A) and of the hexagonal lattice (composed of ideal circles; see Figure 31B). Since these authors have eliminated the diffusion and convection parts of the ion flux from the model and left only the migration component, it is not clear which mechanism is responsible now for the formation of such ideal pore morphology and arrangement. The theory of self-organization requires specifying the mechanism of irreversible processes because the laws of nature in a state far from equilibrium become mechanism dependent [47]. Thus, proposing a new hexagonal pattern formation mechanism after the migration transport has been assigned a leading role in the pore formation would be highly desirable. By employing hexagonally arranged cylindrical pores as the initially set condition without any explanation of their emergence, the Heinschke–Schneider model practically maintains the same approach as was used in the Parkhutik–Shershulsky model in 1992 and later criticized by some other authors as unfounded [91,137].
- In the introduction section of this review, special attention was paid to the nonlinearity of self-organizing systems. Ohm’s law is not met in a cell for PAA synthesis, and the electrical current does not linearly scale with an applied voltage (i.e., the electrical resistivity or conductivity are not constant, according to the character of j–∆U curves and possible oscillations of j–t curves) [5]. Nevertheless, Heinschke and Schneider have used Ohm’s law in their model to calculate the specific conductivity of a small volume of electrolyte directly at the cathode (denoted in that work as σλc). No special reservations as to why this should be possible in a nonlinear system were made.
- Heinschke and Schneider have also addressed the issue of incorporation of electrolyte anions from the bulk solution into PAA. The authors state that: “To compensate the excess positive charge which evolves by the loss of anions in the bulk, an additional amount of H+ have to be reduced at the cathode.” [150]. It can easily be demonstrated that such statement is unfortunately misleading. Let us consider a system consisting of two electrodes immersed into an electrolyte solution. Since such a system obviously stays macroscopically electro-neutral during the entire anodizing process, the electrical current passing through the cathode/solution interface must be equal to the current passing through the anode/solution interface (see Figure 32A). This is dictated by the charge conservation principle: if the total charge within the cell remains equal to 0, the amount of charge flowing into the cell must be equal to the amount of charge flowing out of the cell. Now, let us follow the considerations of Heinschke and Schneider and suppose that some internal redistribution of ions within such cell (e.g., transfer of anions from the solution into the solid film on the anode) indeed results in the reduction of an additional amount of H+ at the cathode. This would require an additional amount of electrons transferred from the external circuit into the cell through the cathode (Figure 32B). Due to the charge conservation principle, an equivalent amount of charge must leave the cell through the anode/solution interface (Figure 32C). This is only possible through electrode reactions summarized by the half-equations 7 and 8 (see Figure 32D): according to Faraday laws, the amounts of additionally produced H+ and Al3+ ions at the anode will be proportional to this additional charge leaving the system to maintain its electro-neutrality (both Faraday laws are summarized as n = Q/F, where n is the number of gram equivalents of a reacted substance, Q is the charge that passed through the interface, and F is the Faraday constant). To sum up, the consumption of an additional positive charge at the cathode (see Figure 32B) will result in the production of an equivalent amount of the same charge at the anode (see Figure 32D), and the mechanism of charge reduction proposed by Heinschke and Schneider is invalid because it violates the charge conservation principle and two Faraday laws at the same time. In addition, if the transfer of anions into PAA is supposed to create excess positive charge within the solution, the PAA layers must be charged negatively for the sake of electro-neutrality of the entire system. No mechanism for the negative charge compensation in PAA was proposed in the Heinschke–Schneider model. As was discussed in Section 5.3 of this review, the actual mechanism of anionic impurity incorporation into PAA is very likely akin to ion exchange when electrolyte anions simply replace OH− ions within PAA (see Figure 16). Thus, no excess charge is created within the solution, and therefore, no electrode processes need to be additionally involved.
7.2. Possible Prospects for Experimental Study of PAA Formation: Toward the Smallest and the Largest Achievable Cell Sizes and Non-Trivial Morphologies of Anodic Alumina Nanostructures
7.2.1. The Smallest Achievable Cell Size in Self-Organized PAA and its Relationship to the Debye–Hückel Length
