Abstract
A computational approach is used on MOF materials to predict the structures showing the best performances for I2 adsorption as a function of the functionalization, the pore size, the presence of the compensating ions, and the flexibility on which to base future improvements in selected materials in view of their targeted application. Such an approach can be generalized for the adsorption of other gases or vapors. Following the results from the simulations, it was evidenced that the maximum capacity of I2 adsorption by MOF solids with longer organic moieties and larger pores could exceed that of previously tested materials. In particular, the best retention performance was evidenced for MIL-100-BTB. However, if the capacity to retain traces of gaseous I2 on the surface is considered, MIL-101-2CH3, MIL-101-2CF3, and UiO-66-2CH3 appear more promising. Furthermore, the impact of temperature is also investigated.
1. Introduction
The energy production in nuclear power plants during normal operating conditions induces greenhouse gas emissions, which are lower than those generated by fossil fuel technologies or, at the most, comparable to those accompanying electricity production based on renewable energy sources [1]. A more serious problem related to safe nuclear power concerns the potential radiological release of fission gases that are formed during fuel reprocessing or nuclear fallout [2]. Among others, the capture of gaseous I2 to avoid its easy spread into the environment remains a real challenge. The impact of this element on the human health can be harmful for long periods since, e.g., 129I has a half-life of 15.7 million years. Furthermore, the high volatility of iodine and its eventual involvement in human metabolic processes, representing an immediate threat for the population, have inspired the quick development of a variety of removal technologies [3].
The selective removal of iodine directly from the gaseous phase by adsorption onto porous materials, thereby limiting the presence of its radioactive vapors in atmosphere, may be a good alternative to the more classical absorption of gaseous constituents by scrubbing into a liquid solvent [4]. The main research strategy here is to propose solid adsorbents that exhibit a strong affinity towards iodine at low surface coverages (valuable solution in the case of I2 traces in the off-gas) or adsorbents offering a high retention capacity necessary to store I2 molecules in large quantities. Solid materials that have already been tested for such uses include zeolites [4], mesoporous silicas functionalized with silver nanoparticles [5], and clathrates [6]. The strong binding affinity of gaseous iodine units for metal surface sites has motivated research interest in metal–organic frameworks (MOFs) that have proven to be highly efficient in various applications where sorption phenomena underlie retention mechanisms [7,8,9,10,11,12]. These materials are composed of metal centers organized in chains or clusters and linked to one another through organic linkers. Due to their high structural and chemical versatility, they have shown remarkable performances when capturing gas components such as H2, CO2, CH4, and their mixtures [13,14,15,16]. Other applications such as catalysis, medical technology, energy storage, and conversion also deserve to be mentioned, especially if the use of composites is accompanied by a special effort to propose green synthesis to reduce environmental impacts during their preparation [17,18,19,20]. The surface activity of MOFs upon adsorption and separation of gases can be altered through different approaches, as has been already reported in the literature [21,22,23,24,25]. For example, it was possible to carry out functionalization of the organic part and/or modification of the metal center in order to tune interactions occurring between the adsorbate and the solid surface, thereby modelling the adsorption power of the adsorbent [26,27,28,29,30,31,32] and its flexibility capacity [33,34]. Furthermore, the usefulness of these porous solids for nuclear applications has been already publicized, since some MOF materials are considered for the adsorption of a radioactive gas [35,36,37,38,39,40] or ion sequestration [41,42].
From an experimental viewpoint, correctly assessing the real decontamination performance of adsorbents is not an easy task because of the potential health risks following exposure to radiation. Moreover, this task may appear fastidious if one takes into account the large number of various MOF samples that can be achieved and tested. In such circumstances, molecular simulation methods offer a powerful and fast tool to evaluate the sorption capacity of different hybrid solids on which to base their subsequent optimization and the selection of the best candidate for their targeted uses. Such a strategy has already been followed when studying the adsorption of H2, CO2, and other gases [25,43,44,45]. By far the greatest added value provided by molecular simulations is the possibility of describing, at a microscopic level, the mechanisms involved [46,47,48,49,50,51].
Concerning the retention of I2 from the gas phase, ZIF-8 is the most frequently investigated adsorbent of the MOF family [52,53]. Strong interactions between the I2 molecules and the imidazolate moieties were indicated to account for an adsorbate retention four times higher than that of activated carbons [53]. Considering various MOF materials with aluminum as a metal center, Loiseau et al. showed that the sorption of I2 in combination with liquid cyclohexane was the highest on samples containing a grafted NH2 group when compared to pristine materials [54]. On the other hand, computational studies have also been performed to screen the performances of other selected MOFs [55,56,57]. Furthermore, some sorbents for radionuclide species have also been designed from UiO-66 and ZIFs series with enhanced retention performances [58].
The intention of the present paper is to consider the impact of: (i) structure functionalization, (ii) the size of the pores, and (iii) the presence of extra-framework counter-ions on both the adsorption performance of selected MOF materials towards gaseous I2 and their adsorption mechanisms. The starting MOF materials included a microporous MIL-53 series [33], a mesoporous MIL-100 and MIL-101 series [59], MIL-127 [60], Zn-BTeC [61], and a UiO-66 series [62]. The different structures investigated are shown together in the Materials and Methods section. As it has been shown that the capture of radioactive contaminants using dry processes (namely, using porous solids without trace of water) can be an important solution for industry [63], we mainly focus here on the adsorption of pure I2 in porous solids.
2. Materials and Methods
Prior to our calculation of the adsorption isotherms, our main effort was focused on determining the structure of each MOF solid considered in this work. For this purpose, we considered the following structures already published in the literature: (a) the MIL-53 series bearing a H, 1CH3, NH2, Br, or Cl functional group grafted on the phenyl ring of benzenedicarboxylate linker (bdc)) [33]; (b) an MIL-100 series possessing a bdc or 1,3,5-tris(4-carboxyphenyl)benzene (BTB) group as an organic moiety) [59]; (c) an MIL-101 series with a H, NH2, Br, CF3, Cl, or 2CH3 group on the phenyl ring of a bdc linker, or those containing a 2,6-naphthalenedicarboxylate (NDC), 4,4′-biphenyldicarboxylate (BPDC) group as an organic moiety) [59]; (d) a UiO-66 series with a H, Br, CH3, Cl, NH2, NO2, or 2CH3 group grafted on the phenyl ring of a bdc part [62].
Concerning the MIL-53 series investigated here, the ‘open’ forms (or large-pore forms) of these structures were considered to determine the maximum amount of I2 adsorbed. In this particular case, the experimental parameters of the unit cell were fully imposed.
All structural models were then energy-minimized within the P1 space group by keeping the cell parameters fixed, using the universal force field (UFF) and charges calculated from the qEq method, as implemented in Materials Studio software [64]. Such a strategy has already been successfully employed to construct plausible structures of various MOFs [33,59,65]. The Ewald summation was considered for calculating electrostatic interactions while short-range interactions were evaluated using a cut-off distance of 12 Å. The convergence criteria were set at: 1.0 × 10−4 kcal mol−1 (energy), 0.005 kcal mol−1 Å−1 (forces), and 5.0 × 10−5 Å (displacement). All geometry optimizations converged to provide a plausible crystallographic structure for each MOF investigated in this paper.
