
A Multifunctional Au/CeO2-Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters



Abstract
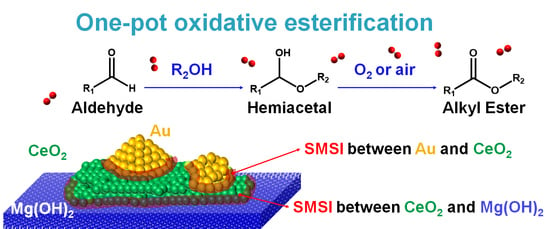
1. Introduction
2. Materials and Methods
2.1. Material Preparation
2.2. Material Characterization
2.3. Catalytic Reaction
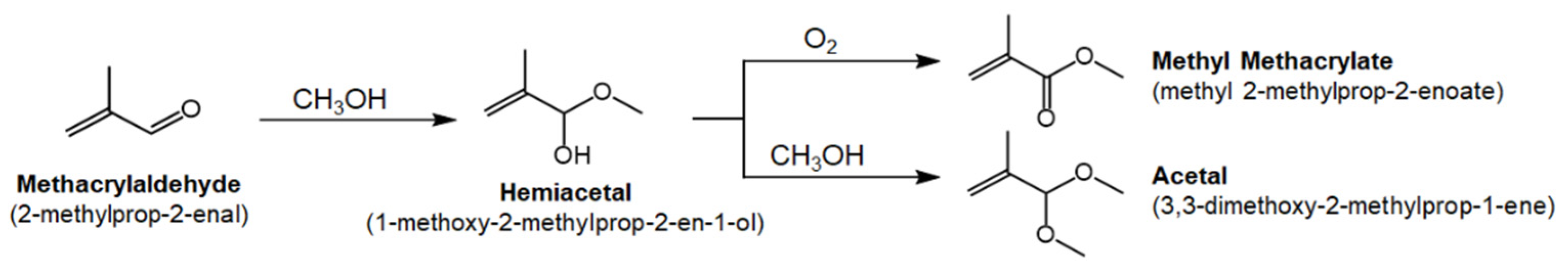
3. Results and Discussion
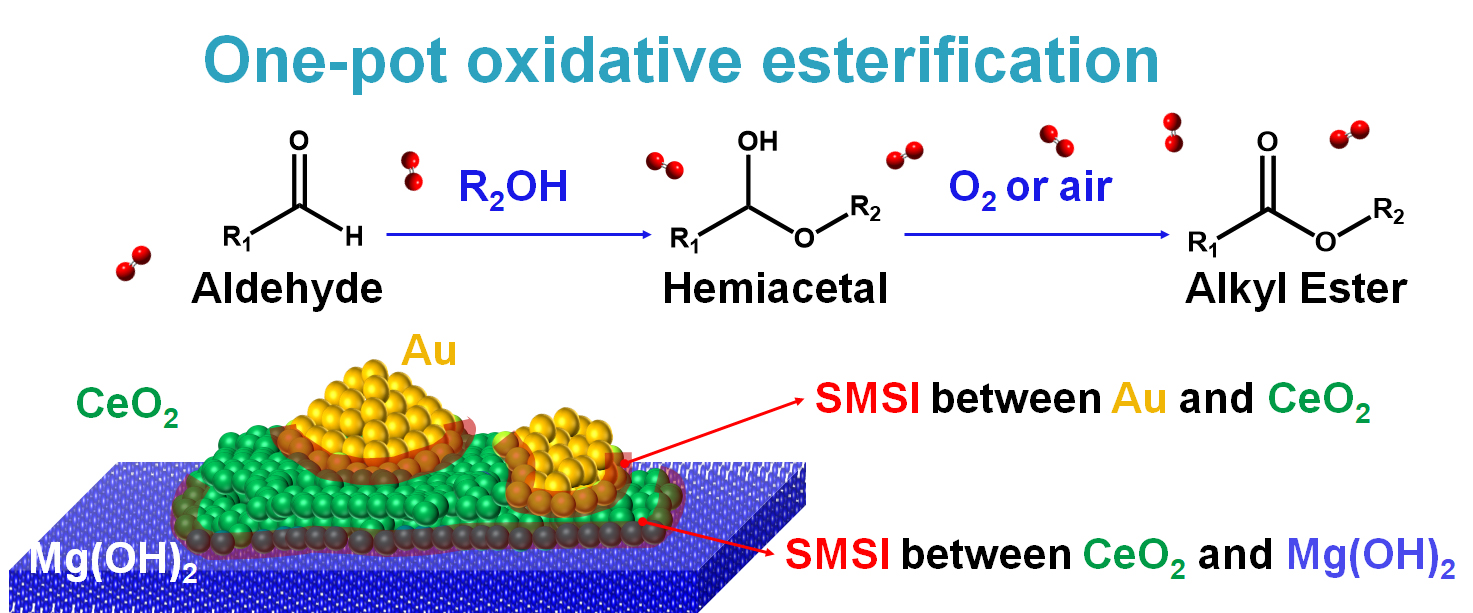
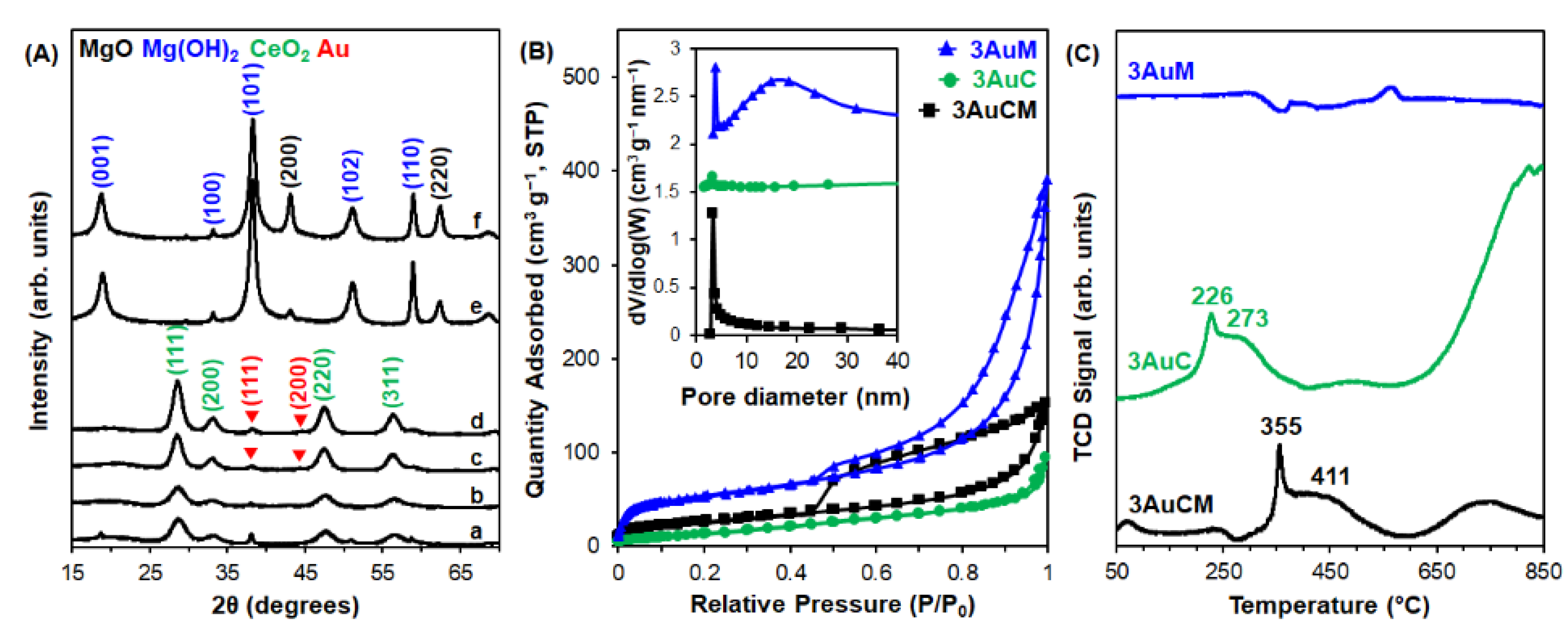
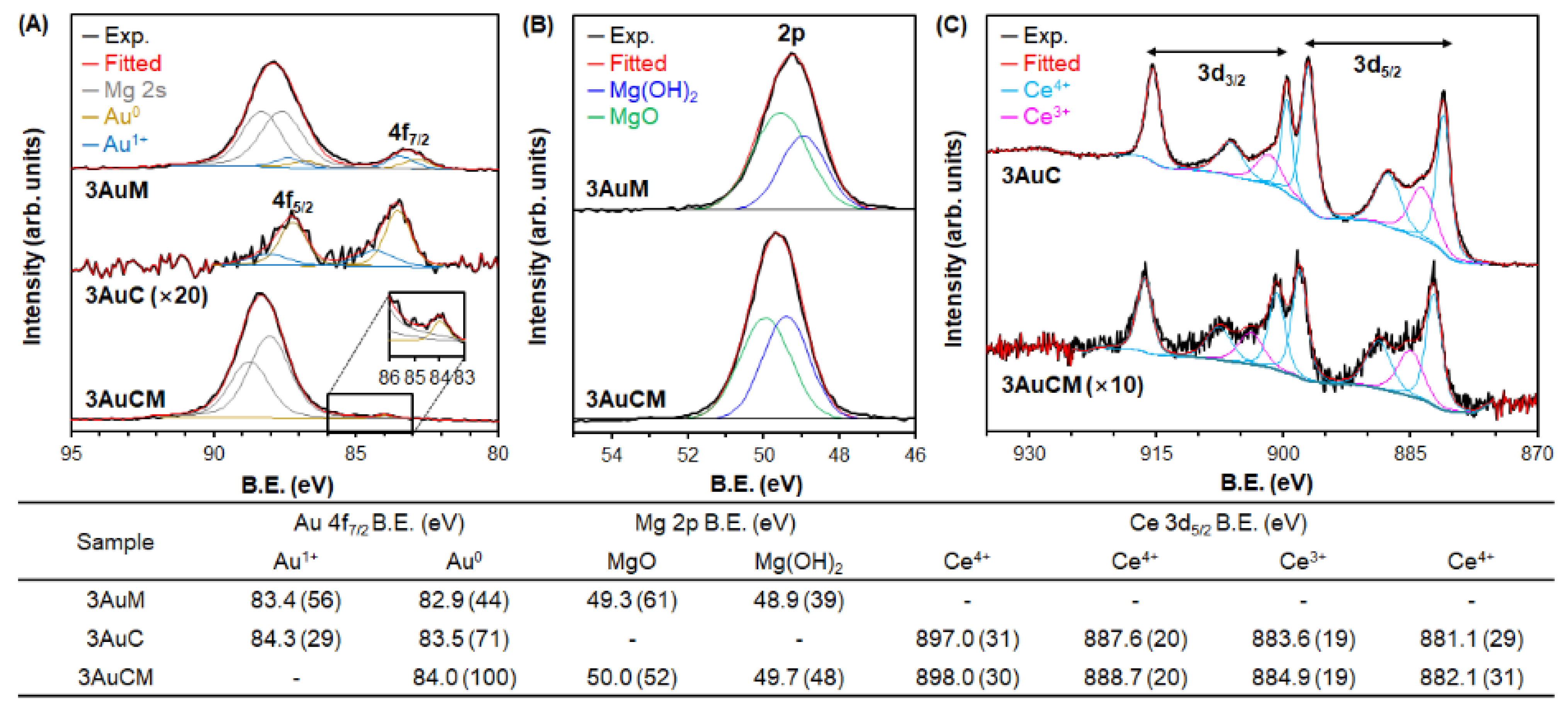
3.1. Materials Characterization
3.2. Reaction Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zope, B.N.; Hibbitts, D.D.; Neurock, M.; Davis, R.J. Reactivity of the Gold/Water Interface During Selective Oxidation Catalysis. Science 2010, 330, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, L.; Tiruvalam, R.; Rahim, M.H.A.; Saiman, M.I.B.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; et al. Solvent-Free Oxidation of Primary Carbon-Hydrogen Bonds in Toluene Using Au-Pd Alloy Nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Yan, R.; Han, J.; Zhang, S. Promoting effects of MgO, (NH4)2SO4 or MoO3 modification in oxidative esterification of methacrolein over Au/Ce0.6Zr0.4O2-based catalysts. Catal. Sci. Technol. 2016, 6, 5453–5463. [Google Scholar] [CrossRef]
- Mallet, T.; Baiker, A. Oxidation of Alcohols with Molecular Oxygen on Solid Catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef]
- Matsumoto, T.; Ueno, M.; Wang, N.; Kobayashi, S. Recent Advances in Immobilized Metal Catalysts for Environmentally Benign Oxidation of Alcohols. Chem. Asian J. 2008, 3, 196–214. [Google Scholar] [CrossRef]
- Kaizuka, K.; Miyamura, H.; Kobayashi, S. Remarkable Effect of Bimetallic Nanocluster Catalysts for Aerobic Oxidation of Alcohols: Combining Metals Changes the Activities and the Reaction Pathways to Aldehydes/Carboxylic Acids or Esters. J. Am. Chem. Soc. 2010, 132, 15096–15098. [Google Scholar] [CrossRef]
- Zhong, W.; Liu, H.; Bai, C.; Liao, S.; Li, Y. Base-Free Oxidation of Alcohols to Esters at Room Temperature and Atmospheric Conditions using Nanoscale Co-Based Catalysts. ACS Catal. 2015, 5, 1850–1856. [Google Scholar] [CrossRef]
