
Influence of Calcium Silicate and Hydrophobic Agent Coatings on Thermal, Water Barrier, Mechanical and Biodegradation Properties of Cellulose


Abstract
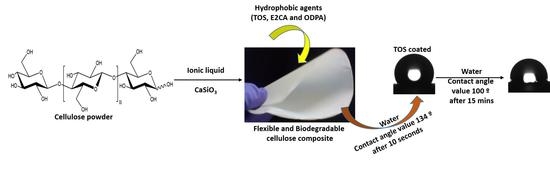
1. Introduction
2. Materials and Methods
2.1. Experimental Details
2.1.1. Materials
2.1.2. Preparation of Cellulose–CaSiO3 Composites
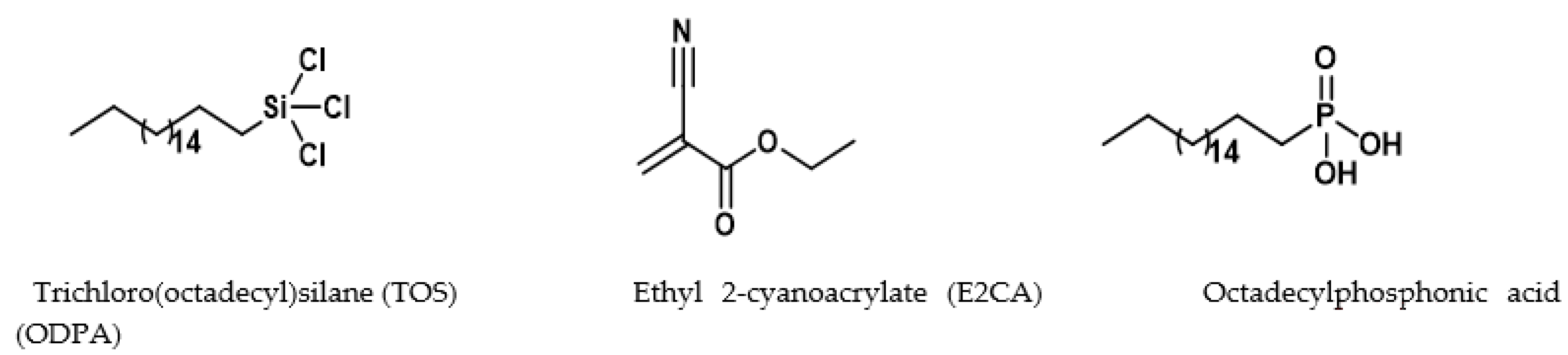
2.2. Surface Modification of Cellulose–CaSiO3 Composite Film via Hydrophobic Agent Coating
- (i)
- Cellulose–CaSiO3 composite surface coating with TOS
- (ii)
- Cellulose–CaSiO3 composite surface coating with ODPA and E2CA
2.3. Characterization Techniques
2.4. Biodegradability Studies
3. Results and Discussion
3.1. Preparation of Cellulose–CaSiO3 Composites
3.2. Characterization of Cellulose–CaSiO3 Composites
Effect of CaSiO3 on Flame Retardant Properties of Cellulose–CaSiO3 Composites
3.3. Surface Coatings of Cellulose–CaSiO3 Composites
3.3.1. Preparation of Coated Cellulose–CaSiO3 Composites
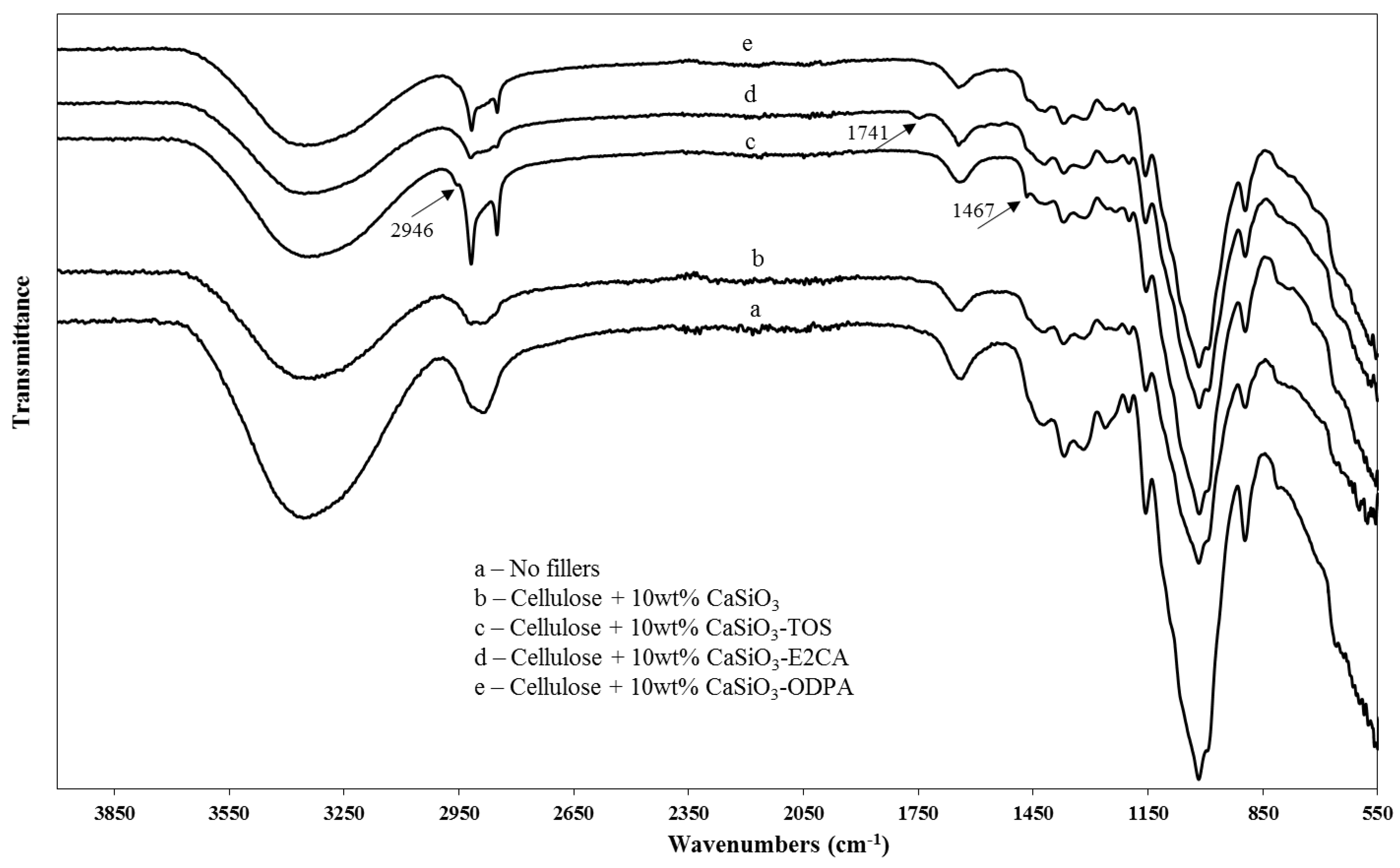
3.3.2. ATR-FTIR of Uncoated and Coated Cellulose–CaSiO3 Composites
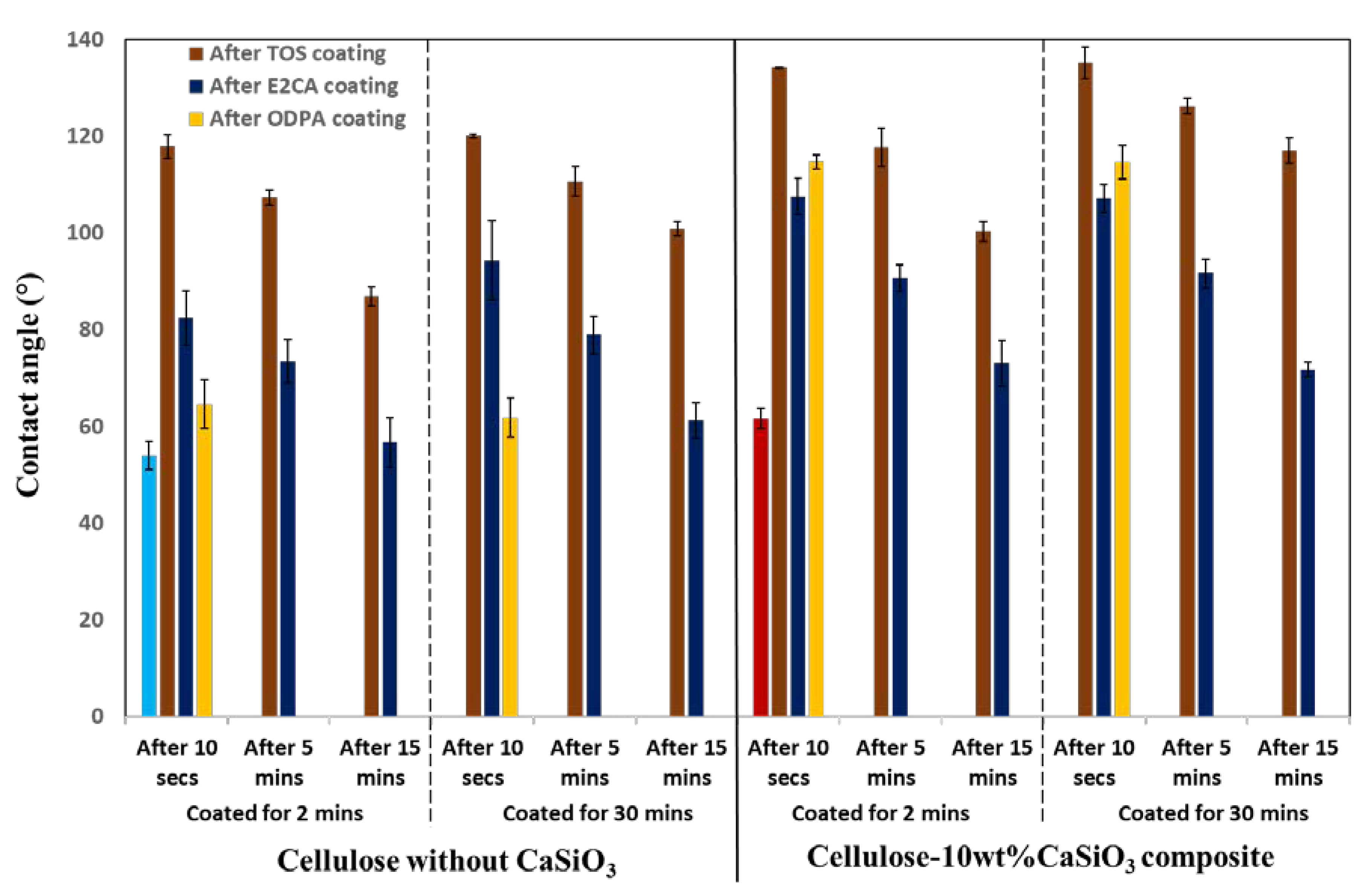
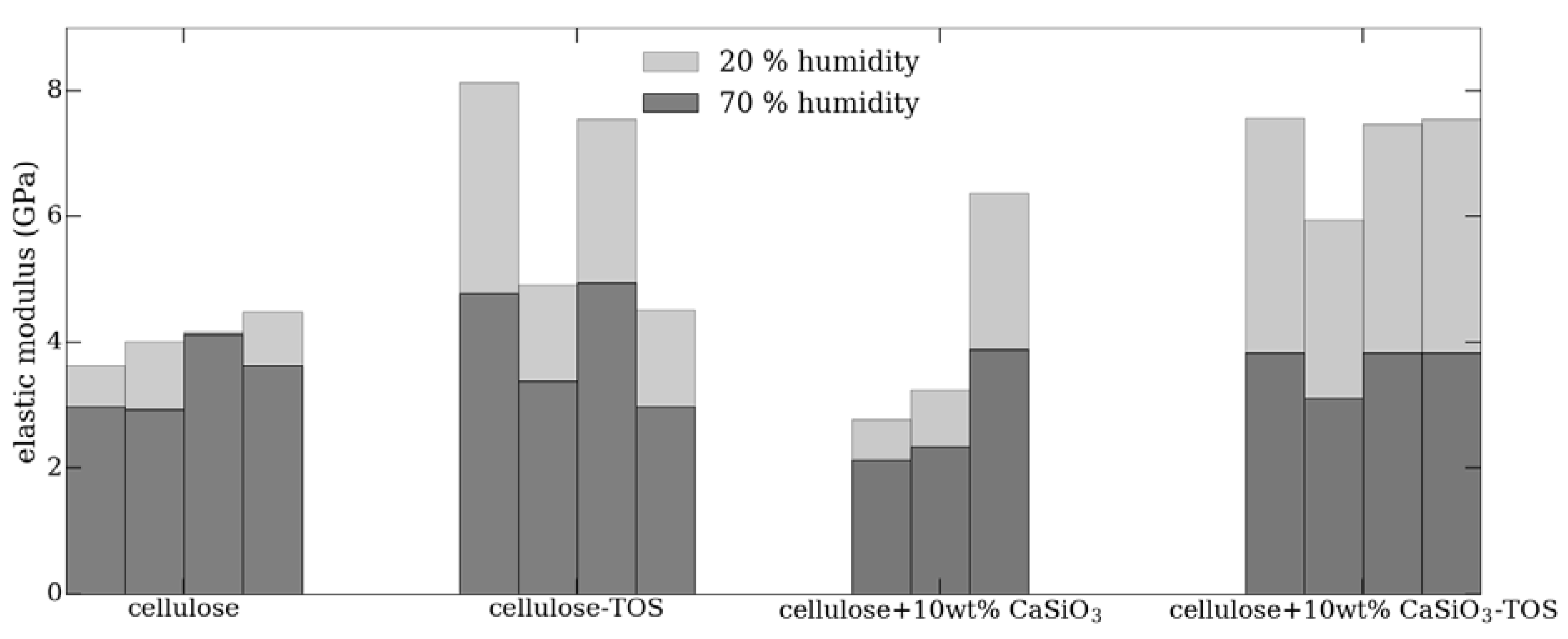
3.3.3. Water Resistance Characteristics of Cellulose–CaSiO3 Composites
3.3.4. Surface Morphology Analysis of Uncoated and Coated Cellulose–CaSiO3 Composites by SEM
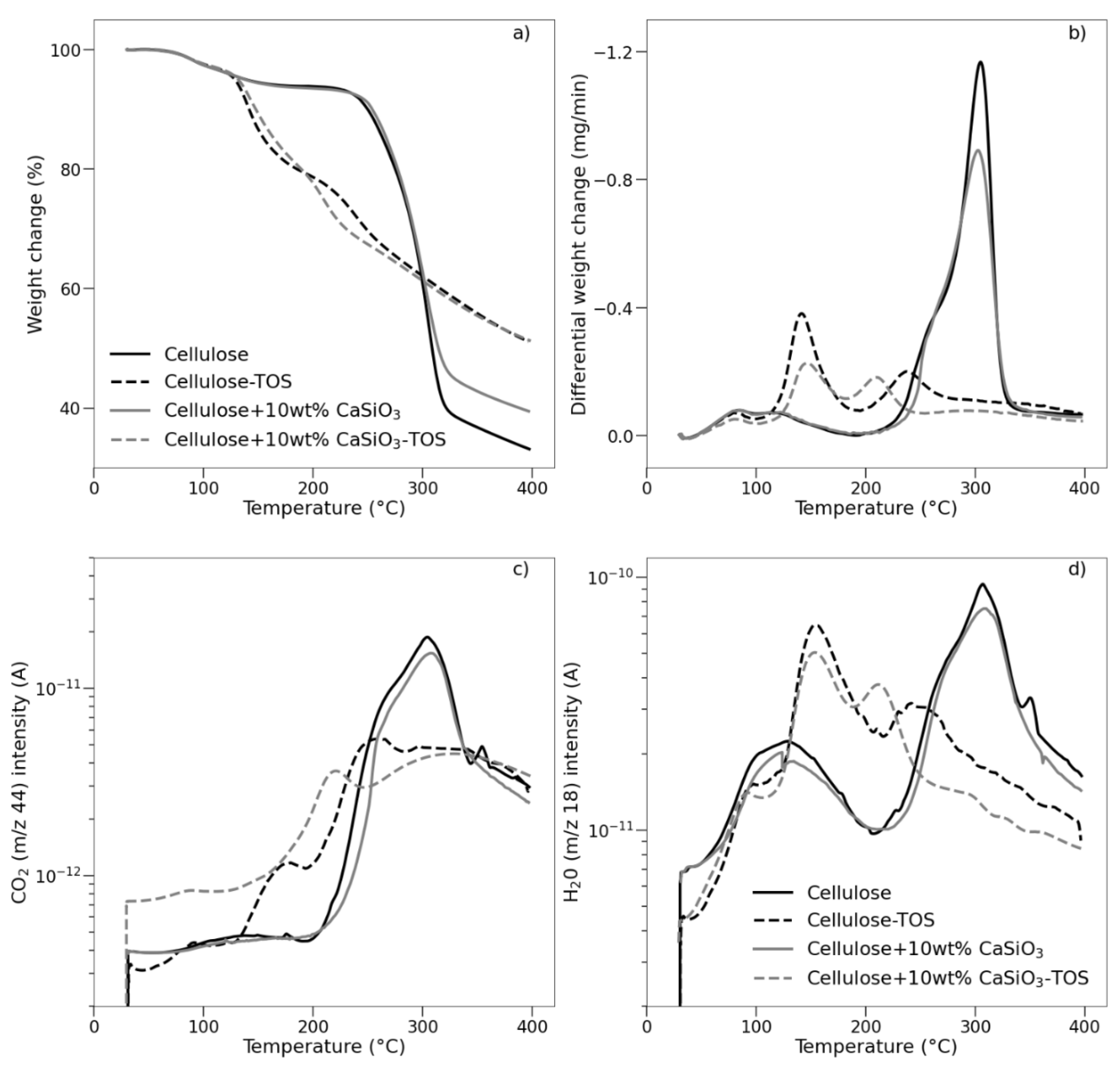
3.3.5. Thermal Stability Studies of Uncoated and Coated Cellulose–CaSiO3 Composites
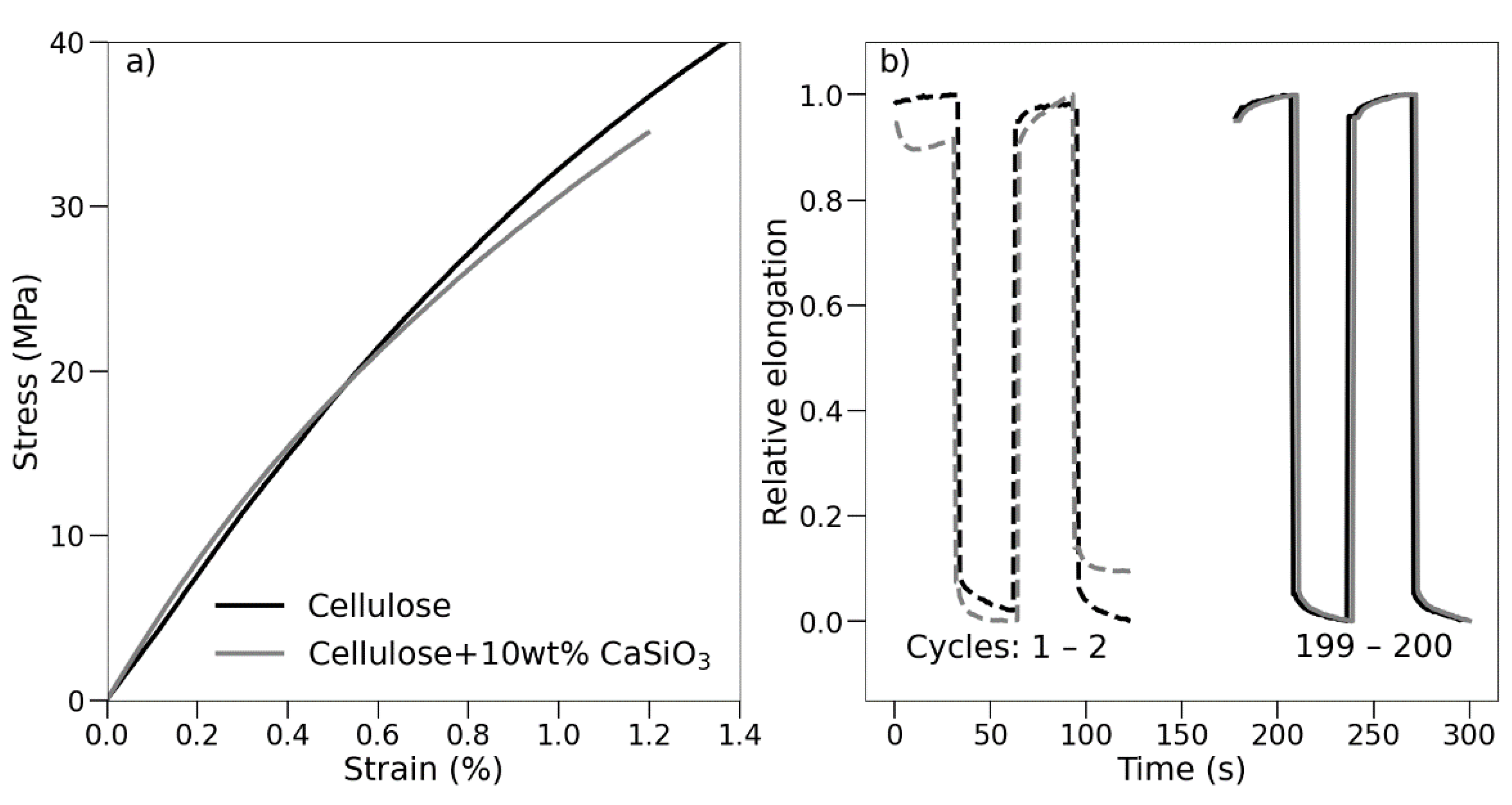
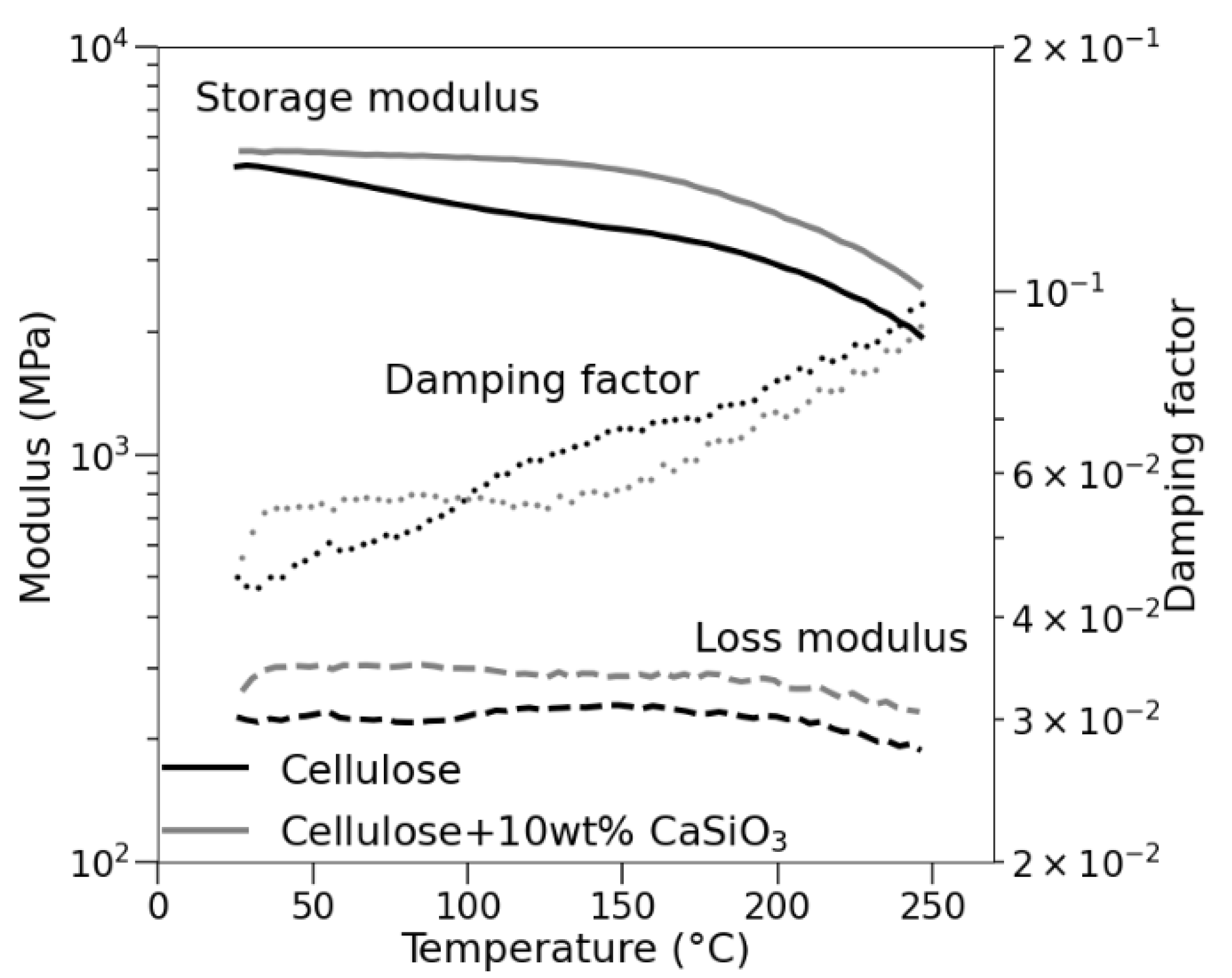
3.3.6. Mechanical Property Studies of Uncoated and Coated Cellulose–CaSiO3 Composites
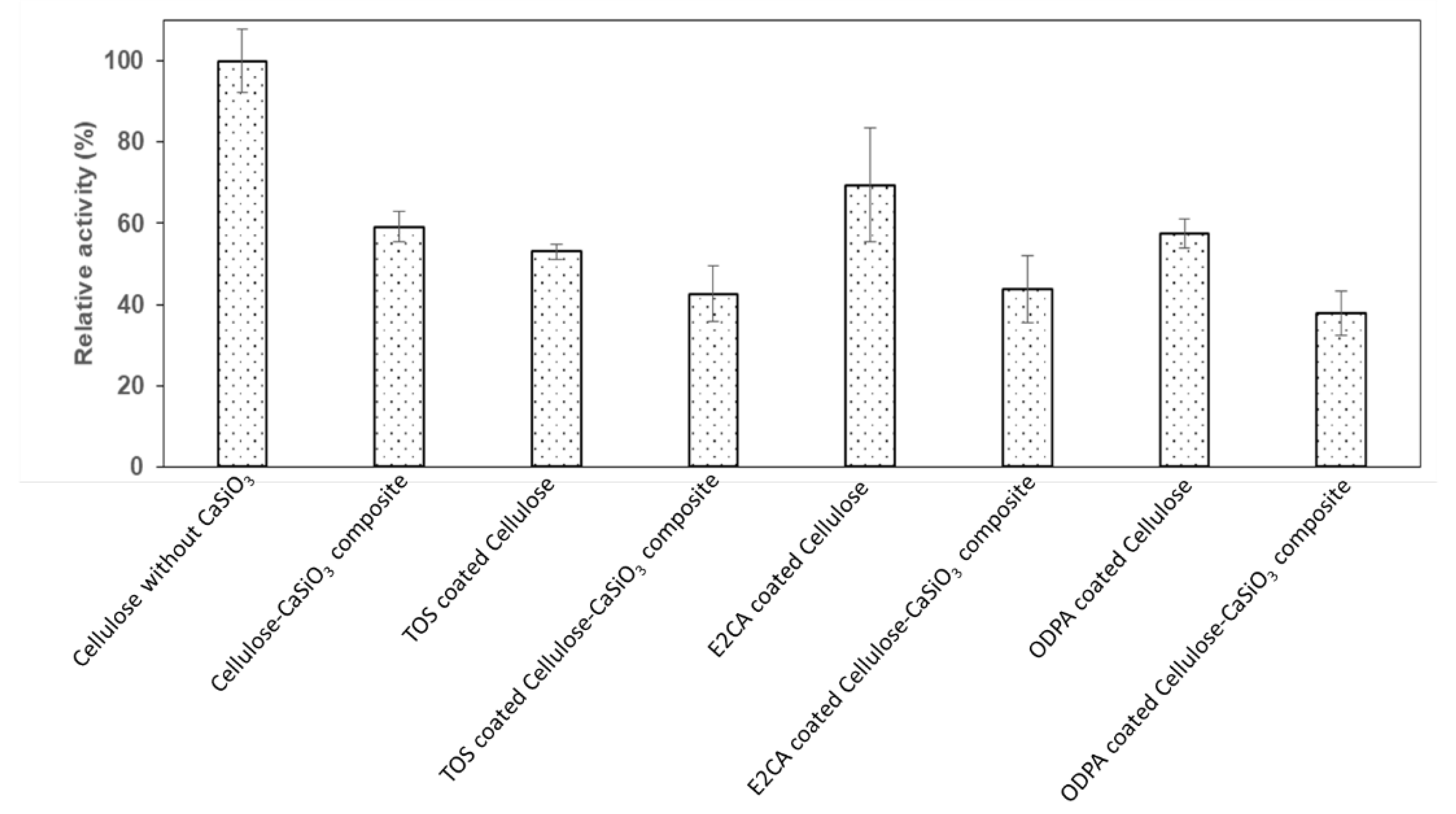
3.4. Biodegradability Studies of Uncoated and Coated Cellulose–CaSiO3 Composites