A classical molecular simulation was subsequently carried out to calculate both the adsorption isotherm for I2 and the adsorption enthalpy at low coverages. Grand Canonical Monte Carlo (GCMC) simulations were performed making use of the SORPTION (from Materials Studio) or home-made code, typically with 2.0 × 106 Monte Carlo steps for production, followed by 5 × 106 steps for equilibration [66]. The Ewald summation was also used for calculating electrostatic interactions while short-range contributions were computed with a cut-off distance of 12 Å. The simulations were conducted at 300 K using the previously simulated structures considered as rigid and with cell parameters allowing for the use of a 12 Å cut-off distance. Again, a UFF force field was used in regard to the framework atoms and I2 molecules, as proposed by Nenoff et al., who developed a diatomic model for I2 without explicitly considering polarizability [53]. Our interest in this force field was to consider that the electrostatic part of the energy had only a small influence, and that a van der Waals interactions played a main role in the adsorption phenomenon, as already proven in the case of various porous solids [67,68,69,70]. It can also be noted that the force field developed by Nenoff was able to reproduce the interactions of I2 with Ag+-mordenite, even without taking into account electrostatic interactions. This justifies the use of this force field to investigate the selected solids in the present case. Furthermore, DFT calculations provided strong indications that the van der Waals forces played a predominant role in the interactions between I2 and MOF, thereby suggesting that the electrostatic effect may be neglected as a first approximation [69].
A similar calculation strategy was repeated at 353 K so as to verify whether the observed trends would be transposable to other conditions of temperature reported in previously published experimental studies.
From the Monte Carlo simulations, it was possible to extract the most plausible configurations that corresponded to the most probable distribution of the guest I2 molecules inside the pores of the different solids investigated within the framework of the present study. The distances reported in the snapshots included in the manuscript are consistent with distances obtained from radial distribution functions. They allow us to elucidate typical interactions and, therefore, to clarify the main interaction sites for I2 adsorption.
Using these structures, textural properties (specific surface area, SSA, and pore volume, PV) were calculated by considering the strategy previously developed by Düren et al. [71]. From the area defined by the motion of the center of a nitrogen molecule rolling along the surface, it was possible to calculate SSA values. The diameter of the probe molecule was considered to be equal to 3.681 Å, whereas the diameter of each atom constituting the MOF structure was taken from the UFF force field data [64]. The PV was calculated for each simulated structure using a similar method of trial insertions within the entire volume of the unit cell. A 0 Å-size probe was used for the determination of the free volume of the unit cell unoccupied by framework atoms [71].
3. Results and Discussion
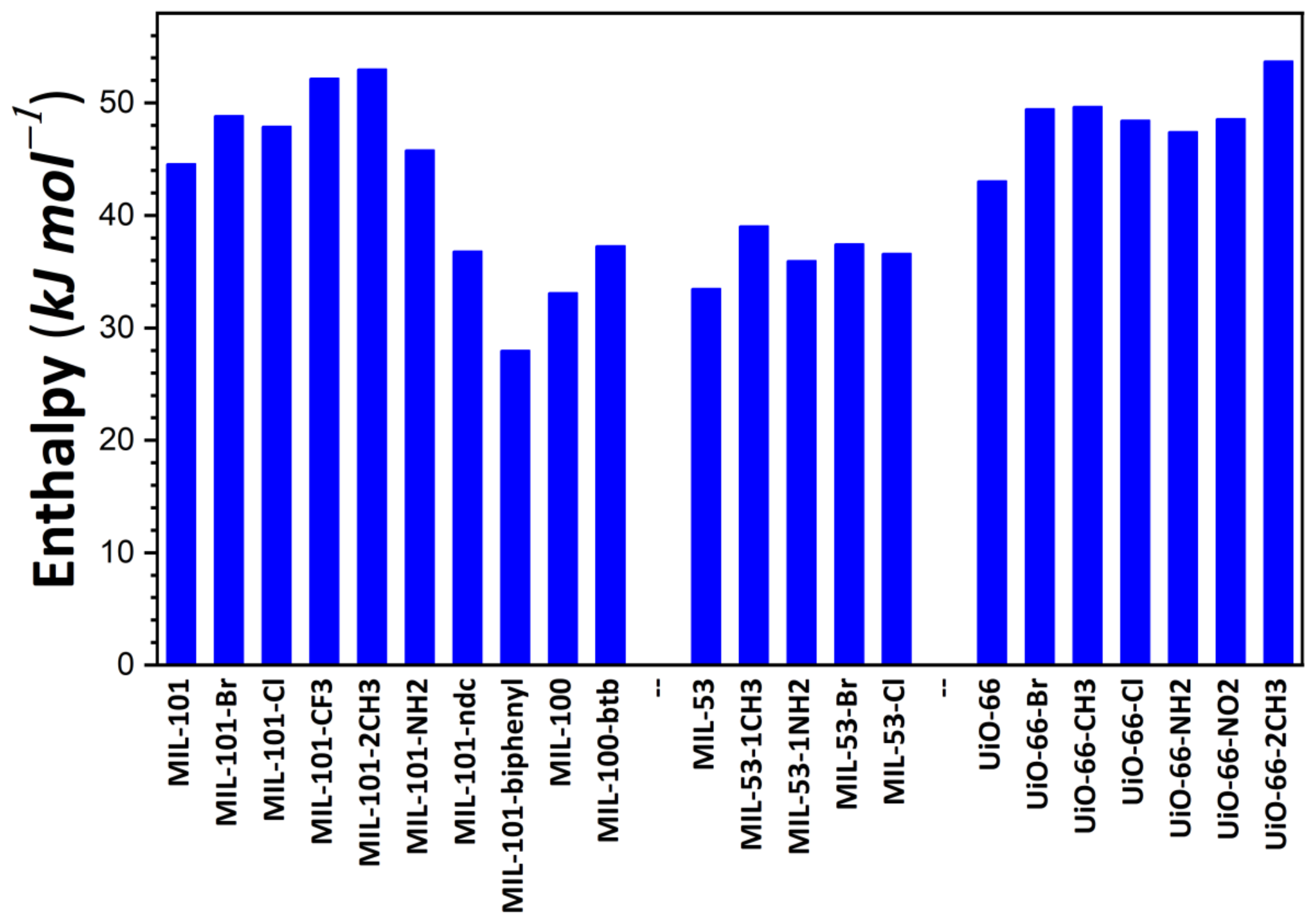
The energy released during the adsorption of the first I2 molecule within the pore space was taken as the first criterion to estimate the performance of the adsorbents. This particular energy value was considered as the measure of adsorbate–adsorbent interaction strength in the presence of traces of I2 vapor. Figure 1 illustrates the variations in this energy as a function of the chemical and structural features of the studied materials. Concerning the MIL-100/MIL-101 family, energy values range between 28 kJ·mol−1 for MIL-101-biphenyl and 52 kJ·mol−1 for MIL-101-2CH3. Such a variation can mainly be explained by the introduction of chemical functionalization (organic moieties). It is likely induced by a modification of pore volume due to the confinement effect as a consequence of the introduction of various chemical moieties. A comparison with the energy variation at low surface coverages recorded on samples belonging to the MIL-53 family clearly shows that the effect of functionalization is drastically decreased when one considers the solid structures with lower pore volumes. This can be explained by the fact that the modification of pore volume through the introduction of chemical functions is strongly limited. Indeed, the MIL-100/MIL-101 structures have larger pores than those of the MIL-53 series. It is worth noting that, in the present calculations, the MIL-53 series was taken in an ‘open’, large-pore form. For such structures, energy changes between 34 kJ·mol−1 for MIL-53 and 48 kJ·mol−1 for MIL-53-1CH3 were observed, in good agreement with the experimental trend reported in [54]. In the case of the UiO-66 series, an intermediary effect was recorded for MIL-53 and the MIL-100/MIL-101 series, with energy values of 42 kJ·mol−1 for UiO-66 and 53 kJ·mol−1 for UiO-66-2CH3. This description may be completed by taking into account the observation that MIL-127 saturated with Cl−, I−, or NO3− releases the same amounts of energy (i.e., close to 35 kJ·mol−1), while the energy values obtained with Zn-BTeC saturated with alkali cations (Li+ to Cs+) range from 41 to 45 kJ·mol−1. All values reported here are higher than the vaporization enthalpy of I2 at ambient temperature, which is close to 20 kJ·mol−1. Given these results of energy calculation, MOFs characterized by a neutral framework appear effective in capturing traces of gaseous I2 released upon nuclear and radiation accidents and incidents; however, ion-containing structures do not involve strong interactions with this vapor, but only relatively strong interactions that are weakly influenced by the nature of the compensating ions.