- Prati, L.; Rossi, M. Gold on Carbon as a New Catalyst for Selective Liquid Phase Oxidation of Diols. J. Catal. 1998, 176, 552–560. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, R.J. Inhibition of gold and platinum catalysts by reactive intermediates produced in the selective oxidation of alcohols in liquid water. Green Chem. 2011, 13, 3484–3491. [Google Scholar] [CrossRef]
- Liu, X.; He, L.; Liu, Y.M.; Cao, Y. Supported Gold Catalysis: From Small Molecule Activation to Green Chemical Synthesis. Acc. Chem. Res. 2014, 47, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Falletta, E.; Pina, C.D.; Teles, J.H.; Pagliaro, M. Industrial Applications of Gold Catalysis. Angew. Chem. Int. Ed. 2016, 55, 14210–14217. [Google Scholar] [CrossRef]
- Wang, B.; Li, H.; Zhu, J.; Sun, W.; Chen, S. Preparation and characterization of mono-/multi-metallic hydrophobic catalysts for the oxidative esterification of methacrolein to methyl methacrylate. J. Mol. Catal. A Chem. 2013, 379, 322–326. [Google Scholar] [CrossRef]
- Paul, B.; Khatun, R.; Sharma, S.K.; Adak, S.; Singh, G.; Das, D.; Siddiqui, N.; Bhandari, S.; Joshi, V.; Sasaki, T.; et al. Fabrication of Au Nanoparticles Supported on One-Dimensional La2O3 Nanorods for Selective Esterification of Methacrolein to Methyl Methacrylate with Molecular Oxygen. ACS Sustain. Chem. Eng. 2019, 7, 3982–3994. [Google Scholar] [CrossRef]
- Sankar, M.; He, Q.; Engle, R.V.; Sainna, M.A.; Logsdail, A.J.; Roldan, A.; Willock, D.J.; Agarwal, N.; Kiely, C.J.; Hutchings, G.J. Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts. Chem. Rev. 2020, 120, 3890–3938. [Google Scholar] [CrossRef]
- Schwarz, J.A.; Contescu, C.; Contescu, A. Methods for Preparation of Catalytic Materials. Chem. Rev. 1995, 95, 477–510. [Google Scholar] [CrossRef]
- Munnik, P.; Jongh, P.E.D.; Jong, K.P.D. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; Iborra, S.; Mifsud, M.; Velty, A. New one-pot multistep process with multifunctional catalysts: Decreasing the E factor in the synthesis of fine chemicals. Green Chem. 2010, 12, 99–107. [Google Scholar] [CrossRef]