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.; Zhu, R.; Yang, Y. Polymer solar cells. Nat. Photonics 2012, 6, 153–161. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; Chen, D.; Shen, G. Flexible electronics based on inorganic nanowires. Chem. Soc. Rev. 2015, 44, 161–192. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh-Nguyen, B.-C.; Ko, E.; Kim, J.H.; Seong, G.H. Fabrication of flexible, transparent silver nanowire electrodes for amperometric detection of hydrogen peroxide. Sens. Actuators B Chem. 2016, 224, 789–797. [Google Scholar] [CrossRef]
- Zeng, W.; Shu, L.; Li, Q.; Chen, S.; Wang, F.; Tao, X.-M. Fiber-Based Wearable Electronics: A Review of Materials, Fabrication, Devices, and Applications. Adv. Mater. 2014, 26, 5310–5336. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Y.; Zhang, G.; Chu, Y.; Du, J.; Zhang, Y.; Li, Z.; Duan, Y.; Fan, Z.; Huang, J. Thermally Stable, Biocompatible, and Flexible Organic Field-Effect Transistors and Their Application in Temperature Sensing Arrays for Artificial Skin. Adv. Funct. Mater. 2015, 25, 2138–2146. [Google Scholar] [CrossRef]
- Forrest, S.R. The path to ubiquitous and low-cost organic electronic appliances on plastic. Nature 2004, 428, 911–918. [Google Scholar] [CrossRef]
- Wong, W.S.; Salleo, A. (Eds.) Electronic Materials: Science & Technology. In Flexible Electronics; Springer: Boston, MA, USA, 2009; Volume 11, ISBN 978-0-387-74362-2. [Google Scholar]
- Aulin, C.; Karabulut, E.; Tran, A.; Wågberg, L.; Lindström, T. Transparent Nanocellulosic Multilayer Thin Films on Polylactic Acid with Tunable Gas Barrier Properties. ACS Appl. Mater. Interfaces 2013, 5, 7352–7359. [Google Scholar] [CrossRef]
- Fujisaki, Y.; Koga, H.; Nakajima, Y.; Nakata, M.; Tsuji, H.; Yamamoto, T.; Kurita, T.; Nogi, M.; Shimidzu, N. Transparent Nanopaper-Based Flexible Organic Thin-Film Transistor Array. Adv. Funct. Mater. 2014, 24, 1657–1663. [Google Scholar] [CrossRef]