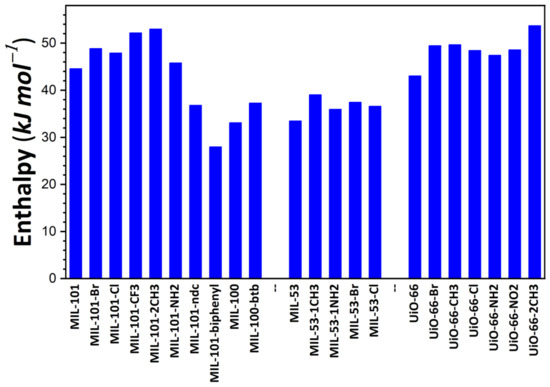
Figure 1.
Energy release during the adsorption of the first I2 molecule onto MOF structures studied in the present paper, as calculated based on the Monte Carlo simulations.
A comparison of the present results with those reported on the basis of other computational studies shows a good agreement with other classical simulations. Indeed Assfour et al. [55] evaluated isosteric heat using different adsorption isotherms calculated at different temperatures and found 30 kJ·mol−1 for MIL-100 and 55 kJ·mol−1 for MIL-53(Al).
To better evaluate the impact of structural functionalization, the interactions between the adsorbate and the adsorbent were investigated by considering the plausible molecular configurations within the pores for each sample. It should be emphasized that plausible molecular configurations correspond to the distribution of guest I2 molecules in porous solids as a function of the statistical distribution extracted from Monte Carlo simulations. The related snapshots are reported in the Supplementary Materials. In the case of the MIL-53 series, the observed impact of the functional groups grafted on the organic moieties is rather weak. Indeed, all distances reported in Figure S1 are more or less identical. Only small changes can be noted for the NH2 group. The same behavior can be observed for the MIL-101 series (Figure S2), where the NH2 function likely interacts with the I2 molecule. However, the effect is only slightly noticeable, which is probably due to the fact that pore sizes are relatively large in line with the open form of the MIL-53 solids. On the contrary, the samples belonging to the UiO-66 series possess smaller pores, and stronger interactions are detected at smaller distances between I2 and the organic linker (Figure S3). This means that, even for the first adsorbing molecules, the nature of the ligand has an effect only when small pores are considered. It is also important to note at this level that the choice of the force field implemented in the calculation code may have some impact on the results obtained. Electrostatic contributions to the adsorbate–adsorbent interactions were neglected here. The development of a force field including such a contribution may certainly be of interest to elucidate the full impact of structural functionalization. However, to date, no calorimetric data are available in the literature, thereby leaving such a force field essentially untested. Based on the present results of our calculations, it is possible to hypothesize that the NH2 functional group is promising in view of use of MOF materials in nuclear safety applications.
The affinity of the solid surface towards I2 and the saturation plateau are two other parameters to investigate. To determine them, the I2 adsorption isotherms were calculated for each solid sample; the results are reported in the Supplementary Materials (Figures S4–S9). The affinity parameter corresponds to the slope of the isotherm at low pressures. A comparison between all solids clearly indicates that the MIL-53 and UiO-66 series present a strong affinity, whereas the MIL-100/MIL-101 ones are characterized by a drastically lower affinity towards I2. This result is thus in good agreement with the conclusions presented before with respect to the adsorption enthalpy calculated at low I2 loadings.
If we consider the saturation plateau for each isotherm, some trends may be inferred from the results of our calculations. In the case of MIL-53, UiO-66, and the MIL-101 series, the MOF structures containing H or NH2 groups appear the most performant in adsorbing large quantities of I2. The order of increasing I2 adsorption capacity was as follows: Cl ≈ NH2 ≈ H > CH3 > Br, MIL-53 series (in agreement with trends obtained from experimental data in the liquid phase [54]); NH2 ≈ H > 2CH3 > Cl > Br > CF3, MIL-101 series; H > NH2 > CH3 > Cl > 2CH3 > NO2 > Br, UiO-66 series. A comparison of these theoretical values with the experimental results shows a relative good agreement, as UiO-66 saturation reaches 0.7 g·g−1 [70]. The difference can be explained by the accessibility of the entire porosity achieved in the theoretical model in contrast with the experiment. The maximum amount of I2 adsorbed was reported to be about 4 g·g−1 in the cases of MIL-101-NH2 and MIL-101-H (c.f., Figure S3). However, grafting a longer organic linker should result in a much higher adsorption capacity, e.g., the 10 g·g−1 for MIL-100-BTB at ambient temperature. In contrast, much lower adsorption capacities were obtained with ionic MOFs (i.e., Zn-BTeC and MIL-127), and they were found to be only slightly dependent on the nature of the compensating ion present in the pores. Such predicted values are still higher than the best reports, such as in Zr-based MOF-808 at 80 °C (2.18 g·g−1) without water [70], or MOF-808 covered by poly(vinylidene fluoride) (1.42 g·g−1) [72]. It should be noted that a comparison with experimental data is difficult due to the scarcity of the experimental reports. Taking as an example the reference [54], the saturation measured in MIL-53(Al)-NH2, MIL-100, and MIL-101 at room temperature (respectively 150, 50 and 350 mg·g−1) is much lower than the theoretical saturations obtained in the present study. However, such experimental values are not consistent with the pore volume (respectively, 0.56, 1 and 1.8 cm3·g−1), thereby suggesting that other phenomena occur upon I2 adsorption (adsorption of other molecules, swelling or degradation of the solid under the conditions applied, or others). Other theoretical and experimental studies are more in line with our simulations, as the saturation of MIL-53(Al) is close to 1 g·g−1, MIL-101 reaches 5 g·g−1 [55], while UiO-66 can adsorb 1.17 g·g−1.
