
Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity

 ,
, 
 , , ,
, , , 
Abstract
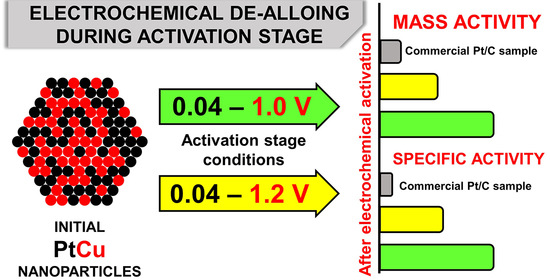
1. Introduction
2. Materials and Methods
3. Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banham, D.W.H.; Ye, S. Current Status and Future Development of Catalyst Materials and Catalyst Layers for Proton Exchange Membrane Fuel Cells: An Industrial Perspective. ACS Energy Lett. 2017, 2, 629–638. [Google Scholar] [CrossRef]
- Kongkanand, A.; Mathias, M.F. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Holton, O.T.; Stevenson, J.W. The Role of Platinum in Proton Exchange Membrane Fuel Cells. Platin. Met. Rev. 2013, 57, 259–271. [Google Scholar] [CrossRef]
- Strasser, P. Free Electrons to Molecular Bonds and Back: Closing the Energetic Oxygen Reduction (ORR)−Oxygen Evolution (OER) Cycle Using Core−Shell Nanoelectrocatalysts. Acc. Chem. Res. 2016, 49, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.K.; Hong, Y.; Kim, B.; Choi, S.-I.; Lee, K. Pt-Cu Based Nanocrystals as the Promising Catalysts for Various Electrocatalytic Reactions. J. Mat. Chem. A 2019, 7, 17183–17203. [Google Scholar] [CrossRef]
- Hussain, S.; Erikson, H.; Kongi, N.; Sarapuu, A.; Solla-Gullón, J.; Maia, G.; Kannan, A.M.; Alonso-Vante, N.; Tammeveski, K. Oxygen reduction reaction on nanostructured Pt-based electrocatalysts: A review. Int. J. Hydrogen Energy 2020, 45, 31775–31797. [Google Scholar] [CrossRef]
- Zhu, F.; Wu, A.; Luo, L.; Wang, C.; Yang, F.; Wei, G.; Xia, G.; Yin, J.; Zhang, J. The Asymmetric Effects of Cu2+ Contamination in a Proton Exchange Membrane Fuel Cell (PEMFC). Fuel Cells 2020, 2, 196–202. [Google Scholar] [CrossRef]
- Moriau, L.J.; Hrnjić, A.; Pavlišič, A.; Kamšek, A.R.; Petek, U.; Ruiz-Zepeda, F.; Šala, M.; Pavko, L.; Šelih, V.S.; Bele, M.; et al. Resolving the nanoparticles’ structure-property relationships at the atomic level: A study of Pt-based electrocatalysts. iScience 2021, 24, 102. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.Y.; Yoo, J.M.; Sung, Y.-E. Highly Durable and Active Pt-Based Nanoscale Design for Fuel-Cell Oxygen-Reduction Electrocatalysts. Adv. Mater. 2018, 30, 1704123. [Google Scholar] [CrossRef]
- Yang, H. Platinum-Based Electrocatalysts with Core-Shell Nanostructures. Angew. Chem. Int. Ed. 2011, 50, 2674–2676. [Google Scholar] [CrossRef]
- Neergat, M.; Rahul, R. Unsupported Cu-Pt Core-Shell Nanoparticles: Oxygen Reduction Reaction (ORR) Catalyst with Better Activity and Reduced Precious Metal Content. J. Electrochem. Soc. 2012, 159, 234–241. [Google Scholar] [CrossRef]
- Lyu, X.; Jia, Y.; Mao, X.; Li, D.; Li, G.; Zhuang, L.; Wang, X.; Yang, D.; Wang, Q.; Du, A.; et al. Gradient-Concentration Design of Stable Core–Shell Nanostructure for Acidic Oxygen Reduction Electrocatalysis. Adv. Mater. 2020, 32, 2003493. [Google Scholar] [CrossRef]
- Alekseenko, A.A.; Guterman, V.E.; Belenov, S.V.; Menshikov, V.S.; Tabachkova, N.Y.; Safronenko, O.I.; Moguchikh, E.A. Pt/C electrocatalysts based on the nanoparticles with the gradient structure. Int. J. Hydrogen Energy 2018, 43, 3676–3687. [Google Scholar] [CrossRef]