7.2.2. The Largest Possible Cell Sizes on the Anodized Aluminum Surface: The Multiscale Nature of Electroconvection
7.2.3. Anodic Alumina Nano- and Microstructures of Unusual and Complex Shapes
8. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masuda, H.; Fukuda, K. Ordered metal nanohole arrays made by a 2-step replication of honeycomb structures of anodic alumina. Science 1995, 268, 1466–1468. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Hasegwa, F.; Ono, S. Self-ordering of cell arrangement of anodic porous alumina formed in sulfuric acid solution. J. Electrochem. Soc. 1997, 144, L127–L130. [Google Scholar] [CrossRef]
- Lee, W.; Ji, R.; Gösele, U.; Nielsch, K. Fast fabrication of long-range ordered porous alumina membranes by hard anodization. Nat. Mater. 2006, 5, 741–747. [Google Scholar] [CrossRef]
- Schwirn, K.; Lee, W.; Hillebrand, R.; Steinhart, M.; Nielsch, K.; Gösele, U. Self-ordered anodic aluminum oxide formed by H2SO4 hard anodization. ACS Nano 2008, 2, 302–310. [Google Scholar] [CrossRef]
- Vega, V.; Garcia, J.; Montero-Moreno, J.M.; Hernando, B.; Bachmann, J.; Prida, V.M.; Nielsch, K. Unveiling the hard anodization regime of aluminum: Insight into nanopores self-organization and growth mechanism. ACS Appl. Mater. Interfaces 2015, 7, 28682–28692. [Google Scholar] [CrossRef] [PubMed]
- Orlik, M. Self-Organization in Electrochemical Systems II; Springer: Berlin/Heidelberg, Germany, 2012; p. 448. [Google Scholar]
- Orlik, M. Introduction to self-organization in chemical and electrochemical systems. J. Solid State Electr. 2015, 19, 3203–3206. [Google Scholar] [CrossRef]
- Li, A.P.; Müller, F.; Birner, A.; Nielsch, K.; Gösele, U. Hexagonal pore arrays with a 50-420 nm interpore distance formed by self-organization in anodic alumina. J. Appl. Phys. 1998, 84, 6023–6026. [Google Scholar] [CrossRef]
- Mozalev, A.; Mozaleva, I.; Sakairi, M.; Takahashi, H. Anodic film growth on Al layers and Ta-Al metal bilayers in citric acid electrolytes. Electrochim. Acta 2005, 50, 5065–5075. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.W. Analysis on porous aluminum anodic oxide film formed in Re-OA-H3PO4 Solution. Mater. Chem. Phys. 2006, 97, 213–218. [Google Scholar] [CrossRef]
- Kikuchi, T.; Nishinaga, O.; Nakajima, D.; Kawashima, J.; Natsui, S.; Sakaguchi, N.; Suzuki, R.O. Ultra-high density single nanometer-scale anodic alumina nanofibers fabricated by pyrophosphoric acid anodizing. Sci. Rep. 2014, 4, 7411. [Google Scholar] [CrossRef]
- Lee, W.; Kim, J.C.; Gösele, U. Spontaneous current oscillations during hard anodization of aluminum under potentiostatic conditions. Adv. Funct. Mater. 2010, 20, 21–27. [Google Scholar] [CrossRef]
- O’Sullivan, J.P.; Wood, G.C. The morphology and mechanism of formation of porous anodic films on aluminium. Proc. R. Soc. London. A. Math. Phys. Sci. 1970, 317, 511–543. [Google Scholar]
- Singh, G.K.; Golovin, A.A.; Aranson, I.S. Formation of self-organized nanoscale porous structures in anodic aluminum oxide. Phys. Rev. B 2006, 73, 205422. [Google Scholar] [CrossRef]
- Apolinário, A.; Quitério, P.; Sousa, C.T.; Ventura, J.; Sousa, J.B.; Andrade, L.; Mendes, A.M.; Araújo, J.P. Modeling the growth kinetics of anodic TiO2 nanotubes. J. Phys. Chem. Lett. 2015, 6, 845–851. [Google Scholar] [CrossRef]
- Pashchanka, M.; Schneider, J.J. Origin of self-organisation in porous anodic alumina films derived from analogy with Rayleigh-Bénard convection cells. J. Mater. Chem. 2011, 21, 18761–18767. [Google Scholar] [CrossRef]
- Pashchanka, M.; Schneider, J.J. Experimental validation of the novel theory explaining self-organization in porous anodic alumina films. Phys. Chem. Chem. Phys. 2013, 15, 7070–7074. [Google Scholar] [CrossRef]
- Konno, Y.; Tsuji, E.; Skeldon, P.; Thompson, G.E.; Habazaki, H. Factors influencing the growth behaviour of nanoporous anodic films on iron under galvanostatic anodizing. J. Solid State Electr. 2012, 16, 3887–3896. [Google Scholar] [CrossRef]
- Xue, X.Y.; Wang, S.H.; Guo, W.X.; Zhang, Y.; Wang, Z.L. Hybridizing energy conversion and storage in a mechanical-to-electrochemical process for self-charging power cell. Nano Lett. 2012, 12, 5048–5054. [Google Scholar] [CrossRef]
- Shimizu, K.; Brown, G.M.; Habazaki, H.; Kobayashi, K.; Skeldon, P.; Thompson, G.E.; Wood, G.C. Selective oxidation of aluminium and interfacial enrichment of iron during anodic oxide growth on an Al6Fe phase. Corros. Sci. 2000, 42, 831–840. [Google Scholar] [CrossRef]
- Masuda, H.; Ohya, M.; Asoh, H.; Nishio, K. Photonic band gap in naturally occurring ordered anodic porous alumina. Jpn. J. Appl. Phys. 2001, 40, L1217–L1219. [Google Scholar] [CrossRef]
- Zhang, Y.; Son, S.J.; Ju, H. Anodized aluminum oxide membranes of tunable porosity with platinum nanoscale-coating for photonic application. Curr. Appl. Phys. 2012, 12, 1561–1565. [Google Scholar] [CrossRef]
- On, C.; Tanyi, E.K.; Pashchanka, M.; Skuza, J.R.; Peters, V.N.; Noginov, M.A. Spectroscopic studies of dye-doped porous alumina membranes. J. Opt. Soc. Am. B 2018, 35, 1785–1790. [Google Scholar] [CrossRef]