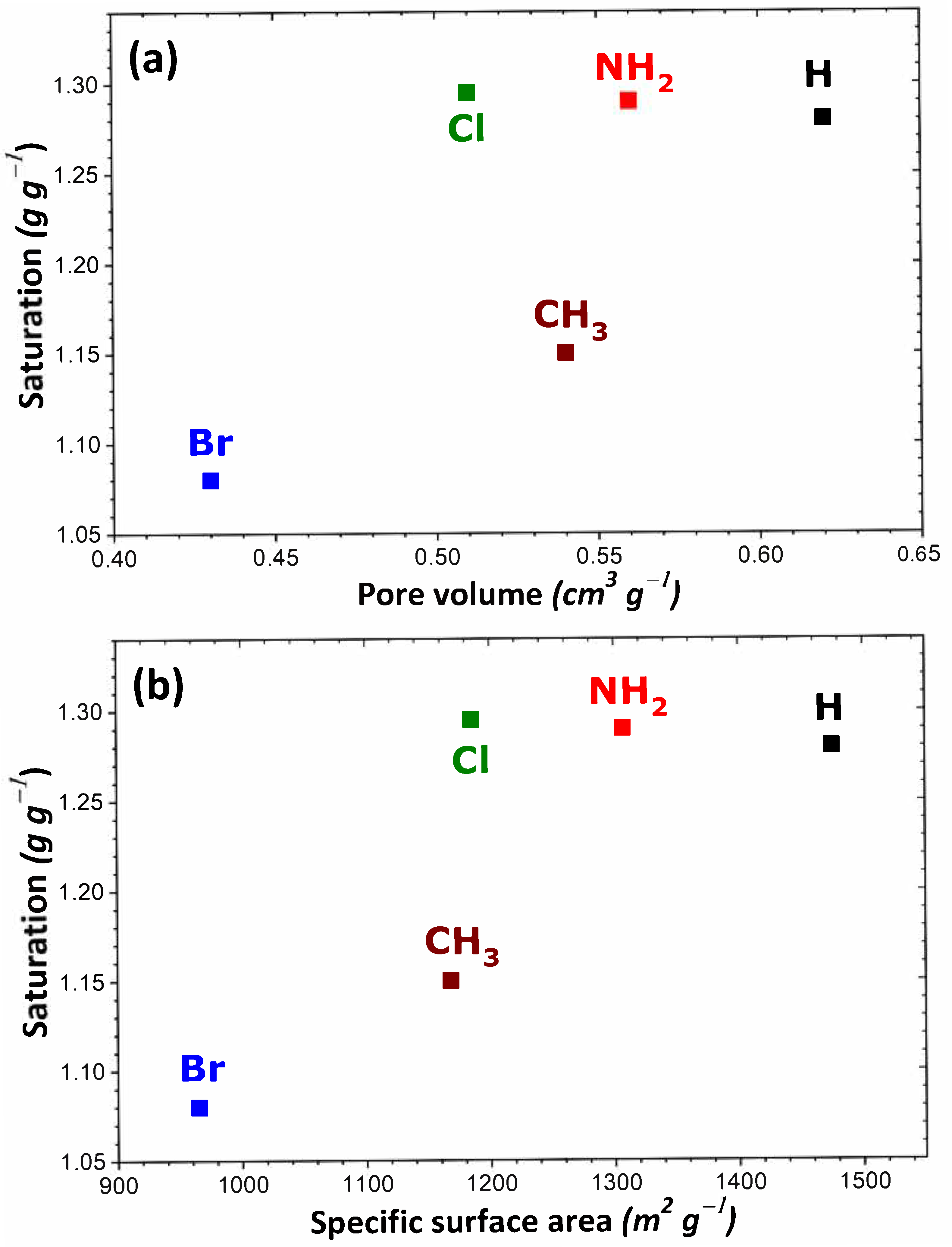
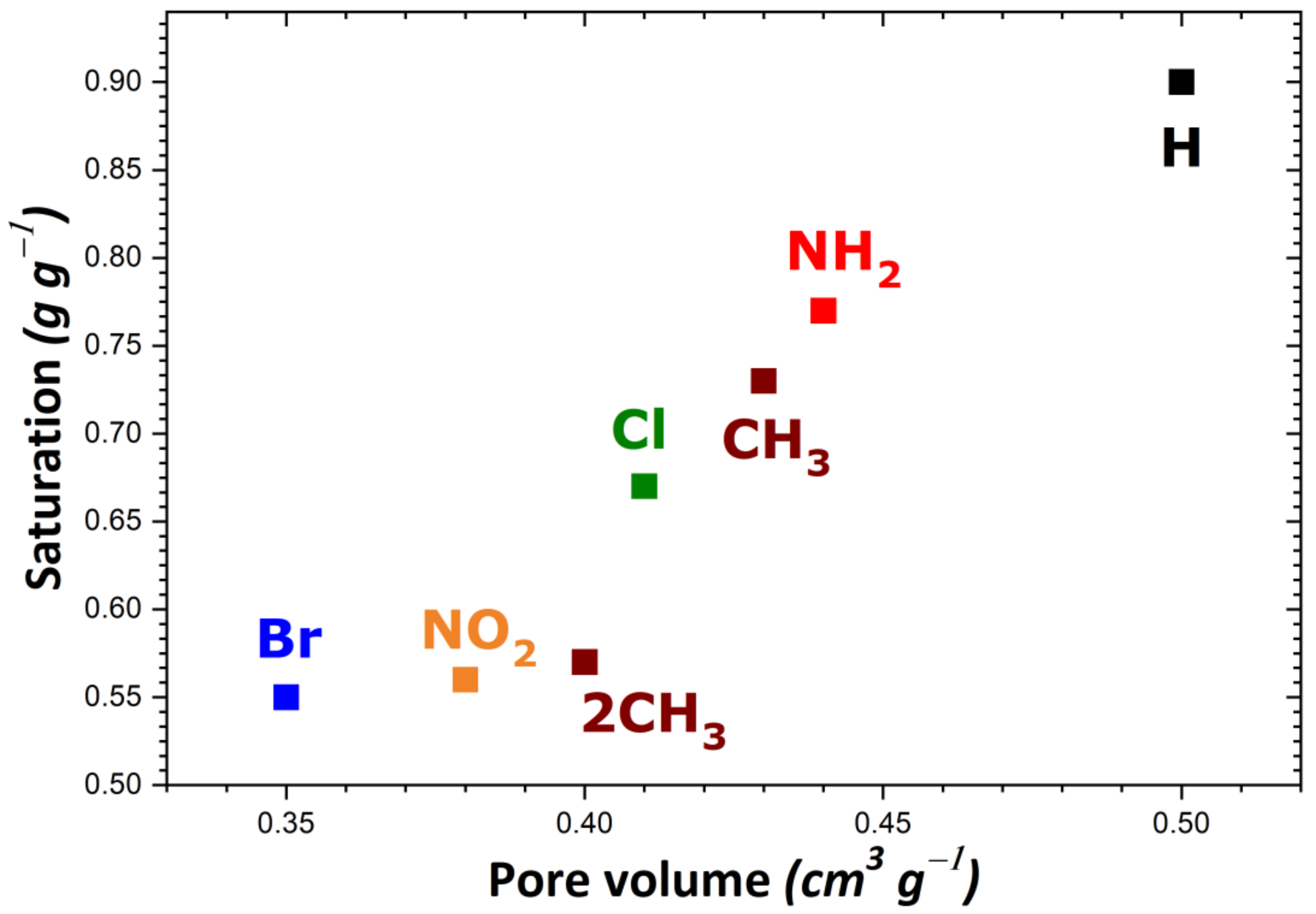
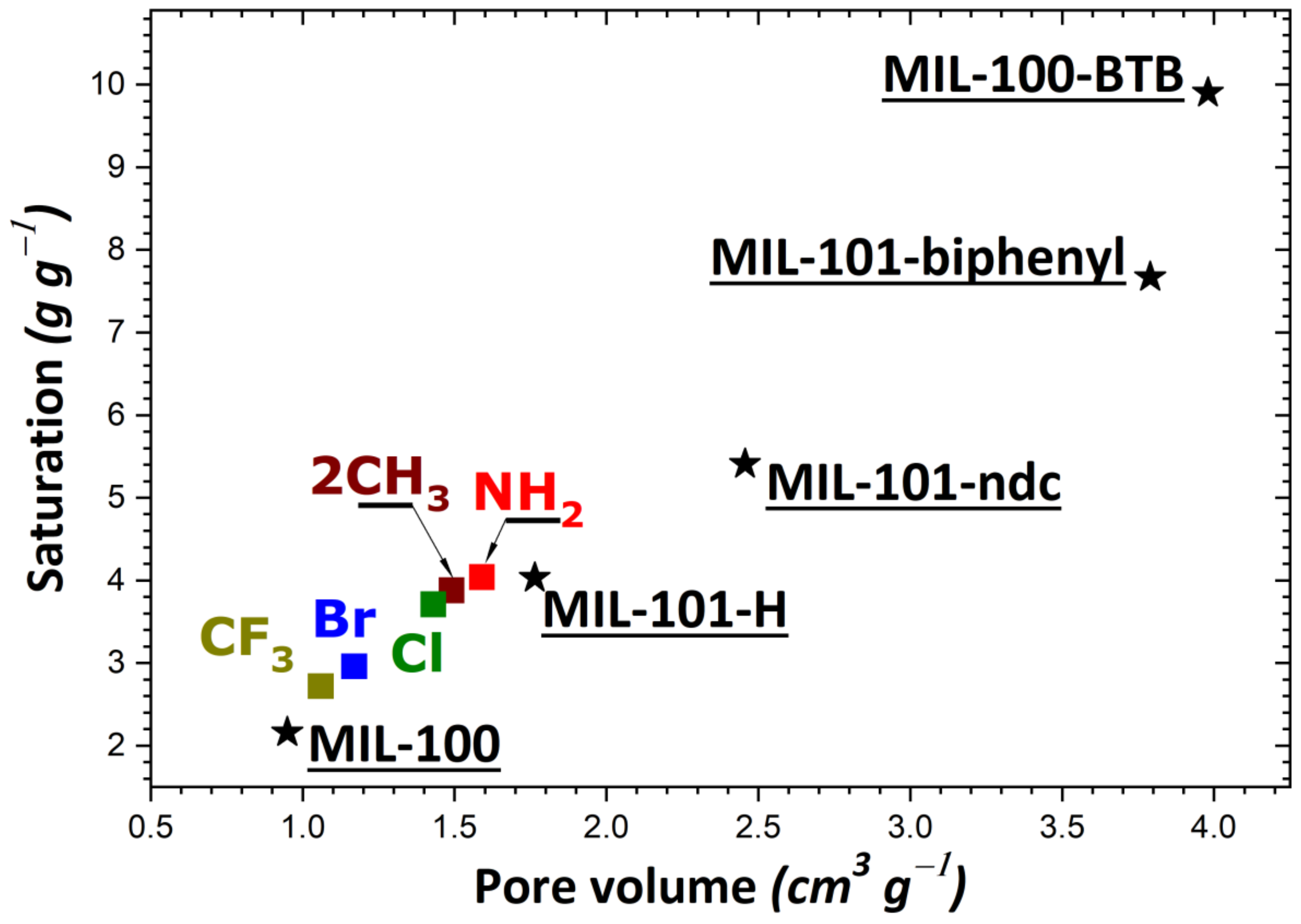
To rationalize these trends, the impact of the theoretical specific surface area and pore volume (obtained following the procedure described in Section 2) was investigated as a result of structural functionalization. For the MIL-53 series, plots of adsorption capacity versus pore volume and specific surface area are given in Figure 2a,b, respectively. No correlation was established between saturation capacity and both textural parameters. On the contrary, clearer trends could be inferred from the analysis of the corresponding plots in Figure 3 for the UiO-66 series. It may be argued that the main difference between the two MOF series lies in the pore network: namely, linear pores in MIL-53 and interconnected cages in UiO-66. To provide further support for this hypothesis, some structures of the MIL-100 and MIL-101 series possessing interconnected cages were taken into consideration. A noticeable direct correlation was achieved this time, as can be seen in Figure 4.
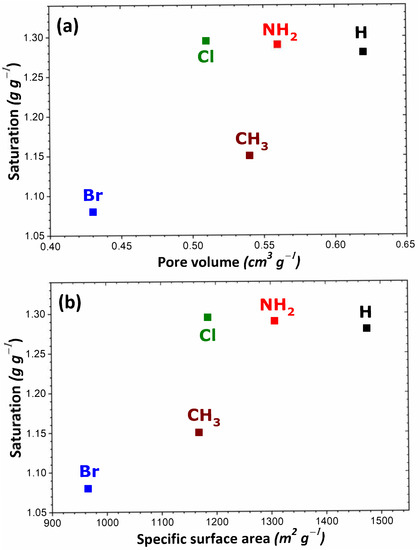
Figure 2.
Evolution of the adsorption capacity of the adsorbents belonging to the MIL-53 series towards gaseous I2 at saturation as a function of pore volume (a) and specific surface area (b).
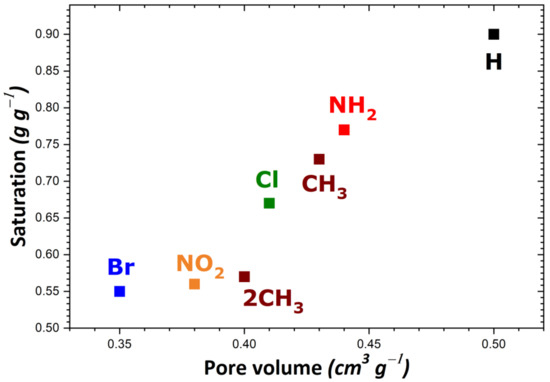
Figure 3.
Evolution of the adsorption capacity of the adsorbents belonging to the UiO-66 series towards gaseous I2 at saturation as a function of pore volume.
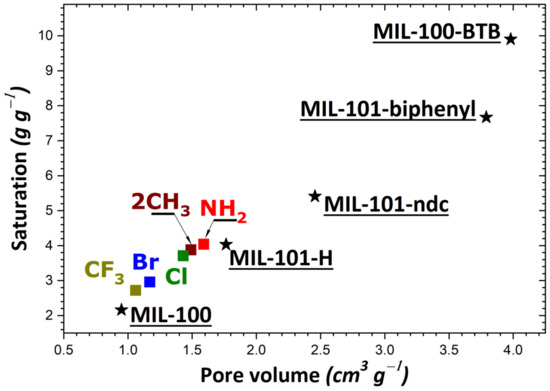
Figure 4.
Evolution of the adsorption capacity of the adsorbents belonging to the MIL-100/MIL-101 series (MIL-100; MIL-101-ndc; MIL-101-biphenyl; MIL-100-BTB; MIL-101-X with X = H, NH2, 2CH3, Cl, Br, CF3) towards gaseous I2 at saturation as a function of pore volume.