- Denard, C.A.; Hartwig, J.F.; Zhao, H. Multistep One-Pot Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. ACS Catal. 2013, 3, 2856–2864. [Google Scholar] [CrossRef]
- Nagai, K. New developments in the production of methyl methacrylate. Appl. Catal. A 2001, 221, 367–377. [Google Scholar] [CrossRef]
- Guan, Y.; Ma, H.; Chen, W.; Li, M.; Qian, G.; Chen, D.; Zhou, X.; Duan, X. Methyl Methacrylate Synthesis: Thermodynamic Analysis for Oxidative Esterification of Methacrolein and Aldol Condensation of Methyl Acetate. Ind. Eng. Chem. Res. 2020, 59, 17408–17416. [Google Scholar] [CrossRef]
- Yamamatsu, S.; Yamaguchi, T.; Yokota, K.; Nagano, O.; Chono, M.; Aoshima, A. Development of Catalyst Technology for Producing Methyl Methacrylate (MMA) by Direct Methyl Esterification. Catal. Surv. Asia 2010, 14, 124–131. [Google Scholar] [CrossRef]
- Zuo, C.; Tian, Y.; Zheng, Y.; Wang, L.; Fu, Z.; Jiao, T.; Wang, M.; Huang, H.; Li, Y. One step oxidative esterification of methacrolein with methanol over Au-CeO2/γ-Al2O3 catalysts. Catal. Commun. 2019, 124, 51–55. [Google Scholar] [CrossRef]
- Trimpalis, A.; Giannakakis, G.; Cao, S.; Stephanopoulos, M.F. NiAu single atom alloys for the selective oxidation of methacrolein with methanol to methyl methacrylate. Catal. Today 2020, 355, 804–814. [Google Scholar] [CrossRef]
- Gao, J.; Fan, G.; Yang, L.; Cao, X.; Zhang, P.; Li, F. Oxidative Esterification of Methacrolein to Methyl Methacrylate over Gold Nanoparticles on Hydroxyapatite. ChemCatChem 2017, 9, 1230–1241. [Google Scholar] [CrossRef]
- Diao, Y.; He, H.; Yang, P.; Wang, L.; Zhang, S. Optimizing the structure of supported Pd catalyst for direct oxidative esterification of methacrolein with methanol. Chem. Eng. Sci. 2015, 135, 128–136. [Google Scholar] [CrossRef]