- Pashchanka, M.; Yadav, S.; Cottre, T.; Schneider, J.J. Porous alumina-metallic Pt/Pd, Cr or Al layered nanocoatings with fully controlled variable interference colors. Nanoscale 2014, 6, 12877–12883. [Google Scholar] [CrossRef]
- Das, G.; Patra, N.; Gopalakrishnan, A.; Zaccaria, R.P.; Toma, A.; Thorat, S.; Di Fabrizio, E.; Diaspro, A.; Salerno, M. Fabrication of large-area ordered and reproducible nanostructures for SERS biosensor application. Analyst 2012, 137, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Choi, Y.; Hong, S.; Kang, T.; Lee, L.P. Self-organized hexagonal-nanopore SERS array. Small 2010, 6, 1741–1744. [Google Scholar] [CrossRef]
- Walsh, R.J.; Chumanov, G. Silver coated porous alumina as a new substrate for surface-enhanced Raman scattering. Appl. Spectrosc. 2001, 55, 1695–1700. [Google Scholar] [CrossRef]
- Shan, D.Z.; Huang, L.Q.; Li, X.; Zhang, W.W.; Wang, J.; Cheng, L.; Feng, X.H.; Liu, Y.; Zhu, J.P.; Zhang, Y. Surface plasmon resonance and interference coenhanced SERS substrate of AAO/Al-based Ag nanostructure arrays. J. Phys. Chem. C 2014, 118, 23930–23936. [Google Scholar] [CrossRef]
- Wang, J.; Huang, L.Q.; Yuan, L.; Zhao, L.H.; Feng, X.H.; Zhang, W.W.; Zhai, L.P.; Zhu, J. Silver nanostructure arrays abundant in sub-5 nm gaps as highly Raman-enhancing substrates. Appl. Surf. Sci. 2012, 258, 3519–3523. [Google Scholar] [CrossRef]
- Pashchanka, M.; Okeil, S.; Schneider, J.J. Long-range hexagonal pore ordering as the key to controlling SERS efficiency in substrates based on porous alumina. J. Phys. Chem. C 2020, 124, 25931–25943. [Google Scholar] [CrossRef]
- Santos, A.; Balderrama, V.S.; Alba, M.; Formentín, P.; Ferré-Borrull, J.; Pallarès, J.; Marsal, L.F. Tunable Fabry-Pérot interferometer based on nanoporous anodic alumina for optical biosensing purposes. Nanoscale Res. Lett. 2012, 7, 370. [Google Scholar] [CrossRef] [PubMed]
- Macias, G.; Hernandez-Eguia, L.P.; Ferre-Borrull, J.; Pallares, J.; Marsal, L.F. Gold-coated ordered nanoporous anodic alumina bilayers for future label-free interferometric biosensors. ACS Appl. Mater. Interfaces 2013, 5, 8093–8098. [Google Scholar] [CrossRef]
- Wu, S.M.; Wildhaber, F.; Vazquez-Mena, O.; Bertsch, A.; Brugger, J.; Renaud, P. Facile fabrication of nanofluidic diode membranes using anodic aluminium oxide. Nanoscale 2012, 4, 5718–5723. [Google Scholar] [CrossRef] [PubMed]
- Gorman, C.B.; Petrie, R.J.; Genzer, J. Effect of substrate geometry on polymer molecular weight and polydispersity during surface-initiated polymerization. Macromolecules 2008, 41, 4856–4865. [Google Scholar] [CrossRef]
- Osmanbeyoglu, H.U.; Hur, T.B.; Kim, H.K. Thin alumina nanoporous membranes for similar size biomolecule separation. J. Membr. Sci. 2009, 343, 1–6. [Google Scholar] [CrossRef]
- Ingham, C.J.; ter Maat, J.; de Vos, W.M. Where bio meets nano: The many uses for nanoporous aluminum oxide in biotechnology. Biotechnol. Adv. 2012, 30, 1089–1099. [Google Scholar] [CrossRef]
- Liu, X.; Wei, M.; Liu, Y.J.; Lv, B.J.; Wei, W.; Zhang, Y.J.; Liu, S.Q. Label-free detection of telomerase activity in urine using telomerase-responsive porous anodic alumina nanochannels. Anal. Chem. 2016, 88, 8107–8114. [Google Scholar] [CrossRef]
- Kim, S.; Zhou, Y.; Cirillo, J.D.; Polycarpou, A.A.; Liang, H. Bacteria repelling on highly-ordered alumina-nanopore structures. J. Appl. Phys. 2015, 117, 155302. [Google Scholar] [CrossRef]
- Porta-i-Batalla, M.; Xifre-Perez, E.; Eckstein, C.; Ferre-Borrull, J.; Marsal, L.F. 3D nanoporous anodic alumina structures for sustained drug release. Nanomaterials 2017, 7, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cheng, F.Y.; Tao, Z.L.; Gao, F.; Chen, J. Removal of nickel ions from wastewater by Mg(OH)2/MgO nanostructures embedded in Al2O3 membranes. J. Alloy. Compd. 2006, 426, 281–285. [Google Scholar] [CrossRef]
- Pashchanka, M.; Engstler, J.; Schneider, J.J.; Siozios, V.; Fasel, C.; Hauser, R.; Kinski, I.; Riedel, R.; Lauterbach, S.; Kleebe, H.J.; et al. Polymer-derived SiOC nanotubes and nanorods via a template approach. Eur. J. Inorg. Chem. 2009, 23, 3496–3506. [Google Scholar] [CrossRef]
- Yanagishita, T.; Nishio, K.; Masuda, H. Fabrication of metal nanohole arrays with high aspect ratios using two-step replication of anodic porous alumina. Adv. Mater. 2005, 17, 2241–2243. [Google Scholar] [CrossRef]
- Lakshmi, B.B.; Patrissi, C.J.; Martin, C.R. Sol-gel template synthesis of semiconductor oxide micro- and nanostructures. Chem. Mater. 1997, 9, 2544–2550. [Google Scholar] [CrossRef]
- Domagalski, J.T.; Xifre-Perez, E.; Marsal, L.F. Recent advances in nanoporous anodic alumina: Principles, engineering, and applications. Nanomaterials 2021, 11, 430. [Google Scholar] [CrossRef]
- Ruiz-Clavijo, A.; Caballero-Calero, O.; Martín-González, M. Revisiting anodic alumina templates: From fabrication to applications. Nanoscale 2021, 13, 2227–2265. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Wessling, M.; Mani, A. On the dynamical regimes of pattern-accelerated electroconvection. Sci. Rep. 2016, 6, 22505. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I. The End of Certainty: Time, Chaos, and the New Laws of Nature; The Free Press: New York, NY, USA, 1997; p. 240. [Google Scholar]