Given the observed trends and correlations, it was possible to propose solid structures that presented high efficiency in adsorbing I2 from the gas phase. The members of the MIL-53 series were characterized by limited adsorption capacity, irrespective of the functional group grafted on the benzene ring. Contrary to this, the MIL-100 and MIL-101 series with large pores exhibited adsorption performances that were promising for efficient I2 capture.
For some selected MOFs with promising sorption performances, the impact of temperature was studied at 353 K (i.e., 80 °C) by determining enthalpy at low coverage and adsorption capacity at 1 bar. The resulting values are reported in Table 1 and compared with those obtained previously at 300 K.

Table 1.
Comparison of theoretical values obtained from Monte Carlo simulations for enthalpy at low coverage (1 molecule per unit cell) and adsorption capacity at 1 bar as a function of temperature.
A detailed analysis of the results reported in Table 1 reveals that adsorption enthalpy is only slightly modified by temperature, and values corresponding to 300 K and 353 K are fairly similar. In contrast, the amount of I2 adsorbed onto these porous solids is strongly dependent on temperature, especially in the case of mesoporous solids. For microporous solids (namely, the selected sampleswithin the UiO-66 and MIL-53 series), the amount adsorbed is relatively constant if one compares the two temperatures. Meanwhile, the amount of I2 uptaken by selected samples from the MIL-101/MIL-100 series is drastically modified as a function of temperature, and it strongly decreases when passing from 300 K to 353 K. From these results, it appears that the choice of the best performant samples may be also dependent on the operating conditions applied.