- Gangwal, V.R.; Schaaf, J.V.D.; Kuster, B.F.M.; Schouten, J.C. Influence of pH on noble metal catalysed alcohol oxidation: Reaction kinetics and modelling. J. Catal. 2005, 229, 389–403. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Meng, L.; Deng, Y.; Li, Y.; Lei, Y. Palladium-Catalyzed Aerobic Oxidative Direct Esterification of Alcohols. Angew. Chem. Int. Ed. 2011, 50, 5144–5148. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yamaguchi, T.; Matsushita, K.; Iitsuka, C.; Miura, J.; Akaogi, T.; Ishida, H. Aerobic Oxidative Esterification of Aldehydes with Alcohols by Gold−Nickel Oxide Nanoparticle Catalysts with a Core−Shell Structure. ACS Catal. 2013, 3, 1845–1849. [Google Scholar] [CrossRef]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Espriu, B.S.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Solvent-Free Oxidation of Primary Alcohols to Aldehydes Using Au-Pd/TiO2 Catalysts. Science 2006, 311, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Siler, C.G.F.; Madix, R.J.; Friend, C.M. Ag/Au Mixed Sites Promote Oxidative Coupling of Methanol on the Alloy Surface. Chem. Eur. J. 2014, 20, 4646–4652. [Google Scholar] [CrossRef]
- Xiao, Q.; Liu, Z.; Bo, A.; Zavahir, S.; Sarina, S.; Bottle, S.; Riches, J.D.; Zhu, H. Catalytic Transformation of Aliphatic Alcohols to Corresponding Esters in O2 under Neutral Conditions Using Visible-Light Irradiation. J. Am. Chem. Soc. 2015, 137, 1956–1966. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wang, A.; Zhang, T.; Mou, C.Y. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8, 403–416. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Norskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 233, 232–235. [Google Scholar] [CrossRef]
- Haruta, M.; Data, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Murdoch, M.; Waterhouse, G.I.N.; Nadeem, M.A.; Metson, J.B.; Keane, M.A.; Howe, R.F.; Llorca, J.; Idriss, H. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nat. Chem. 2011, 3, 489–492. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Yan, R.; Han, J.; Zhang, S. Gold nanoparticles supported on Ce–Zr oxides for the oxidative esterification of aldehydes to esters. Catal. Sci. Technol. 2015, 5, 3682–3692. [Google Scholar] [CrossRef]
- Hayashi, T.; Inagaki, T.; Itayama, N.; Baba, H. Selective oxidation of alcohol over supported gold catalysts: Methyl glycolate formation from ethylene glycol and methanol. Catal. Today 2006, 117, 210–213. [Google Scholar] [CrossRef]