- Chen, G.; Yang, X.; Xie, Z.; Zhao, F.; Zhou, Z.; Yuan, Q. Hollow PtCu octahedral nanoalloys: Efficient bifunctional electrocatalysts towards oxygen reduction reaction and methanol oxidation reaction by regulating near-surface composition. J. Coll. Inter. Sci. 2020, 562, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.S.; Zhang, G.; Lu, W.T.; Qin, X.P.; Shao, Z.G.; Yi, B.L. Preparation of hollow PtCu nanoparticles as high-performance electrocatalysts for oxygen reduction reaction in the absence of a surfactant. RSC Adv. 2016, 6, 39993–40001. [Google Scholar] [CrossRef]
- Zhou, X.; Gan, Y.; Du, J.; Tian, D.; Zhang, R.; Yang, C.; Dai, Z. A review of hollow Pt-based nanocatalysts applied in proton exchange membrane fuel cells. J. Power Source 2013, 232, 310–322. [Google Scholar] [CrossRef]
- Niu, H.J.; Chen, H.Y.; Wen, G.L.; Feng, J.J.; Zhang, Q.L.; Wang, A.J. One-pot solvothermal synthesis of three-dimensional hollow PtCu alloyed dodecahedron nanoframes with excellent electrocatalytic performances for hydrogen evolution and oxygen reduction. J. Coll. Inter. Sci. 2018, 539, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, L.; Li, H.-H.; Yu, S.-H.; Heggen, M.; Strasser, P. Octahedral PtNi Nanoparticle Catalysts: Exceptional Oxygen Reduction Activity by Tuning the Alloy Particle Surface Composition. Nano Lett. 2012, 12, 5885–5889. [Google Scholar] [CrossRef]
- Song, T.; Xue, H.; Guo, N.; Sun, J.; Qin, L.; Guo, L.; Huang, K.; He, F.; Wang, Q. Dual-Modulation of Electronic Structure and Active Sites of PtCu Nanodendrites by Surface Nitridation to Achieve Efficient Methanol Electrooxidation and Oxygen Reduction Reaction. Chem. Comm. 2020, 56, 7136–7139. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Pukazhselvan, D.; da Silva, E.L.; Santos, M.C.; Meng, L.; Ramasamy, D.; Jothi, S.; Graca, V.; Shi, S. Highly branched Pt Cu nanodandelion with high activity for oxygen reduction reaction. Int. J. Hydrogen Energy 2019, 44, 174–179. [Google Scholar] [CrossRef]
- Alia, S.M.; Jensen, K.; Contreras, C.; Garzon, F.; Pivovar, B.; Yan, Y. Platinum Coated Copper Nanowires and Platinum Nanotubes as Oxygen Reduction Electrocatalysts. ACS Catal. 2013, 3, 358–362. [Google Scholar] [CrossRef]
- Hodnik, N.; Jeyabharathi, C.; Meier, J.C.; Kostka, A.; Phani, K.L.; Rečnik, A.; Bele, M.; Hočevar, S.; Gaberšček, M.; Mayrhofer, K.J.J. Effect of ordering of PtCu3 nanoparticle structure on the activity and stability for the oxygen reduction reaction. Phys. Chem. Chem. Phys. 2014, 16, 13610–13615. [Google Scholar] [CrossRef] [PubMed]
- Barim, S.B.; Bozbag, S.E.; Deljoo, B.; Aindow, M.; Erkey, C. Highly Active Carbon Supported PtCu Electrocatalysts for PEMFCs by in situ Supercritical Deposition Coupled with Electrochemical Dealloying. Fuel Cells 2020, 20, 285–299. [Google Scholar] [CrossRef]
- Gan, L.; Heggen, M.; O’Malley, R.; Theobald, B.; Strasser, P. Understanding and Controlling Nanoporosity Formation for Improving the Stability of Bimetallic Fuel Cell Catalysts. Nano Lett. 2013, 13, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, Y.; Zhu, J.; Liu, S.; Muller, D.A.; Abruña, H.D. Morphology and Activity Tuning of Cu3Pt/C Ordered Intermetallic Nanoparticles by Selective Electrochemical Dealloying. Nano Lett. 2015, 15, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Zepeda, F.; Gatalo, M.; Pavlišič, A.; Dražić, G.; Jovanovič, P.; Bele, M.; Gaberšček, M.; Hodnik, N. Atomically Resolved Anisotropic Electrochemical Shaping of Nano-Electrocatalyst. Nano Lett. 2019, 19, 4919–4927. [Google Scholar] [CrossRef]