4. Conclusions
In conclusion, a research strategy based on a molecular simulation was tested to elucidate the effects of structural functionalization, the nature of pores, flexibility, and the presence of compensating cations on MOF retention capacity for I2 from the gas phase. This strategy aims to replace fastidious experiments while avoiding the potential health risks caused by radiation exposure. This study offers a rational approach to select the best adsorbents in view of the industrial uses envisaged here. These results may contribute to orienting choices of water-stable porous solids (within the framework of the present selection of materials) for applications involving the capture of either traces or large amounts of vapor (such as those released during nuclear accidents). As far as all adsorption properties are considered, MIL-100-BTB has been proven to exhibit the most promising performance at room temperature. MIL-100 possessing longer organic moieties and thus larger pores can even exceed the maximum adsorption capacity of the former sample and, as such, it may be used in the case of massive adsorption. In contrast, the capacity of MOF materials to retain traces of gaseous I2 on their surface argues in favor of MIL-101-2CH3, MIL-101-CF3, or UiO-66-CH3. They may be of greater interest in separation uses of porous materials subjected to the poisoning effect of water. In fact, they carry organic linkers that are generally not affected by the adsorption of water molecules. Calculations also show that the choice of solids is strongly dependent on the conditions of use of these solids. Indeed, mesoporous solids are greatly impacted by an increase in temperature, while microporous solids (UiO-66 and MIL-53 series) appear more stable. It follows that, at temperatures higher than room temperature, such solids could present an interesting alternative for I2 capture.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/nano11092245/s1, Figure S1: Molecular conformations of the first I2 molecule adsorbed onto MOF structures belonging to the MIL-53 series functionalized by: (a) H, (b) CH3, (c) NH2, (d) Cl, (e) Br, as obtained based on the Monte Carlo simulations; Figure S2: Molecular conformations of the first I2 molecule adsorbed onto MOF structures belonging to the MIL-100/101 series: (a) MIL-100, (b) MIL-101-Cl, (c) MIL-101-Br, (d) MIL-101-2CF3, (e) MIL-101-NH2, (f) MIL-101-biphenyl, (g) MIL-100-BTB, as obtained based on the Monte Carlo simulations; Figure S3: Molecular conformations of the first I2 molecule adsorbed onto MOF structures belonging to the UiO-66 series: (a) H, (b) Cl, (c) Br, (d) NH2, (e) NO2, (f) CH3, (g) 2CH3, as obtained based on the Monte Carlo simulations; Figure S4: Adsorption isotherms for I2 onto MOF structures belonging to the MIL-53 series, as calculated based on the Monte Carlo simulations; Figure S5: Adsorption isotherms for I2 onto MOF structures belonging to the UiO-66 series, as calculated based on the Monte Carlo simulations; Figure S6: Adsorption isotherms for I2 onto MOF structures belonging to the MIL-101 series, as calculated based on the Monte Carlo simulations; Figure S7: Adsorption isotherms for I2 onto MOF structures belonging to the modified MIL-100/MIL-101 solids, as calculated based on the Monte Carlo simulations; Figure S8: Adsorption isotherms for I2 onto MOF structures belonging to the cation-saturated Zn-BTeC family, as calculated based on the Monte Carlo simulations; Figure S9: Adsorption isotherms for I2 onto MOF structures belonging to the anion-saturated MIL-127, as calculated based on the Monte Carlo simulations.
Author Contributions
Conceptualization, methodology, calculations F.S.; formal analysis, writing—original draft preparation F.S. and J.Z. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lenzen, M. Life cycle energy and greenhouse gas emissions of nuclear energy: A review. Energy Convers. Manag. 2008, 49, 2178–2199. [Google Scholar] [CrossRef]
- Cohen, B.L. High-level radioactive waste from light-water reactors. Rev. Mod. Phys. 1977, 49, 1–20. [Google Scholar] [CrossRef]
- Küpper, F.C.; Feiters, M.C.; Olofsson, B.; Kaiho, T.; Yanagida, S.; Zimmermann, M.B.; Carpentier, L.J.; Luther, G.W.; Lu, Z.; Jonsson, M.; et al. Commemorating Two Centuries of Iodine Research: An Interdisciplinary Overview of Current Research. Angew. Chem. Int. Ed. 2011, 50, 11598–11620. [Google Scholar] [CrossRef]
- Haefner, D.; Tranter, T. Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey; Report INL/EXT-07-12299, 16; Idaho National Laboratory: Idaho Falls, ID, USA, 2007.
- Mnasri, N.; Charnay, C.; de Ménorval, L.C.; Moussaoui, Y.; Elaloui, E.; Zajac, J. Silver nanoparticle-containing submicron-in-size mesoporous silica-based systems for iodine entrapment and immobilization from gas phase. Microporous Mesoporous Mater. 2014, 196, 305–313. [Google Scholar] [CrossRef]
- Massasso, G.; Long, J.; Haines, J.; Devautour-Vinot, S.; Maurin, G.; Grandjean, A.; Onida, B.; Donnadieu, B.; Larionova, J.; Guérin, C.; et al. Iodine Capture by Hofmann-Type Clathrate NiII(pz)[NiII(CN)4]. Inorg. Chem. 2014, 53, 4269–4271. [Google Scholar] [CrossRef]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal–Organic Framework Materials as Chemical Sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef]
- Horcajada, P.; Gref, R.; Baati, T.; Allan, P.K.; Maurin, G.; Couvreur, P.; Férey, G.; Morris, R.E.; Serre, C. Metal–Organic Frameworks in Biomedicine. Chem. Rev. 2012, 112, 1232–1268. [Google Scholar] [CrossRef]
- Czaja, A.U.; Trukhan, N.; Müller, U. Industrial applications of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1284–1293. [Google Scholar] [CrossRef]
- Lee, J.Y.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-Organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Canivet, J.; Fateeva, A.; Guo, Y.; Coasne, B.; Farrusseng, D. Water adsorption in MOFs: Fundamentals and applications. Chem. Soc. Rev. 2014, 43, 5594–5617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, P.; Wong, N.E.; Shimizu, G.K.H. MOFs as proton conductors—challenges and opportunities. Chem. Soc. Rev. 2014, 43, 5913–5932. [Google Scholar] [CrossRef]
- Getman, R.B.; Bae, Y.S.; Wilmer, C.E.; Snurr, R.Q. Review and Analysis of Molecular Simulations of Methane, Hydrogen, and Acetylene Storage in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 703–723. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.H.; Long, J.R. Carbon Dioxide Capture in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.W. Hydrogen Storage in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal–Organic Frameworks for Separations. Chem. Rev. 2012, 112, 869–932. [Google Scholar] [CrossRef]