- Glansdorff, P.; Prigogine, I. Thermodynamic Theory of Structure, Stability and Fluctuations; Wiley-Interscience: London, UK, 1971; p. 306. [Google Scholar]
- Haken, H. Synergetics: Introduction and Advanced Topics; Springer: Berlin/Heidelberg, Germany, 2004; Volume 1, p. 764. [Google Scholar]
- Masuda, H.; Yada, K.; Osaka, A. Self-ordering of cell configuration of anodic porous alumina with large-size pores in phosphoric acid solution. Jpn. J. Appl. Phys. 1998, 37, L1340–L1342. [Google Scholar] [CrossRef]
- Ono, S.; Saito, M.; Asoh, H. Self-ordering of anodic porous alumina induced by local current concentration: Burning. Electrochem. Solid State Lett. 2004, 7, B21–B24. [Google Scholar] [CrossRef]
- Zaraska, L.; Sulka, G.D.; Jaskula, M. The effect of n-alcohols on porous anodic alumina formed by self-organized two-step anodizing of aluminum in phosphoric acid. Surf. Coat. Technol. 2010, 204, 1729–1737. [Google Scholar] [CrossRef]
- Stępniowski, W.J.; Bojar, Z. Synthesis of anodic aluminum oxide (AAO) at relatively high temperatures. Study of the influence of anodization conditions on the alumina structural features. Surf. Coat. Technol. 2011, 206, 265–272. [Google Scholar] [CrossRef]
- Pashchanka, M. Multilevel self-organization on anodized aluminium: Discovering hierarchical honeycomb structures from nanometre to sub-millimetre scale. Phys. Chem. Chem. Phys. 2020, 22, 15867–15875. [Google Scholar] [CrossRef]
- Mata-Zamora, M.E.; Saniger, J.M. Thermal evolution of porous anodic aluminas: A comparative study. Rev. Mex. Fis. 2005, 51, 502–509. [Google Scholar]
- Mardilovich, P.P.; Govyadinov, A.N.; Mazurenko, N.I.; Paterson, R. New and modified anodic alumina membranes part II. Comparison of solubility of amorphous (normal) and polycrystalline anodic alumina membranes. J. Membr. Sci. 1995, 98, 143–155. [Google Scholar] [CrossRef]
- Pashchanka, M. Controllable reduction of anionic contamination in degradable amorphous anodic alumina nanoporous membranes. ACS Appl. Nano Mater. 2020, 3, 10531–10542. [Google Scholar] [CrossRef]
- Sulka, G.D.; Parkola, K.G. Anodising potential influence on well-ordered nanostructures formed by anodisation of aluminium in sulphuric acid. Thin Solid Film. 2006, 515, 338–345. [Google Scholar] [CrossRef]
- Masuda, H.; Takenaka, K.; Ishii, T.; Nishio, K. Long-range-ordered anodic porous alumina with less-than-30 nm hole interval. Jpn. J. Appl. Phys. 2006, 45, L1165–L1167. [Google Scholar] [CrossRef]
- Kikuchi, T.; Nakajima, D.; Kawashima, J.; Natsui, S.; Suzuki, R.O. Fabrication of anodic porous alumina via anodizing in cyclic oxocarbon acids. Appl. Surf. Sci. 2014, 313, 276–285. [Google Scholar] [CrossRef]
- Kikuchi, T.; Nishinaga, O.; Natsui, S.; Suzuki, R.O. Fabrication of anodic nanoporous alumina via acetylenedicarboxylic acid anodizing. ECS Electrochem. Lett. 2014, 3, C25–C28. [Google Scholar] [CrossRef]
- Pashchanka, M.; Schneider, J.J. Formation of porous anodic alumina under unstable electroconvection flow regimes: A case study of tartronic acid electrolyte. J. Phys. Chem. C 2017, 121, 23683–23692. [Google Scholar] [CrossRef]
- Ma, Y.J.; Wen, Y.H.; Li, J.; Li, Y.X.; Zhang, Z.Y.; Feng, C.C.; Sun, R.G. Fabrication of self-ordered alumina films with large interpore distance by Janus anodization in citric acid. Sci. Rep. 2016, 6, 39165. [Google Scholar] [CrossRef]
- Surganov, V.F.; Gorokh, G.G. Anodic oxide cellular structure formation on aluminum films in tartaric acid electrolyte. Mater. Lett. 1993, 17, 121–124. [Google Scholar] [CrossRef]
- Pashchanka, M.; Schneider, J.J. Evidence for electrohydrodynamic convection as a source of spontaneous self-ordering in porous anodic alumina films. Phys. Chem. Chem. Phys. 2016, 18, 6946–6953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kikuchi, T.; Nishinaga, O.; Natsui, S.; Suzuki, R.O. Self-ordering behavior of anodic porous alumina via selenic acid anodizing. Electrochim. Acta 2014, 137, 728–735. [Google Scholar] [CrossRef]
- Nishinaga, O.; Kikuchi, T.; Natsui, S.; Suzuki, R.O. Rapid fabrication of self-ordered porous alumina with 10-/sub-10-nm-scale nanostructures by selenic acid anodizing. Sci. Rep. 2013, 3, 2748. [Google Scholar] [CrossRef] [PubMed]
- Pashchanka, M.; Schneider, J.J. Self-ordering regimes of porous anodic alumina layers formed in highly diluted sulfuric acid electrolytes. J. Phys. Chem. C 2016, 120, 14590–14596. [Google Scholar] [CrossRef]
- Asoh, H.; Nishio, K.; Nakao, M.; Yokoo, A.; Tamamura, T.; Masuda, H. Fabrication of ideally ordered anodic porous alumina with 63 nm hole periodicity using sulfuric acid. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. Process. Meas. Phenom. 2001, 19, 569–572. [Google Scholar] [CrossRef]
- Akiya, S.; Kikuchi, T.; Natsui, S.; Sakaguchi, N.; Suzuki, R.O. Self-ordered porous alumina fabricated via phosphonic acid anodizing. Electrochim. Acta 2016, 190, 471–479. [Google Scholar] [CrossRef]
- Kikuchi, T.; Nishinaga, O.; Natsui, S.; Suzuki, R.O. Fabrication of self-ordered porous alumina via etidronic acid anodizing and structural color generation from submicrometer-scale dimple array. Electrochim. Acta 2015, 156, 235–243. [Google Scholar] [CrossRef]