- Taarning, E.; Madsen, A.T.; Marchetti, J.M.; Egeblad, K.; Christensen, C.H. Oxidation of glycerol and propanediols in methanol over heterogeneous gold catalysts. Green Chem. 2008, 10, 408–414. [Google Scholar] [CrossRef]
- Su, F.Z.; Ni, J.; Sun, H.; Cao, Y.; He, H.Y.; Fan, K.N. Gold Supported on Nanocrystalline b-Ga2O3 as a Versatile Bifunctional Catalyst for Facile Oxidative Transformation of Alcohols, Aldehydes, and Acetals into Esters. Chem. Eur. J. 2008, 14, 7131–7135. [Google Scholar] [CrossRef] [PubMed]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: One pot-base free oxidative esterification of 5-hydroxymethyl-2-furfural into 2,5-dimethylfuroate with gold on nanoparticulated ceria. J. Catal. 2009, 265, 109–116. [Google Scholar] [CrossRef]
- Oliveira, R.L.; Kiyohara, P.K.; Rossi, L.M. Clean preparation of methyl esters in one-step oxidative esterification of primary alcohols catalyzed by supported gold nanoparticles. Green Chem. 2009, 11, 1366–1370. [Google Scholar] [CrossRef]
- Miyamura, H.; Yasukawa, T.; Kobayashi, S. Aerobic oxidative esterification of alcohols catalyzed by polymer-incarcerated gold nanoclusters under ambient conditions. Green Chem. 2010, 12, 776–778. [Google Scholar] [CrossRef]
- Parreira, L.A.; Bogdanchikova, N.; Pestryakov, A.; Zepeda, T.A.; Tuzovskaya, I.; Farias, M.H.; Gusevskaya, E.V. Nanocrystalline gold supported on Fe-, Ti- and Ce-modified hexagonal mesoporous silica as a catalyst for the aerobic oxidative esterification of benzyl alcohol. Appl. Catal. A 2011, 397, 145–152. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, G.; Zou, H.; Cao, Y.; Zhang, Y.; Zhang, R.; Zhang, F.; Xian, M. The base-free and selective oxidative transformation of 1,3-propanediol into methyl esters by different Au/CeO2 catalysts. Green Chem. 2011, 13, 2690–2695. [Google Scholar] [CrossRef]
- Liu, P.; Li, C.; Hensen, E.J.M. Efficient Tandem Synthesis of Methyl Esters and Imines by Using Versatile Hydrotalcite-Supported Gold Nanoparticles. Chem. Eur. J. 2012, 18, 12122–12129. [Google Scholar] [CrossRef] [PubMed]