- Kumar, S.; Jain, S.; Nehra, M.; Dilbaghi, N.; Marrazza, G.; Kim, K.H. Green synthesis of metal-organic frameworks: A state-of-the-art review of potential environmental and medical applications. Coord. Chem. Rev. 2020, 420, 213407. [Google Scholar] [CrossRef]
- Zhang, S.; Xia, W.; Yang, Q.; Kaneti, Y.V.; Xu, X.; Alshehri, S.M.; Ahamad, T.; Hossain, S.A.; Na, J.; Tang, J.; et al. Core-shell motif construction: Highly graphitic nitrogen-doped porous carbon electrocatalysts using MOF-derived carbon@COF heterostructures as sacrificial templates. Chem. Eng. J. 2020, 396, 125154. [Google Scholar] [CrossRef]
- Gumilar, G.; Kaneti, Y.V.; Henzie, J.; Chatterjee, S.; Na, J.; Yuliarto, B.; Nugraha, N.; Patah, A.; Bhaumik, A.; Yamauchi, Y. General synthesis of hierarchical sheet/plate-like M-BDC (M=Cu, Mn, Ni and Zr) metal-organic frameworks fpr electrochemical non-enzymatic glucose sensing. Chem. Sci. 2020, 11, 3644–3655. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kaneti, Y.V.; Bando, Y.; Lin, J.; Liu, C.; Li, J.; Yamauchi, Y. Metal-Organic framework-derived one-dimensional porous or hollow carbon-based nanofibres for energy storage and conversion. Mater. Horiz. 2018, 5, 394. [Google Scholar] [CrossRef] [Green Version]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Lu, W.; Wei, Z.; Gu, Z.Y.; Liu, T.F.; Park, J.; Park, J.; Tian, J.; Zhang, M.; Zhang, Q.; Gentle, T., III; et al. Tuning the structure and function of metal–organic frameworks via linker design. Chem. Soc. Rev. 2014, 43, 5561–5593. [Google Scholar] [CrossRef]
- Nouar, F.; Breeze, M.I.; Campo, B.C.; Vimont, A.; Clet, G.; Daturi, M.; Devic, T.; Walton, R.I.; Serre, C. Tuning the properties of the UiO-66 metal organic framework by Ce substitution. Chem. Comm. 2015, 51, 14458–14461. [Google Scholar] [CrossRef]
- Devic, T.; Serre, C. High valence 3p and transition metal based MOFs. Chem. Soc. Rev. 2014, 43, 6097–6115. [Google Scholar] [CrossRef]
- Zhai, Q.G.; Bu, X.H.; Mao, C.Y.; Zhao, X.; Feng, P.Y. Systematic and Dramatic Tuning on Gas Sorption Performance in Heterometallic Metal–Organic Frameworks. J. Am. Chem. Soc. 2016, 138, 2524–2527. [Google Scholar] [CrossRef]
- Xiong, Y.; Fan, Y.Z.; Damasceno Borges, D.; Chen, C.X.; Wei, Z.W.; Wang, H.P.; Pan, M.; Jiang, J.J.; Maurin, G.; Su, C.Y. Ligand and Metal Effects on the Stability and Adsorption Properties of an Isoreticular Series of MOFs Based on T-Shaped Ligands and Paddle-Wheel Secondary Building Units. Chem. Eur. J. 2016, 22, 16147–16156. [Google Scholar] [CrossRef] [PubMed]
- Yot, P.; Yang, K.; Guillerm, V.; Ragon, F.; Dmitriev, V.; Parisiades, P.; Elkaim, E.; Devic, T.; Horcajada, P.; Serre, C.; et al. Impact of the Metal Centre and Functionalization on the Mechanical Behaviour of MIL-53 Metal–Organic Frameworks. Eur. J. Inorg. Chem. 2016, 27, 4424–4429. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.K.; Cohen, S.M. Postsynthetic modification of metal–organic frameworks—A progress report. Chem. Soc. Rev. 2011, 40, 498–519. [Google Scholar] [CrossRef] [PubMed]
- Dau, P.V.; Tanabe, K.K.; Cohen, S.M. Functional group effects on metal–organic framework topology. Chem. Comm. 2012, 48, 9370–9372. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Cohen, S.M. Modulating Metal−Organic Frameworks to Breathe: A Postsynthetic Covalent Modification Approach. J. Am. Chem. Soc. 2009, 131, 16675–16677. [Google Scholar] [CrossRef]
- He, Y.; Li, B.; O’Keeffe, M.; Chen, B. Multifunctional metal–organic frameworks constructed from meta-benzenedicarboxylate units. Chem. Soc. Rev. 2014, 43, 5618–5656. [Google Scholar] [CrossRef] [PubMed]
- Devic, T.; Horcajada, P.; Serre, C.; Salles, F.; Maurin, G.; Moulin, B.; Heurtaux, D.; Clet, G.; Vimont, A.; Greneche, J.M.; et al. Functionalization in flexible porous solids: Effects on the pore opening and the host-guest interactions. J. Am. Chem. Soc. 2010, 132, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Salles, F.; Wuttke, S.; Devic, T.; Heurtaux, D.; Maurin, G.; Vimont, A.; Daturi, M.; David, O.; Magnier, E.; et al. How Linker’s Modification Controls Swelling Properties of Highly Flexible Iron(III) Dicarboxylates MIL-88. J. Am. Chem. Soc. 2011, 133, 17839–17847. [Google Scholar] [CrossRef] [PubMed]
- Sava, D.F.; Garino, T.J.; Nenoff, T.M. Iodine Confinement into Metal–Organic Frameworks (MOFs): Low-Temperature Sintering Glasses To Form Novel Glass Composite Material (GCM) Alternative Waste Forms. Ind. Eng. Chem. Res. 2012, 51, 614–620. [Google Scholar] [CrossRef]
- Banerjee, D.; Simon, C.M.; Plonka, A.M.; Motkuri, R.K.; Liu, J.; Chen, X.; Smit, B.; Parise, J.B.; Haranczyk, M.; Thallapally, P.K. Metal–organic framework with optimally selective xenon adsorption and separation. Nat. Comm. 2016, 7, 11831. [Google Scholar] [CrossRef]
- Riley, B.J.; Vienna, J.D.; Strachan, D.M.; McCloy, J.S.; Jerden, J.L., Jr. Materials and processes for the effective capture and immobilization of radioiodine: A review. J. Nucl. Mater. 2016, 470, 307–326. [Google Scholar] [CrossRef] [Green Version]
- Munn, A.S.; Millange, F.; Frigoli, M.; Guillou, N.; Falaise, C.; Stevenson, V.; Volkringer, C.; Loiseau, T.; Cibin, G.; Walton, R.I. Iodine sequestration by thiol-modified MIL-53(Al). CrystEngComm 2016, 18, 8108–8114. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Qi, B.; Liu, Y.; Zhao, L.; Jiao, Y.; Zhang, Y.; Jiang, Z.; Li, Q.; Wang, J.; Chen, S.; et al. AgII doped MIL-101 and its adsorption of iodine with high speed in solution. J. Solid State Chem. 2016, 237, 274–283. [Google Scholar] [CrossRef]
- Chen, Y.; Li, L.; Yang, J.; Wang, S.; Li, J. Reversible flexible structural changes in multidimensional MOFs by guest molecules (I2, NH3) and thermal stimulation. J. Solid State Chem. 2015, 226, 114–119. [Google Scholar] [CrossRef]
- Abney, C.W.; Taylor-Pashow, M.L.; Russell, S.R.; Chen, Y.; Samantaray, R.; Lockard, J.V.; Lin, W. Topotactic Transformations of Metal–Organic Frameworks to Highly Porous and Stable Inorganic Sorbents for Efficient Radionuclide Sequestration. Chem. Mater. 2014, 26, 5231–5243. [Google Scholar] [CrossRef]
- Shi, P.F.; Hu, H.C.; Zhang, Z.Y.; Xiong, G.; Zhao, B. Heterometal–organic frameworks as highly sensitive and highly selective luminescent probes to detect I− ions in aqueous solutions. Chem. Commun. 2015, 51, 3985–3988. [Google Scholar] [CrossRef] [PubMed]