- Zhang, W.; Fei, L.; Zhang, J.; Chen, K.; Yin, Y.; Wang, C. Durable and tunable temperature responsive silk fabricated with reactive thermochromic pigments. Prog. Org. Coat. 2020, 147, 105697. [Google Scholar] [CrossRef]
- Mathur, P.; Sheikh, J.N.; Sen, K. Durable flame-retardant wool using sulphamic acid. Polym. Degrad. Stab. 2020, 174, 109101. [Google Scholar] [CrossRef]
- Shokri, J.; Adibki, K. Application of Cellulose and Cellulose Derivatives in Pharmaceutical Industries. In Cellulose—Medical, Pharmaceutical and Electronic Applications; InTech: London, UK, 2013. [Google Scholar]
- Song, Z.; Xiao, H.; Zhao, Y. Hydrophobic-modified nano-cellulose fiber/PLA biodegradable composites for lowering water vapor transmission rate (WVTR) of paper. Carbohydr. Polym. 2014, 111, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, L.; Bongiovanni, R.; Chaussy, D.; Gerbaldi, C.; Beneventi, D. Cellulose-based Li-ion batteries: A review. Cellulose 2013, 20, 1523–1545. [Google Scholar] [CrossRef]
- Danafar, F.; Kalantari, M. A Review of Natural Rubber Nanocomposites Based on Carbon Nanotubes. J. Rubber Res. 2018, 21, 293–310. [Google Scholar] [CrossRef]
- Huang, W.; Ling, S.; Li, C.; Omenetto, F.G.; Kaplan, D.L. Silkworm silk-based materials and devices generated using bio-nanotechnology. Chem. Soc. Rev. 2018, 47, 6486–6504. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Kim, J.; Ide, Y.; Ho Kim, J.; Hossain, M.S.A.; Bando, Y.; Yamauchi, Y.; Wu, K.C.W. 3D network of cellulose-based energy storage devices and related emerging applications. Mater. Horiz. 2017, 4, 522–545. [Google Scholar] [CrossRef]
- Zhao, D.; Zhu, Y.; Cheng, W.; Chen, W.; Wu, Y.; Yu, H. Cellulose-Based Flexible Functional Materials for Emerging Intelligent Electronics. Adv. Mater. 2020, 2000619. [Google Scholar] [CrossRef]