- Brett, G.L.; Miedziak, P.J.; Dimitratos, N.; Sanchez, J.A.L.; Dummer, N.F.; Tiruvalam, R.; Kiely, C.J.; Knight, D.W.; Taylor, S.H.; Morgan, D.J.; et al. Oxidative esterification of 1,2-propanediol using gold and gold-palladium supported nanoparticles. Catal. Sci. Technol. 2012, 2, 97–104. [Google Scholar] [CrossRef]
- Hutchings, G.J. Catalysis: A Golden Future. Gold Bull. 1996, 29, 123–130. [Google Scholar] [CrossRef]
- Villa, A.; Dimitratos, N.; Thaw, C.E.C.; Hammond, C.; Veith, G.M.; Wang, D.; Manzoli, M.; Prati, L.; Hutchings, G.J. Characterisation of gold catalysts. Chem. Soc. Rev. 2016, 45, 4953–4994. [Google Scholar] [CrossRef]
- Wan, X.; Deng, W.; Zhang, Q.; Wang, Y. Magnesia-supported gold nanoparticles as efficient catalysts for oxidative esterification of aldehydes or alcohols with methanol to methyl esters. Catal. Today 2014, 233, 147–154. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Gulati, U.; Rajesh, U.C.; Rawat, D.S.; Zaleski, J.M. Development of magnesium oxide–silver hybrid nanocatalysts for synergistic carbon dioxide activation to afford esters and heterocycles at ambient pressure. Green Chem. 2020, 22, 3170–3177. [Google Scholar] [CrossRef]
- Wang, Y.; Widmann, D.; Wittmann, M.; Lehnert, F.; Gu, D.; Schuth, F.; Behm, R.J. High activity and negative apparent activation energy in low-temperature CO oxidation—Present on Au/Mg(OH)2, absent on Au/TiO2. Catal. Sci. Technol. 2017, 7, 4145–4161. [Google Scholar] [CrossRef]
- Feng, G.; Han, W.; Wang, Z.; Li, F.; Xue, W. Highly Reducible Nanostructured CeO2 for CO Oxidation. Catalysts 2018, 8, 535. [Google Scholar] [CrossRef]
- Fu, Q.; Kudriavtseva, S.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Gold–ceria catalysts for low-temperature water-gas shift reaction. Chem. Eng. J. 2003, 93, 41–53. [Google Scholar] [CrossRef]
- Saito, M.; Itoh, M.; Iwamoto, J.; Li, C.-Y.; Machida, K. Synergistic effect of MgO and CeO2 as a support for ruthenium catalysts in ammonia synthesis. Catal. Lett. 2006, 106, 107–110. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Z.; Zhu, Q.; Han, H.; Yang, Y.; Han, B. Highly efficient hydrogenation of carbon dioxide to methyl formate over supported gold catalysts. Green Chem. 2015, 17, 1467–1472. [Google Scholar] [CrossRef]