- Wang, D.; Yu, Y.; Xin, H.L.; Hovden, R.; Ercius, P.; Mundy, J.A.; Chen, H.; Richard, J.H.; Muller, D.A.; DiSalvo, F.J.; et al. Tuning Oxygen Reduction Reaction Activity via Controllable Dealloying: A Model Study of Ordered Cu3Pt/C Intermetallic Nanocatalysts. Nano Lett. 2012, 12, 5230–5238. [Google Scholar] [CrossRef] [PubMed]
- Marcu, A.; Toth, G.; Srivastava, R.; Strasser, P. Preparation, characterization and degradation mechanisms of PtCu alloy nanoparticles for automotive fuel cells. J. Power Source 2012, 208, 288–295. [Google Scholar] [CrossRef]
- Lopes, P.P.; Tripkovic, D.; Martins, P.F.B.D.; Strmcnik, D.; Ticianelli, E.A.; Stamenkovic, V.R.; Markovic, N.M. Dynamics of electrochemical Pt dissolution at atomic and molecular levels. J. Electroanal. Chem. 2018, 819, 123–129. [Google Scholar] [CrossRef]
- Sharma, R.; Simonsen, S.B.; Morgen, P.; Andersen, S.M. Inhibition of Ostwald ripening through surface switching species during potentiodynamic dissolution of platinum nanoparticles as an efficient strategy for platinum group metal (PGM) recovery. Electrochim. Acta 2019, 321, 134662. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaker, D.E.; Ziegelbauer, J.M.; Ramaswamy, N.; Halder, A.; Mukerjee, S. Fundamental Aspects of ad-Metal Dissolution and Contamination in Low and Medium Temperature Fuel Cell Electrocatalysis: A Cu Based Case Study Using In Situ Electrochemical X-ray Absorption Spectroscopy. J. Phys. Chem. C 2013, 117, 4585–4596. [Google Scholar] [CrossRef]
- Cai, X.; Lin, R.; Shen, D.D.; Zhu, Y. Gram-scale Synthesis of Well-dispersed Shape-controlled Pt-Ni/C as High-performance Catalysts for the Oxygen Reduction Reaction. ACS Appl. Mat. Interfaces 2019, 11, 29689–29697. [Google Scholar] [CrossRef] [PubMed]
- Menshchikov, V.S.; Alekseenko, A.A.; Guterman, V.E.; Nechitailov, A.; Glebova, N.B.; Tomasov, A.A.; Spiridonova, O.A.; Belenov, S.V.; Zelenina, N.K.; Safronenko, O.I. Effective Platinum-Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell. Nanomaterials 2020, 10, 742. [Google Scholar] [CrossRef] [PubMed]
- Pavlets, A.S.; Alekseenko, A.A.; Tabachkova, N.Y.; Safronenko, O.I.; Nikulin, A.Y.; Alekseenko, D.V.; Guterman, V.E. A novel strategy for the synthesis of Pt–Cu uneven nanoparticles as an efficient electrocatalyst toward oxygen reduction. Int. J. Hydrogen Energy 2021, 46, 5355–5368. [Google Scholar] [CrossRef]
- Garcia-Cardona, J.; Sires, I.; Alcaide, F.; Brillas, E.; Centellas, F.; Cabot, P.L. Electrochemical performance of carbon-supported Pt(Cu) electrocatalysts for low-temperature fuel cells. Int. J. Hydrogen Energy 2020, 45, 20582–20593. [Google Scholar] [CrossRef]
- El-Deeb, H.; Bron, M. Electrochemical Dealloying of PtCu/CNT Electrocatalysts Synthesized by NaBH4-Assisted Polyol-Reduction: Influence of Preparation Parameters on Oxygen Reduction Activity. Electrochim. Acta 2015, 164, 315–322. [Google Scholar] [CrossRef]
- Liu, Z.; Koh, S.; Yu, C.; Strasser, P. Synthesis, Dealloying, and ORR Electrocatalysis of PDDA-Stabilized Cu-Rich Pt Alloy Nanoparticles. J. Electrochem. Soc. 2007, 154, B1192. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, D.; Yamamoto, Y.; Ichino, R.; Okido, M. Preparation of Cu nanoparticles with NaBH4 by aqueous reduction method. Transactions Nonferr. Met. Soc. China 2012, 2, 117–123. [Google Scholar]