- Gao, L.; Chao, L.; Hou, M.; Liang, J.; Chen, Y.; Yu, H.-D.; Huang, W. Flexible, transparent nanocellulose paper-based perovskite solar cells. NPJ Flex. Electron. 2019, 3, 4. [Google Scholar] [CrossRef]
- Zhu, H.; Xiao, Z.; Liu, D.; Li, Y.; Weadock, N.J.; Fang, Z.; Huang, J.; Hu, L. Biodegradable transparent substrates for flexible organic-light-emitting diodes. Energy Environ. Sci. 2013, 6, 2105. [Google Scholar] [CrossRef]
- Khan, A.; Khan, F.R.; Kim, H.S. Electro-Active Paper as a Flexible Mechanical Sensor, Actuator and Energy Harvesting Transducer: A Review. Sensors 2018, 18, 3474. [Google Scholar] [CrossRef]
- Vicente, A.T.; Araújo, A.; Mendes, M.J.; Nunes, D.; Oliveira, M.J.; Sanchez-Sobrado, O.; Ferreira, M.P.; Águas, H.; Fortunato, E.; Martins, R. Multifunctional cellulose-paper for light harvesting and smart sensing applications. J. Mater. Chem. C 2018, 6, 3143–3181. [Google Scholar] [CrossRef]
- Jose, J.; Thomas, V.; Vinod, V.; Abraham, R.; Abraham, S. Nanocellulose based functional materials for supercapacitor applications. J. Sci. Adv. Mater. Devices 2019, 4, 333–340. [Google Scholar] [CrossRef]
- Sirviö, J.A.; Heiskanen, J.P. Room-temperature dissolution and chemical modification of cellulose in aqueous tetraethylammonium hydroxide–carbamide solutions. Cellulose 2020, 27, 1933–1950. [Google Scholar] [CrossRef]
- Xu, A.; Cao, L.; Wang, B.; Ma, J. Dissolution Behavior of Cellulose in IL + DMSO Solvent: Effect of Alkyl Length in Imidazolium Cation on Cellulose Dissolution. Adv. Mater. Sci. Eng. 2015, 2015, 1–4. [Google Scholar] [CrossRef]
- Isogai, A.; Atalla, R.H. Alkaline Method for Dissolving Cellulose. U.S. Patent US5410034A, 25 April 1995. [Google Scholar]
- Olsson, C.; Westman, G. Direct Dissolution of Cellulose: Background, Means and Applications. In Cellulose—Fundamental Aspects; InTech: London, UK, 2013. [Google Scholar]