- Chen, Y.W.; Brichkov, A.S.; Kozik, V.V. Nanosized Au Catalysts Supported on Mg(OH)2-CeO2 for Preferential Oxidation of CO in Hydrogen Stream. Mod. Res. Catal. 2019, 8, 11–23. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Y.; Zheng, Y.; Wang, M.; Zuo, C.; Huang, H.; Yin, D.; Fu, Z.; Tan, J.; Zhou, Z. Nano-Au/MCeOx Catalysts for the Direct Oxidative Esterification of Methylacrolein to Methyl Esters. Ind. Eng. Chem. Res. 2019, 58, 19397–19405. [Google Scholar] [CrossRef]
- Montero, S.G.; Alshammari, H.; Dalebout, R.; Nowicha, E.; Morgan, D.J.; Shaw, G.; He, Q.; Sankar, M. Deactivation studies of bimetallic AuPd nanoparticles supported on MgO during selective aerobic oxidation of alcohols. Appl. Catal. A 2017, 546, 58–66. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Zou, G.; Zhang, T.; Wei, D.; Xu, X.; Guan, Y.; Zheng, A. A controlled synthesis method of alkyl methacrylate block copolymers via living anionic polymerization at ambient temperature. RSC Adv. 2019, 9, 16049–16056. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Au Content (wt. %) (a) | Au Size (nm) (b) | Au Dispersion (%) (c) | SBET (m2 g−1) (d) | Dp (nm) (e) | Vtot (cm3 g−1) (f) | Amount of Acidic Sites (mmol g−1) (g) | Amount of Basic Sites (mmol g−1) (h) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AT | AW | AS | BT | Bw | BM | BS | |||||||
| 3AuM | 2.80 | 3.8 | 30.8 | 187 | 3.8 | 0.58 | 0.401 | 0 | 0.401 | 0.028 | 0.007 | 0.014 | 0.007 |
| 3AuC | 2.63 | 5.0 | 23.9 | 60 | 3.4 | 0.13 | 0 | 0 | 0 | 0.018 | 0.006 | 0.003 | 0.009 |
| 3AuCM | 2.36 | 4.1 | 28.5 | 97 | 3.3 | 0.23 | 0.154 | 0.009 | 0.145 | 0.030 | 0.013 | 0.010 | 0.007 |
| Entry | Catalyst | Weight (g) | M/R (b) Ratio (%) | MACR Conv. (%) | MMA Sel. (%) | Hemiacetal Sel. (%) | Acetal Sel. (%) | YMMA (%) (c) | Carbon Balance (%) (d) | TON (e) | STY (f) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MgO | 1.5 | - | 30.9 | 0.0 | 40.2 | 59.8 | 0 | 22 | - | - |