- Takenaga, A.; Kikuchi, T.; Natsui, S.; Suzuki, R.O. Exploration for the self-ordering of porous alumina fabricated via anodizing in etidronic acid. Electrochim. Acta 2016, 211, 515–523. [Google Scholar] [CrossRef]
- Yakovleva, N.M.; Anicai, L.; Yakovlev, A.N.; Dima, L.; Khanina, E.Y.; Buda, M.; Chupakhina, E.A. Structural study of anodic films formed on aluminum in nitric acid electrolyte. Thin Solid Film. 2002, 416, 16–23. [Google Scholar] [CrossRef]
- Lee, W.; Nielsch, K.; Gösele, U. Self-ordering behavior of nanoporous anodic aluminum oxide (AAO) in malonic acid anodization. Nanotechnology 2007, 18, 475713. [Google Scholar] [CrossRef]
- Ono, S.; Saito, M.; Asoh, H. Self-ordering of anodic porous alumina formed in organic acid electrolytes. Electrochim. Acta 2005, 51, 827–833. [Google Scholar] [CrossRef]
- Chu, S.Z.; Wada, K.; Inoue, S.; Isogai, M.; Katsuta, Y.; Yasumori, A. Large-scale fabrication of ordered nanoporous alumina films with arbitrary pore intervals by critical-potential anodization. J. Electrochem. Soc. 2006, 153, B384–B391. [Google Scholar] [CrossRef]
- Shingubara, S.; Morimoto, K.; Sakaue, H.; Takahagi, T. Self-organization of a porous alumina nanohole array using a sulfuric/oxalic acid mixture as electrolyte. Electrochem. Solid State Lett. 2004, 7, E15–E17. [Google Scholar] [CrossRef]
- Stępniowski, W.J.; Norek, M.; Michalska-Domanska, M.; Bombalska, A.; Nowak-Stępniowska, A.; Kwaśny, M.; Bojar, Z. Fabrication of anodic aluminum oxide with incorporated chromate ions. Appl. Surf. Sci. 2012, 259, 324–330. [Google Scholar] [CrossRef]
- Zhao, L.R.; Wang, J.; Li, Y.; Wang, C.W.; Zhou, F.; Liu, W.M. Anodic aluminum oxide films formed in mixed electrolytes of oxalic and sulfuric acid and their optical constants. Phys. B Condens. Matter 2010, 405, 456–460. [Google Scholar] [CrossRef]
- Chen, W.; Wu, J.S.; Xia, X.H. Porous anodic alumina with continuously manipulated pore/cell size. ACS Nano 2008, 2, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Stepniowski, W.J.; Norek, M.; Michalska-Domanska, M.; Forbot, D.; Krol, A. Study on the correlation between criterion number derived from Rayleigh-Bénard convective cells and arrangement of nanoporous anodic aluminum oxide. Mater. Lett. 2014, 125, 124–127. [Google Scholar] [CrossRef]
- Li, Y.; Ling, Z.; Wang, J.; Chen, S.; Hu, X.; He, X. Fabrication of porous alumina templates with a large-scale tunable interpore distance in a H2C2O4-C2H5OH-H2O solution. Chin. Sci. Bull. 2008, 53, 1608–1612. [Google Scholar] [CrossRef]
- Martin, J.; Manzano, C.V.; Caballero-Calero, O.; Martin-Gonzalez, M. High-aspect-ratio and highly ordered 15-nm porous alumina templates. ACS Appl. Mater. Interfaces 2013, 5, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Manzano, C.V.; Martin, J.; Martin-Gonzalez, M.S. Ultra-narrow 12 nm pore diameter self-ordered anodic alumina templates. Microporous Mesoporous Mater. 2014, 184, 177–183. [Google Scholar] [CrossRef]
- Kashi, M.A.; Ramazani, A. The effect of temperature and concentration on the self-organized pore formation in anodic alumina. J. Phys. D Appl. Phys. 2005, 38, 2396–2399. [Google Scholar] [CrossRef]
- Kikuchi, T.; Yamamoto, T.; Natsui, S.; Suzuki, R.O. Fabrication of anodic porous alumina by squaric acid anodizing. Electrochim. Acta 2014, 123, 14–22. [Google Scholar] [CrossRef]
- Diggle, J.W.; Downie, T.C.; Goulding, C.W. Anodic oxide films on aluminum. Chem. Rev. 1969, 69, 365–405. [Google Scholar] [CrossRef]
- Okeil, S.; Pashchanka, M.; Heinschke, S.; Bruns, M.; Schneider, J.J. Synergistic physical and chemical enhancement effects observed on surface-enhanced Raman spectroscopy substrates of silver-coated, barrier-type anodic alumina. J. Phys. Chem. C 2020, 124, 13316–13328. [Google Scholar] [CrossRef]
- Atkins, P.W. Physical Chemistry, 2nd ed.; Oxford University Press: Oxford, UK, 1982; p. 1095. [Google Scholar]
- Pinching, G.D.; Bates, R.G. Second dissociation constant of oxalic acid from 0 to 50 °C, and the pH of certain oxalate buffer solutions. J. Res. Natl. Bur. Stand. 1948, 40, 405–416. [Google Scholar] [CrossRef]
- Li, F.Y.; Zhang, L.; Metzger, R.M. On the growth of highly ordered pores in anodized aluminum oxide. Chem. Mater. 1998, 10, 2470–2480. [Google Scholar] [CrossRef]
- Nielsch, K.; Choi, J.; Schwirn, K.; Wehrspohn, R.B.; Gösele, U. Self-ordering regimes of porous alumina: The 10% porosity rule. Nano Lett. 2002, 2, 677–680. [Google Scholar] [CrossRef]
- Stępniowski, W.J.; Nowak-Stępniowska, A.; Presz, A.; Czujko, T.; Varin, R.A. The effects of time and temperature on the arrangement of anodic aluminium oxide nanopores. Mater. Charact. 2014, 91, 1–9. [Google Scholar] [CrossRef]
- Matsui, Y.; Nishio, K.; Masuda, H. Highly ordered anodic porous alumina with 13-nm hole intervals using a 2D array of monodisperse nanoparticles as a template. Small 2006, 2, 522–525. [Google Scholar] [CrossRef]
- Beck, G.; Petrikowski, K. Influence of the microstructure of the aluminium substrate on the regularity of the nanopore arrangement in an alumina layer formed by anodic oxidation. Surf. Coat. Technol. 2008, 202, 5084–5091. [Google Scholar] [CrossRef]