- Zhang, S.; Li, F.-X.; Yu, J.; Hsieh, Y.-L. Dissolution behaviour and solubility of cellulose in NaOH complex solution. Carbohydr. Polym. 2010, 81, 668–674. [Google Scholar] [CrossRef]
- Rodríguez-Castellanos, W.; Martínez-Bustos, F.; Rodrigue, D.; Trujillo-Barragán, M. Extrusion blow molding of a starch–gelatin polymer matrix reinforced with cellulose. Eur. Polym. J. 2015, 73, 335–343. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Chen, Q.; Yu, Z.; Wang, C.; Jin, S.; Ding, Y.; Wu, G. Dissolution of cellulose with ionic liquids and its application: A mini-review. Green Chem. 2006, 8, 325. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of Cellose with Ionic Liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef]
- Zavrel, M.; Bross, D.; Funke, M.; Büchs, J.; Spiess, A.C. High-throughput screening for ionic liquids dissolving (ligno-)cellulose. Bioresour. Technol. 2009, 100, 2580–2587. [Google Scholar] [CrossRef]
- Payal, R.S.; Balasubramanian, S. Dissolution of cellulose in ionic liquids: An ab initio molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2014, 16, 17458–17465. [Google Scholar] [CrossRef]
- Sun, N.; Rahman, M.; Qin, Y.; Maxim, M.L.; Rodríguez, H.; Rogers, R.D. Complete dissolution and partial delignification of wood in the ionic liquid 1-ethyl-3-methylimidazolium acetate. Green Chem. 2009, 11, 646. [Google Scholar] [CrossRef]
- Liebert, T.; Heinze, T. Interaction of ionic liquids wlth polysaccharides 5. Solvents and reaction media for the modification of cellulose. BioResources 2008, 3, 576–601. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Fu, F.; Liu, X. Single-faced flame resistance of cotton fabrics modified via mist copolymerization. RSC Adv. 2017, 7, 53871–53877. [Google Scholar] [CrossRef]