| 2 | CeO2 | 1.5 | - | 61.1 | 1.7 | 81.2 | 17.2 | 1.0 | 38 | - | - |
| 3 | CeO2-MgO | 1.5 | - | 33.8 | 2.6 | 54.1 | 43.4 | 0.9 | 10 | - | - |
| 4 | 3AuCM | 1.5 | 0.315 | 82.7 | 97.6 | 1.6 | 0.8 | 80.7 | 88 | 1170 | 1142 |
| 5 (g) | 3AuCM | 1.5 | 0.315 | 52.1 | 96.8 | 2.7 | 0.5 | 50.4 | 88 | 737 | 713 |
| 6 | 3AuCM | 3.0 | 0.630 | 93.3 | 98.2 | 1.8 | 0.0 | 91.6 | 81 | 660 | 648 |
| 7 (h) | 3AuCM | 1.5 | 0.315 | 40.0 | 89.9 | 4.5 | 5.5 | 36.0 | 80 | 566 | 510 |
| 8 | 3AuC | 1.5 | 0.315 | 77.7 | 82.5 | 7.4 | 10.1 | 64.1 | 25 | 1203 | 993 |
| 9 | 3AuC | 3.0 | 0.630 | 86.3 | 80.1 | 7.2 | 12.7 | 69.1 | 20 | 668 | 535 |
| 10 | 3AuM | 1.5 | 0.315 | 66.5 | 94.3 | 2.3 | 3.4 | 62.7 | 71 | 735 | 693 |
| 11 | 3AuM | 3.0 | 0.630 | 83.7 | 96.7 | 2.1 | 1.2 | 80.9 | 82 | 463 | 447 |
| 12 | 2AuM | 1.5 | 0.210 | 62.5 | 87.3 | 2.6 | 10.1 | 54.5 | 75 | 1433 | 1250 |
| 13 | 1AuM | 1.5 | 0.105 | 31.7 | 84.3 | 7.7 | 7.9 | 26.7 | 64 | 1382 | 1164 |
| Entry | Aldehyde | Alcohol | Product | Conversion/Selectivity (%) (a) |
|---|---|---|---|---|
| 1 |  | methanol |  | 82.7/97.6 |
| 2 (b) |  | n-butanol |  | 42.9/96.4 |
| 3 (c) |  | 1-propanol |  | 31.4/87.5 |
| 4 (d) |  | isopropyl alcohol |  | 22.3/80.7 |
| 5 (e) |  | methanol |  | 59.4/100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.; Kwon, S.; Kim, N.; Na, K. A Multifunctional Au/CeO2-Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters. Nanomaterials 2021, 11, 1536. https://doi.org/10.3390/nano11061536
Lim S, Kwon S, Kim N, Na K. A Multifunctional Au/CeO2-Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters. Nanomaterials. 2021; 11(6):1536. https://doi.org/10.3390/nano11061536
Chicago/Turabian StyleLim, Seulgi, Seungdon Kwon, Nagyeong Kim, and Kyungsu Na. 2021. "A Multifunctional Au/CeO2-Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters" Nanomaterials 11, no. 6: 1536. https://doi.org/10.3390/nano11061536
APA StyleLim, S., Kwon, S., Kim, N., & Na, K. (2021). A Multifunctional Au/CeO2-Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters. Nanomaterials, 11(6), 1536. https://doi.org/10.3390/nano11061536