- Düren, T.; Bae, Y.S.; Snurr, R.Q. Using molecular simulation to characterise metal–organic frameworks for adsorption applications. Chem. Soc. Rev. 2009, 38, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Basdogan, Y.; Keskin, S. Simulation and modelling of MOFs for hydrogen storage. CrystEngComm 2015, 17, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Trousselet, F.; Archereau, A.; Boutin, A.; Coudert, F.X. Heterometallic Metal–Organic Frameworks of MOF-5 and UiO-66 Families: Insight from Computational Chemistry. J. Phys. Chem. C 2016, 120, 24885–24894. [Google Scholar] [CrossRef] [Green Version]
- Taddei, M.; Tiana, D.; Casati, N.; van Bokhoven, J.A.; Smit, B.; Ranocchiari, M. Mixed-linker UiO-66: Structure–property relationships revealed by a combination of high-resolution powder X-ray diffraction and density functional theory calculations. Phys. Chem. Chem. Phys. 2017, 19, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Sava, D.F.; Chapman, K.W.; Rodriguez, M.A.; Greathouse, J.A.; Crozier, P.S.; Zhao, H.; Chupas, P.J.; Nenoff, T.M. Competitive I2 Sorption by Cu-BTC from Humid Gas Streams. Chem. Mater. 2013, 25, 2591–2596. [Google Scholar] [CrossRef]
- Ghose, S.K.; Li, Y.; Yakovenko, A.; Dooryhee, E.; Ehm, L.; Ecker, L.E.; Dippel, A.C.; Halder, G.J.; Strachan, D.M.; Thallapally, P.K. Understanding the Adsorption Mechanism of Xe and Kr in a Metal–Organic Framework from X-ray Structural Analysis and First-Principles Calculations. J. Phys. Chem. Lett. 2015, 6, 1790–1794. [Google Scholar] [CrossRef]
- Ryan, P.; Farha, O.K.; Broadbelt, L.; Snurr, R.Q. Computational screening of metal-organic frameworks for xenon/krypton separation. AIChE J. 2011, 57, 1759–1766. [Google Scholar] [CrossRef]
- Sikora, B.J.; Wilmer, C.E.; Greenfield, M.L.; Snurr, R.Q. Thermodynamic analysis of Xe/Kr selectivity in over 137 000 hypothetical metal–organic frameworks. Chem. Sci. 2012, 3, 2217–2223. [Google Scholar] [CrossRef]
- Maurin, G. Role of molecular simulations in the structure exploration of Metal-Organic Frameworks: Illustrations through recent advances in the field. C. R. Chim. 2016, 19, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.T.; Bennett, T.D.; Cheetham, A.K.; Navrotsky, A. Thermochemistry of Zeolitic Imidazolate Frameworks of Varying Porosity. J. Am. Chem. Soc. 2013, 135, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Sava, D.F.; Rodriguez, M.A.; Chapman, K.W.; Chupas, P.J.; Greathouse, J.A.; Crozier, P.S.; Nenoff, T.M. Capture of Volatile Iodine, a Gaseous Fission Product, by Zeolitic Imidazolate Framework-8. J. Am. Chem. Soc. 2011, 133, 12398–12401. [Google Scholar] [CrossRef] [PubMed]
- Falaise, C.; Volkringer, C.; Facqueur, J.; Bousquet, T.; Gasnot, L.; Loiseau, T. Capture of iodine in highly stable metal–organic frameworks: A systematic study. Chem. Commun. 2013, 49, 10320–10322. [Google Scholar] [CrossRef] [PubMed]
- Assfour, B.; Assaad, T.; Odeh, A. In silico screening of metal organic framework for iodine capture and storage. Chem. Phys. Lett. 2014, 610–611, 45–49. [Google Scholar] [CrossRef]
- Yuan, S.; Qin, J.S.; Zou, L.F.; Chen, Y.P.; Wang, X.; Zhang, Q.; Zhou, H.C. Thermodynamically Guided Synthesis of Mixed-Linker Zr-MOFs with Enhanced Tunability. J. Am. Chem. Soc. 2016, 138, 6636–6642. [Google Scholar] [CrossRef]
- Lan, Y.; Tong, M.; Yang, Q.; Zhong, C. Computational screening of covalent organic frameworks for the capture of radioactive iodine and methyl iodide. CrystEngComm 2017, 19, 4920–4926. [Google Scholar] [CrossRef]
- Jin, K.; Lee, B.; Park, J. Metal-Organic frameworks as a versatile platform for radionuclide management. Coord. Chem. Rev. 2020, 424, 213473. [Google Scholar]
- Horcajada, P.; Chevreau, H.; Heurtaux, D.; Benyettou, F.; Salles, F.; Devic, T.; Garcia-Marquez, A.; Yu, C.; Lavrard, H.; Dutson, C.L.; et al. Extended and functionalized porous iron(III) tri- or dicarboxylates with MIL-100/101 topologies. Chem. Commun. 2014, 50, 6872–6874. [Google Scholar] [CrossRef]
- Chevreau, H.; Permyakova, A.; Nouar, F.; Fabry, P.; Livage, C.; Ragon, F.; Garcia-Marquez, A.; Devic, T.; Steunou, N.; Serre, C.; et al. Synthesis of the biocompatible and highly stable MIL-127(Fe): From large scale synthesis to particle size control. CrystEngComm 2016, 18, 4094–4101. [Google Scholar] [CrossRef]
- Yang, E.; Ding, Q.R.; Kang, Y.; Wang, F. Synthesis and characterization of homo- and heterometallic metal–organic frameworks based on 1,2,4,5-benzenetetracarboxylate ligand. Inorg. Chem. Commun. 2013, 36, 195–198. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Wiersum, A.D.; Llewellyn, P.L.; Guillerm, V.; Serre, C.; Maurin, G. Functionalizing porous zirconium terephthalate UiO-66(Zr) for natural gas upgrading: A computational exploration. Chem. Commun. 2011, 47, 9603–9605. [Google Scholar] [CrossRef]
- Nandanwar, S.U.; Coldsnow, K.; Utgikar, V.; Sabharwall, P.; Aston, D.E. Capture of harmful radioactive contaminants from off-gas stream using solid sorbents for clean environment—A review. Chem. Eng. J. 2016, 306, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Salles, F.; Maurin, G.; Serre, C.; Llewellyn, P.L.; Knöfel, C.; Choi, H.J.; Filinchuk, Y.; Oliviero, L.; Vimont, A.; Long, J.R.; et al. Multistep N2 breathing in the Metal-Organic Framework Co(1,4-benzenedipyrazolate). J. Am. Chem. Soc. 2010, 132, 13782–13788. [Google Scholar] [CrossRef]
- Dassault Systèmes. BIOVIA Materials Studio; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- Chebbi, M.; Chibani, S.; Paul, J.F.; Cantrel, L.; Badawi, M. Evaluation of volatile iodine trapping in presence of contaminants: A periodic DFT study on cation exchanged-faujasite. Microporous Mesoporous Mater. 2017, 239, 111–122. [Google Scholar] [CrossRef]
- Sava-Gallis, D.F.; Ermanoski, I.; Greathouse, J.A.; Chapman, K.W.; Nenoff, T.M. Iodine gas adsorption in nanoporous materials: A combined experimental-modeling study. Int. Eng. Chem. Res. 2017, 56, 2331–2338. [Google Scholar] [CrossRef]
- Chibani, S.; Chiter, F.; Cantrel, L.; Paul, J.F. Capture of iodine species in MIL-53(Al), MIL-120(Al) and HKUST-1(Cu) periodic DFT and ab initio Molecular Dynamics studies. J. Phys. Chem. C 2017, 121, 25283–25291. [Google Scholar] [CrossRef]
- Chen, P.; He, X.; Pang, M.; Dong, X.; Zhao, S.; Zhang, W. Iodine capture using Zr-based Metal-Organic Frameworks (Zr-MOFs): Adsorption performance and mechanism. Appl. Mater. Interf. 2020, 12, 20429–20439. [Google Scholar] [CrossRef] [PubMed]
- Düren, T.; Millange, F.; Férey, G.; Walton, K.S.; Snurr, R.Q. Calculating Geometric Surface Areas as a Characterization Tool for Metal−Organic Frameworks. J. Phys. Chem. C 2007, 111, 15350–15356. [Google Scholar] [CrossRef]
- Wang, L.; Chen, P.; Dong, X.; Zhang, W.; Zhao, S.; Xiao, S.; Ouyang, Y. Porous MOF-808@PVDF beads for removal of iodine from gas streams. RSC Adv. 2020, 10, 44679. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).