- Mohamed, A.L.; Hassabo, A.G. Flame Retardant of Cellulosic Materials and Their Composites. In Flame Retardants Electronic; Springer International Publishing: Berlin, Germany, 2015; pp. 247–314. [Google Scholar] [CrossRef]
- Nakanishi, S.; Masuko, F.; Hori, K.; Hashimoto, T. Pyrolytic Gas Generation of Cotton Cellulose With and Without Flame Retardants at Different Stages of Thermal Degradation: Effects of Nitrogen, Phosphorus, and Halogens. Text. Res. J. 2000, 70, 574–583. [Google Scholar] [CrossRef]
- Salmeia, K.; Gaan, S.; Malucelli, G. Recent Advances for Flame Retardancy of Textiles Based on Phosphorus Chemistry. Polymers 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Alongi, J.; Malucelli, G. Cotton flame retardancy: State of the art and future perspectives. RSC Adv. 2015, 5, 24239–24263. [Google Scholar] [CrossRef]
- Gao, W.-W.; Zhang, G.-X.; Zhang, F.-X. Enhancement of flame retardancy of cotton fabrics by grafting a novel organic phosphorous-based flame retardant. Cellulose 2015, 22, 2787–2796. [Google Scholar] [CrossRef]
- Zheng, D.; Zhou, J.; Wang, Y.; Zhang, F.; Zhang, G. A reactive flame retardant ammonium salt of diethylenetriaminepenta(methylene-phosphonic acid) for enhancing flame retardancy of cotton fabrics. Cellulose 2018, 25, 787–797. [Google Scholar] [CrossRef]
- Zheng, C.; Li, D.; Ek, M. Improving fire retardancy of cellulosic thermal insulating materials by coating with bio-based fire retardants. Nord. Pulp Pap. Res. J. 2019, 34, 96–106. [Google Scholar] [CrossRef]
- Teisala, H.; Tuominen, M.; Kuusipalo, J. Superhydrophobic Coatings on Cellulose-Based Materials: Fabrication, Properties, and Applications. Adv. Mater. Interfaces 2014, 1, 1300026. [Google Scholar] [CrossRef]
- Bretel, G.; Rull-Barrull, J.; Nongbe, M.C.; Terrier, J.-P.; Le Grognec, E.; Felpin, F.-X. Hydrophobic Covalent Patterns on Cellulose Paper through Photothiol-X Ligations. ACS Omega 2018, 3, 9155–9159. [Google Scholar] [CrossRef]