- Cao, G.Z. Nanostructures and Nanomaterials: Synthesis, Properties, and Applications; Imperial College Press: London, UK, 2004; p. 434. [Google Scholar]
- Beck, G.; Bretzler, R. Regularity of nanopores in anodic alumina formed on orientated aluminium single crystals. Mater. Chem. Phys. 2011, 128, 383–387. [Google Scholar] [CrossRef]
- Runge, J.M. The Metallurgy of Anodizing Aluminium, Connecting Science to Practice; Springer: Cham, Switzerland, 2018; p. 473. [Google Scholar]
- Runge, J.M.; Pomis, A. Anodic oxide film formation: Relating mechanism to composition and structure. In Proceedings of the Aluminium Surface Science and Technology Conference (ASST), UMIST, Manchester, UK, 21–25 May 2000. [Google Scholar]
- Ghorbani, M.; Nasirpouri, F.; Iraji zad, A.; Saedi, A. On the growth sequence of highly ordered nanoporous anodic aluminium oxide. Mater. Des. 2006, 27, 983–988. [Google Scholar] [CrossRef]
- Napolskii, K.S.; Roslyakov, I.V.; Romanchuk, A.Y.; Kapitanova, O.O.; Mankevich, A.S.; Lebedev, V.A.; Eliseev, A.A. Origin of long-range orientational pore ordering in anodic films on aluminium. J. Mater. Chem. 2012, 22, 11922–11926. [Google Scholar] [CrossRef]
- Roslyakov, I.V.; Koshkodaev, D.S.; Eliseev, A.A.; Hermida-Merino, D.; Petukhov, A.V.; Napolskii, K.S. Crystallography-induced correlations in pore ordering of anodic alumina films. J. Phys. Chem. C 2016, 120, 19698–19704. [Google Scholar] [CrossRef]
- Norek, M.; Szamyjer, Ł. Anodic alumina growth on Al substrates after multi-variant mechanical and heat treatment. Surf. Coat. Technol. 2019, 357, 802–810. [Google Scholar] [CrossRef]
- Stępniowski, W.J.; Michalska-Domanska, M.; Norek, M.; Twardosz, E.; Florkiewicz, W.; Polkowski, W.; Zasada, D.; Bojar, Z. Anodization of cold deformed technical purity aluminum (AA1050) in oxalic acid. Surf. Coat. Technol. 2014, 258, 268–274. [Google Scholar] [CrossRef]
- Kirby, B.H. Micro-and Nanoscale Fluid Mechanics: Transport in Microfluidic Devices, 1st ed.; Cambridge University Press: New York, NY, USA, 2013; p. 512. [Google Scholar]
- Fan, L.X.; Guo, D.L.; Ren, F.; Fu, Q.; Jiang, C.Z. Substrate grain boundary effects on the ordering of nanopores in anodic aluminum oxide. Solid State Commun. 2008, 148, 286–288. [Google Scholar] [CrossRef]
- Sadasivan, V.; Richter, C.P.; Menon, L.; Williams, P.F. Electrochemical self-assembly of porous alumina templates. AIChE J. 2005, 51, 649–655. [Google Scholar] [CrossRef]
- Hebert, K.R.; Albu, S.P.; Paramasivam, I.; Schmuki, P. Morphological instability leading to formation of porous anodic oxide films. Nat. Mater. 2012, 11, 162–166. [Google Scholar] [CrossRef]
- Berg, J.C. An Introduction to Interfaces and Colloids. The Bridge to Nanoscience; World Scientific: New Jersey, NY, USA; London, UK; Singapore, 2010; p. 804. [Google Scholar]
- Keller, F.; Hunter, M.S.; Robinson, D.L. Structural features of oxide coatings on aluminium. J. Electrochem. Soc. 1953, 100, 411–419. [Google Scholar] [CrossRef]
- Michelson, C.E. The current-voltage characteristics of porous anodic oxides on aluminum. J. Electrochem. Soc. 1968, 115, 213–219. [Google Scholar] [CrossRef]
- Murphy, J.F.; Michelson, C.E. A theory for the formation of anodic oxide coatings on aluminium. In Proceedings of the Symposium on Anodizing Aluminum, Nottingham, UK, 12–14 September 1961; pp. 83–95. [Google Scholar]
- Thompson, G.E.; Furneaux, R.C.; Wood, G.C.; Richardson, J.A.; Goode, J.S. Nucleation and growth of porous anodic films on aluminum. Nature 1978, 272, 433–435. [Google Scholar] [CrossRef]
- Thompson, G.E.; Wood, G.C. Porous anodic film formation on aluminium. Nature 1981, 290, 230–232. [Google Scholar] [CrossRef]
- Sulka, G.D. Highly ordered anodic porous alumina formation by self-organized anodizing. In Nanostructured Materials in Electrochemistry; Eftekhari, A., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 1–116. [Google Scholar]
- Christoulaki, A.; Chenneviere, A.; Grillo, I.; Porcar, L.; Dubois, E.; Jouault, N. A novel methodology to study nanoporous alumina by small-angle neutron scattering. J. Appl. Crystallogr. 2019, 52, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mardilovich, P.P.; Govyadinov, A.N.; Mukhurov, N.I.; Rzhevskii, A.M.; Paterson, R. New and modified anodic alumina membranes part I. Thermotreatment of anodic alumina membranes. J. Membr. Sci. 1995, 98, 131–142. [Google Scholar] [CrossRef]
- Shriver, D.F.; Atkins, P.W. Inorganic Chemistry, 3rd ed.; Oxford University Press: New York, NY, USA, 1999; p. 822. [Google Scholar]
- Frolov, Y.G. A Course in Colloid Chemistry: Surface Phenomena and Disperse Systems; Khimiya: Moscow, Russia, 1989; p. 463. [Google Scholar]
- Hunter, M.S.; Fowle, P. Determination of barrier layer thickness of anodic oxide coatings. J. Electrochem. Soc. 1954, 101, 481–485. [Google Scholar] [CrossRef]
- Churms, S.C. The effect of pH on the ion-exchange properties of hydrated alumina: Part I. Capacity and selectivity. S. Afr. J. Chem. 1966, 19, 98–107. [Google Scholar]