- Pryadchenko, V.V.; Srabionyan, V.V.; Kurzin, A.A.; Bulat, N.V.; Shemet, D.B.; Avakyan, L.A.; Belenov, S.V.; Volochaev, V.A.; Zizak, I.; Guterman, V.E.; et al. Bimetallic PtCu core-shell nanoparticles in PtCu/C electrocatalysts: Structural and electrochemical characterization. Appl. Catal. A Gen. 2016, 525, 226–236. [Google Scholar] [CrossRef]
- Favilla, P.C.; Acosta, J.J.; Schvezov, C.E.; Sercovich, D.J.; Collet-Lacos, J.R. Size control of carbon-supported platinum nanoparticles made using polyol method for low temperature fuel cells. Chem. Eng. Sci. 2013, 101, 27–34. [Google Scholar] [CrossRef]
- Wang, C.; van der Vliet, D.; Chang, K.-C.; You, H.; Strmcnik, D.; Schlueter, J.A.; Markovic, N.M.; Stamenkovic, V.R. Monodisperse Pt3Co Nanoparticles as a Catalyst for the Oxygen Reduction Reaction: Size-Dependent Activity. J. Phys. Chem. C 2009, 113, 19365–19368. [Google Scholar] [CrossRef]
- Leontyev, I.N.; Kuriganova, A.B.; Allix, M.; Rakhmatullin, A.; Timoshenko, P.E.; Maslova, O.A.; Mikheykin, A.S.; Smirnova, N.V. On the Evaluation of the Average Crystalline Size and Surface Area of Platinum Catalyst Nanoparticles. Phys. Status Solidi Basic Res. 2018, 255, 1800240. [Google Scholar] [CrossRef]
- Baldizzone, C.; Gan, L.; Hodnik, N.; Keeley, G.P.; Kostka, A.; Heggen, M.; Strasser, P.; Mayrhofer, K.J.J. Stability of Dealloyed Porous Pt/Ni Nanoparticles. ACS Catal. 2015, 5, 5000–5007. [Google Scholar] [CrossRef]
- Inaba, M.; Quinson, J.; Arenz, M. pH matters: The influence of the catalyst ink on the oxygen reduction activity determined in thin film rotating disk electrode measurements. J. Power Source 2017, 353, 19–27. [Google Scholar] [CrossRef]
- Schlögl, K.; Mayrhofer, K.J.J.; Hanzlik, M.; Arenz, M. Identical-location TEM investigations of Pt/C electrocatalyst degradation at elevated temperatures. J. Electroanal. Chem. 2011, 662, 355–360. [Google Scholar] [CrossRef]
- Martens, S.; Asen, L.; Ercolano, G.; Dionigi, F.; Zalitis, C.; Hawkins, A.; Bonastre, A.M.; Seidl, L.; Knool, A.C.; Sharman, J.; et al. A comparison of rotating disc electrode, floating electrode technique and membrane electrode assembly measurements for catalyst testing. J. Power Source 2018, 392, 274–284. [Google Scholar] [CrossRef]
- Zhu, H.; Li, X.; Wang, F. Synthesis and characterization of Cu@Pt/C core-shell structured catalysts for proton exchange membrane fuel cell. Int. J. Hydrogen Energy 2011, 36, 9151–9154. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Zhou, H.-J.; Sun, P.-C.; Chen, T.-H. Exceptional methanol electro-oxidation activity by bimetallic concave and dendritic Pt-Cu nanocrystals catalysts. J. Power Source 2014, 245, 663–670. [Google Scholar] [CrossRef]
- Alekseenko, A.A.; Belenov, S.V.; Menshikov, V.S.; Guterman, V.E. Pt(Cu)/C Electrocatalysts with Low Platinum Content. Russ. J. Electrochem. 2018, 54, 415–425. [Google Scholar] [CrossRef]
- Garsany, Y.; Ge, J.; St-Pierre, J.; Rocheleau, R.; Swider-Lyons, K.E. Analytical Procedure for Accurate Comparison of Rotating Disk Electrode Results for the Oxygen Reduction Activity of Pt/C. J. Electrochem. Soc. 2014, 161, F628–F640. [Google Scholar] [CrossRef]