- Nyström, D.; Lindqvist, J.; Östmark, E.; Hult, A.; Malmström, E. Superhydrophobic bio-fibre surfaces via tailored grafting architecture. Chem. Commun. 2006, 3594–3596. [Google Scholar] [CrossRef]
- Wang, T.; Hu, X.; Dong, S. A general route to transform normal hydrophilic cloths into superhydrophobic surfaces. Chem. Commun. 2007, 1849. [Google Scholar] [CrossRef]
- Ghose, T.K. Measurement of cellulase activities. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, L.; Chen, G. Fabrication of graphene/poly(ethyl 2-cyanoacrylate) composite electrode for amperometric detection in capillary electrophoresis. Sens. Actuators B Chem. 2013, 182, 689–695. [Google Scholar] [CrossRef]
- Han, M.G.; Kim, S.; Liu, S.X. Synthesis and degradation behavior of poly(ethyl cyanoacrylate). Polym. Degrad. Stab. 2008, 93, 1243–1251. [Google Scholar] [CrossRef]
- Bayer, I.S.; Fragouli, D.; Attanasio, A.; Sorce, B.; Bertoni, G.; Brescia, R.; Di Corato, R.; Pellegrino, T.; Kalyva, M.; Sabella, S.; et al. Water-Repellent Cellulose Fiber Networks with Multifunctional Properties. ACS Appl. Mater. Interfaces 2011, 3, 4024–4031. [Google Scholar] [CrossRef]
- Du, X.; Li, J.S.; Li, L.X.; Levkin, P.A. Porous poly(2-octyl cyanoacrylate): A facile one-step preparation of superhydrophobic coatings on different substrates. J. Mater. Chem. A 2013, 1, 1026–1029. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Sotenko, M.; Cruz-Izquierdo, A.; Rymansaib, Z.; Iravani, P.; Kirwan, K.; Scott, J.L. Preparation of Printable and Biodegradable Cellulose-Laponite Composite for Electronic Device Application. J. Polym. Environ. 2021, 29, 17–27. [Google Scholar] [CrossRef]
- Marcinko, S.; Fadeev, A.Y. Hydrolytic Stability of Organic Monolayers Supported on TiO2 and ZrO2. Langmuir 2004, 20, 2270–2273. [Google Scholar] [CrossRef]