- Lee, W.; Schwirn, K.; Steinhart, M.; Pippel, E.; Scholz, R.; Gösele, U. Structural engineering of nanoporous anodic aluminium oxide by pulse anodization of aluminium. Nat. Nanotechnol. 2008, 3, 234–239. [Google Scholar] [CrossRef]
- Han, H.; Park, S.-J.; Jang, J.S.; Ryu, H.; Kim, K.J.; Baik, S.; Lee, W. In situ determination of the pore opening point during wet-chemical etching of the barrier layer of porous anodic aluminum oxide: Nonuniform impurity distribution in anodic oxide. ACS Appl. Mater. Interfaces 2013, 5, 3441–3448. [Google Scholar] [CrossRef]
- Durtschi, J.D.; Erali, M.; Herrmann, M.G.; Elgort, M.G.; Voelkerding, K.V.; Smith, R.E. Optically improved aluminum oxide membrane through electroless Ni modification. J. Membr. Sci. 2005, 248, 81–87. [Google Scholar] [CrossRef]
- Furneaux, R.C.; Rigby, W.R.; Davidson, A.P. The formation of controlled-porosity membranes from anodically oxidized aluminium. Nature 1989, 337, 147–149. [Google Scholar] [CrossRef]
- Hoar, T.P.; Mott, N.F. A mechanism for the formation of porous anodic oxide films on aluminium. J. Phys. Chem. Solids 1959, 9, 97–99. [Google Scholar] [CrossRef]
- Cabrera, N.; Mott, N.F. Theory of the oxidation of metals. Rep. Prog. Phys. 1949, 12, 163–184. [Google Scholar] [CrossRef]
- Verwey, E.J.W. Electrolytic conduction of a solid insulator at high fields. The formation of the anodic oxide film on aluminium. Physica 1935, 2, 1059–1063. [Google Scholar] [CrossRef]
- Dewald, J.F. The effects of space charge on the rate of formation of anode films. Acta Metall. 1954, 2, 340–341. [Google Scholar] [CrossRef]
- Dewald, J.F. A theory of the kinetics of formation of anode films at high fields. J. Electrochem. Soc. 1955, 102, 1–6. [Google Scholar] [CrossRef]
- Hunter, M.S.; Fowle, P. Factors affecting the formation of anodic oxide coatings. J. Electrochem. Soc. 1954, 101, 514–519. [Google Scholar] [CrossRef]
- Csocán, P. Some observations on the growth mechanism of hard anodic oxide coatings on aluminium. Trans. Inst. Met. Finish. 1964, 41, 51–56. [Google Scholar] [CrossRef]
- Dorsey, G.A., Jr. Structural changes during the anodizing and sealing of anodic aluminas: Intermediate and far infrared analysis. J. Electrochem. Soc. 1968, 115, 1053–1056. [Google Scholar] [CrossRef]
- Lee, W.; Park, S.-J. Porous anodic aluminum oxide: Anodization and templated synthesis of functional nanostructures. Chem. Rev. 2014, 114, 7487–7556. [Google Scholar] [CrossRef]
- Thompson, G.E.; Wood, G.C. Anodic films on aluminium. In Treatise on Materials Science and Technology. Corrosion: Aqueous Process and Passive Films; Scully, J.C., Ed.; Academic Press Inc.: New York, NY, USA, 1983; Volume 23, pp. 205–329. [Google Scholar]
- Thompson, G.E. Porous anodic alumina: Fabrication, characterization and applications. Thin Solid Film. 1997, 297, 192–201. [Google Scholar] [CrossRef]
- Parkhutik, V.P.; Shershulsky, V.I. Theoretical modeling of porous oxide growth on aluminum. J. Phys. D Appl. Phys. 1992, 25, 1258–1263. [Google Scholar] [CrossRef]
- Ebihara, K.; Takahashi, H.; Nagayama, M. Structure and density of anodic oxide films formed on aluminum in sulfuric acid solutions. J. Met. Finish. Soc. Jpn. 1982, 33, 156–164. [Google Scholar] [CrossRef]
- Su, Z.X.; Zhou, W.Z. Formation mechanism of porous anodic aluminium and titanium oxides. Adv. Mater. 2008, 20, 3663–3667. [Google Scholar] [CrossRef]
- Su, Z.X.; Hähner, G.; Zhou, W.Z. Investigation of the pore formation in anodic aluminium oxide. J. Mater. Chem. 2008, 18, 5787–5795. [Google Scholar] [CrossRef]
- Randon, J.; Mardilovich, P.P.; Govyadinov, A.N.; Paterson, R. Computer simulation of inorganic membrane morphology part 3. Anodic alumina films and membranes. J. Colloid Interface Sci. 1995, 169, 335–341. [Google Scholar] [CrossRef]
- Masuda, H.; Asoh, H.; Watanabe, M.; Nishio, K.; Nakao, M.; Tamamura, T. Square and triangular nanohole array architectures in anodic alumina. Adv. Mater. 2001, 13, 189–192. [Google Scholar] [CrossRef]
- Roslyakov, I.V.; Gordeeva, E.O.; Napolskii, K.S. Role of electrode reaction kinetics in self-ordering of porous anodic alumina. Electrochim. Acta 2017, 241, 362–369. [Google Scholar] [CrossRef]
- Houser, J.E.; Hebert, K.R. The role of viscous flow of oxide in the growth of self-ordered porous anodic alumina films. Nat. Mater. 2009, 8, 415–420. [Google Scholar] [CrossRef]
- Skeldon, P.; Thompson, G.E.; Garcia-Vergara, S.J.; Iglesias-Rubianes, L.; Blanco-Pinzon, C.E. A tracer study of porous anodic alumina. Electrochem. Solid-State Lett. 2006, 9, B47–B51. [Google Scholar] [CrossRef]
- Lu, S.J.; Su, Z.S.; Sha, J.; Zhou, W.Z. Ionic nano-convection in anodisation of aluminium plate. Chem. Commun. 2009, 5639–5641. [Google Scholar] [CrossRef]
- Jessensky, O.; Müller, F.; Gösele, U. Self-organized formation of hexagonal pore arrays in anodic alumina. Appl. Phys. Lett. 1998, 72, 1173–1175. [Google Scholar] [CrossRef]
- Schneider, J.M.; Watson, P.K. Electrohydrodynamic stability of space-charge-limited currents in dielectric liquids. 1. Theoretical study. Phys. Fluids 1970, 13, 1948–1954. [Google Scholar] [CrossRef]