- Wei, C.; Rao, R.R.; Peng, J.; Huang, B.; Stephens, I.E.L.; Risch, M.; Xu, Z.J.; Shao-Horn, Y. Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Fu, G.; Chen, Z.; Tang, Y.; Wang, Y.; Huang, S. Novel strategy for the synthesis of hollow Pt–Cu tetradecahedrons as an efficient electrocatalyst toward methanol oxidation. CrystEngComm 2019, 21, 1903–1909. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Sun, L.; Wang, W.; Chen, Z. Enhanced electrocatalytic activity of PtCu bimetallic nanoparticles on CeO2/carbon nanotubes for methanol electro-oxidation. Int. J. Hydrogen Energy 2020, 45, 8558–8567. [Google Scholar] [CrossRef]
- Wu, G.; Li, D.; Dai, C.; Wang, D.; Li, N. Well-Dispersed High-Loading Pt Nanoparticles Suppoted by Shell-Core Nanostructured Carbon for Methanol Electrooxidation. Langmuir 2008, 24, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Khorasani-Motlagh, M.; Noroozifar, M.; Ekrami-Kakhki, M.-S. Investigation of the nanometals (Ni and Sn) in platinum binary and ternary electrocatalysts for methanol electrooxidation. Int. J. Hydrogen Energy 2011, 36, 11554–11563. [Google Scholar] [CrossRef]
- Lei, W.; Li, M.; He, L.; Meng, X.; Mu, Z.; Yu, Y.; Ross, F.M.; Yang, W. A general strategy for bimetallic Pt-based nano-branched structures as highly active and stable oxygen reduction and methanol oxidation bifunctional catalysts. Nano Res. 2020, 13, 638–645. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Z.-B.; Li, J.-L.; Zhang, J.-J.; Sui, X.-L.; Zhang, L.-M. Hybrid of carbon-supported Pt nanoparticles and three dimensional graphene aerogel as high stable electrocatalyst for methanol electrooxidation. Electrochim. Acta 2016, 189, 175–183. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial Composition | Potential Range at the Activation Stage, V | Composition after the Activation Stage | ESA, m2g−1 (Pt) | Ik, Ag−1 (Pt) | Ik, Am−2 (Pt) | Number of ē | E1/2, V |
|---|---|---|---|---|---|---|---|---|
| PtCu/C | PtCu1.3 | 0.04–1.20 | PtCu0.25 | 41 | 417 | 10.1 | 4.3 | 0.93 |
| 0.04–1.00 | PtCu0.24 | 43 | 878 | 20.3 | 3.9 | 0.95 | ||
| JM20 | Pt | 0.04–1.20 | - | 80 | 181 | 2.2 | 4.3 | 0.92 |
| 0.04–1.00 | - | 75 | 184 | 2.2 | 4.2 | 0.93 |
| Sample | Potential Range at the Activation Stage, V | QCH3OH, C/g(Pt) | Imax, Ag−1(Pt) | Eonset, V | Iinitial, Ag−1(Pt) | Ifinal, Ag−1(Pt) |
|---|---|---|---|---|---|---|
| PtCu/C | 0.04–1.00 | 18,703 | 1236 | 0.51 | 650 | 352 |
| PtCu/C (1.2 V) | 0.04–1.20 | 16,647 | 1084 | 0.50 | 596 | 328 |
| JM20 | 0.04–1.20 | 5648 | 350 | 0.51 | 320 | 127 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlets, A.; Alekseenko, A.; Menshchikov, V.; Belenov, S.; Volochaev, V.; Pankov, I.; Safronenko, O.; Guterman, V. Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity. Nanomaterials 2021, 11, 1499. https://doi.org/10.3390/nano11061499
Pavlets A, Alekseenko A, Menshchikov V, Belenov S, Volochaev V, Pankov I, Safronenko O, Guterman V. Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity. Nanomaterials. 2021; 11(6):1499. https://doi.org/10.3390/nano11061499
Chicago/Turabian StylePavlets, Angelina, Anastasia Alekseenko, Vladislav Menshchikov, Sergey Belenov, Vadim Volochaev, Ilya Pankov, Olga Safronenko, and Vladimir Guterman. 2021. "Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity" Nanomaterials 11, no. 6: 1499. https://doi.org/10.3390/nano11061499
APA StylePavlets, A., Alekseenko, A., Menshchikov, V., Belenov, S., Volochaev, V., Pankov, I., Safronenko, O., & Guterman, V. (2021). Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity. Nanomaterials, 11(6), 1499. https://doi.org/10.3390/nano11061499