- Chelliah, N.M.; Saxena, A.; Sharma, K.; Singh, H.; Surappa, M.K. Surface characterization of nanoporous aluminium oxide films synthesized by single-step DC and AC anodization. Surf. Interfaces 2017, 7, 139–145. [Google Scholar] [CrossRef]
- Heinschke, S.; Schneider, J.J. On the prediction of well-ordered porous anodic alumina films. J. Phys. Chem. C 2020, 124, 11913–11921. [Google Scholar] [CrossRef]
- Plieth, W. Electrochemistry for Materials Science; Elsevier Science: Amsterdam, The Netherlands, 2007; p. 420. [Google Scholar]
- Entelis, S.G.; Tiger, R.P. Reaction Kinetics in the Liquid Phase; John Wiley and Sons: New York, NY, USA; Toronto, ON, Canada, 1976; p. 362. [Google Scholar]
- Getling, A.V. Rayleigh-Bénard Convection: Structures and Dynamics; World Scientific: River Edge, NJ, USA, 1998; Volume 11, p. 245. [Google Scholar]
- Fan, H.; Lin, T.; Zhang, W.; Ma, J.; Lu, S.; Zhu, X. Formation mechanism of petal-like micropattern and nanofibers in porous anodic alumina. Mater. Res. Bull. 2017, 90, 119–124. [Google Scholar] [CrossRef]
- Garcia-Vergara, S.J.; Skeldon, P.; Thompson, G.E.; Habazaki, H. A flow model of porous anodic film growth on aluminium. Electrochim. Acta 2006, 52, 681–687. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, L.; Song, Y.; Jia, H.; Yu, H.; Xiao, X.; Yang, X. Oxygen bubble mould effect: Serrated nanopore formation and porous alumina growth. Mon. Chem.-Chem. Mon. 2008, 139, 999–1003. [Google Scholar] [CrossRef]
- Han, J.K.; Kim, J.; Choi, Y.C.; Chang, K.-S.; Lee, J.; Youn, H.J.; Bu, S.D. Structure of alumina nanowires synthesized by chemical etching of anodic alumina membrane. Phys. E Low-Dimens. Syst. Nanostructures 2007, 36, 140–146. [Google Scholar] [CrossRef]
- Friedman, A.L.; Menon, L. Dendritic-pattern formation in anodic aluminum oxide templates. J. Appl. Phys. 2007, 101, 084310. [Google Scholar] [CrossRef]
- Riccomagno, E.; Shayganpour, A.; Salerno, M. Optimization of anodic porous alumina fabricated from commercial aluminum food foils: A statistical approach. Materials 2017, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Sanz, O.; Echave, F.J.; Odriozola, J.A.; Montes, M. Aluminum anodization in oxalic acid: Controlling the texture of Al2O3/Al monoliths for catalytic aplications. Ind. Eng. Chem. Res. 2011, 50, 2117–2125. [Google Scholar] [CrossRef]
- Friedman, A.L.; Brittain, D.; Menon, L. Roles of pH and acid type in the anodic growth of porous alumina. J. Chem. Phys. 2007, 127, 154717. [Google Scholar] [CrossRef] [PubMed]





































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte | Voltage ∆U (V) | Temperature (°C) | Pore Diameter dp (nm) | Cell Size Dint (nm) | Reference |
|---|---|---|---|---|---|
| 0.3 M Oxalic acid (H2C2O4) | 40 | 0 | 67 ± 6 (after widening by etching in 5% H3PO4 at 30 °C for 90 min.) | 99 ± 8 | Masuda and Fukuda [1] |
| 1.67 M Malonic acid (CH2(COOH)2) | 110–140 | 0 | — | the Dint/∆U ratio varied from 2.0 to 1.8 nm V−1 upon increased ∆U | Lee et al. [74] |
| 2–5 M Malonic acid (CH2(COOH)2) | 120 | 5 | — | 300 | Ono et al. [75] |
| 1–10 vol % Glycolic acid (HOCH2COOH) | 50–150 | 10–20 | ≈35 | 150 (50 V, 10 vol %, 10 °C); 320 (150 V, 1 vol %, 10 °C); | Chu et al. [76] |
| 2–4 wt % Malic acid (C2H2OH(COOH)2) | 220–450 | 10–20 | — | 550 (4 wt %, 220 V, 10 °C); 950 (2 wt %, 450 V, 10 °C); | Chu et al. [76] |
| 0.3 M Acetylenedicarboxylic acid (HO2C−C≡C−CO2H) | 87.5–97.5 | 0–60 °C | 100–110 | 250 | Kikuchi et al. [61] |
| 0.3 M Tartronic acid (HOOC(CHOH)COOH) | 110 | 0 | 48 ± 13 | 222 ± 24 | Pashchanka and Schneider [62] |
| 0.125 M Citric acid (C3H5O(COOH)3) | 260–450 | 10–30 | 230 | 1100 | Mozalev et al. [9] |
| 3–5 M Tartaric acid (HOOC(CHOH)2COOH) | 195 | 5 | — | 500 | Ono et al. [75] |
| 0.3 M Sulfuric acid (H2SO4) | 27 | 0 | 15–40 | 63 | Masuda et al. [2] Asoh et al. [69] |
| 1:1 mixture of 0.3 M Sulfuric acid (H2SO4) and 0.3 M Oxalic acid (H2C2O4) | 36 | 3 | — | 73 | Shingubara et al. [77] |
| 0.1–3.0 M Selenic acid (H2SeO4) | 37–51 | 0 | ≈40 | 95–110 | Kikuchi et al. [66] |
| 10 wt % Phosphoric acid (H3PO4) | 160 | 3 | 267 | 420 | Li et al. [8] |
| 0.3 M Phosphoric acid (H3PO4) | 195 | 0 | — | 500 | Masuda et al. [50] |
| 0.5–2.0 M Phosphonic acid (H3PO3) | 150–180 | 0–20 | 132 (under the following optimized conditions: 1.0 M, 150 V, and 20 °C) | 370–440 | Akiya et al. [70] |
| 0.3 M Chromic acid (H2CrO4) | 20–50 | 20–50 | 37.5–87.0 (dp variation was achieved by changing both ∆U and temperature) | 66.0–179.5 | Stępniowski et al. [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pashchanka, M. Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces. Nanomaterials 2021, 11, 2271. https://doi.org/10.3390/nano11092271
Pashchanka M. Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces. Nanomaterials. 2021; 11(9):2271. https://doi.org/10.3390/nano11092271
Chicago/Turabian StylePashchanka, Mikhail. 2021. "Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces" Nanomaterials 11, no. 9: 2271. https://doi.org/10.3390/nano11092271
APA StylePashchanka, M. (2021). Conceptual Progress for Explaining and Predicting Self-Organization on Anodized Aluminum Surfaces. Nanomaterials, 11(9), 2271. https://doi.org/10.3390/nano11092271