- Sha, Y.; Deng, C.; Liu, B. Development of C18-functionalized magnetic silica nanoparticles as sample preparation technique for the determination of ergosterol in cigarettes by microwave-assisted derivatization and gas chromatography/mass spectrometry. J. Chromatogr. A 2008, 1198–1199, 27–33. [Google Scholar] [CrossRef]
- Grønli, M.; Antal, M.J.; Várhegyi, G. A Round-Robin Study of Cellulose Pyrolysis Kinetics by Thermogravimetry. Ind. Eng. Chem. Res. 1999, 38, 2238–2244. [Google Scholar] [CrossRef]
- Statheropoulos, M.; Kyriakou, S.A. Quantitative thermogravimetric-mass spectrometric analysis for monitoring the effects of fire retardants on cellulose pyrolysis. Anal. Chim. Acta 2000, 409, 203–214. [Google Scholar] [CrossRef]
- Ruan, D.; Zhang, L.; Zhang, Z.; Xia, X. Structure and properties of regenerated cellulose/tourmaline nanocrystal composite films. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 367–373. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Yano, H.; Nogi, M.; Eichhorn, S.J. An estimation of the Young’s modulus of bacterial cellulose filaments. Cellulose 2008, 15, 507–513. [Google Scholar] [CrossRef]
- Zhou, S.; Tashiro, K.; Hongo, T.; Shirataki, H.; Yamane, C.; Ii, T. Influence of Water on Structure and Mechanical Properties of Regenerated Cellulose Studied by an Organized Combination of Infrared Spectra, X-ray Diffraction, and Dynamic Viscoelastic Data Measured as Functions of Temperature and Humidity. Macromolecules 2001, 34, 1274–1280. [Google Scholar] [CrossRef]
- Peixoto, J.; Oort, A.H. The Climatology of Relative Humidity in the Atmosphere. J. Clim. 1996, 9, 3443–3463. [Google Scholar] [CrossRef]
- Brynn Hibbert, D.; Gooding, J.J. Data analysis for chemistry: An introductory guide for students and laboratory scientists. Choice Rev. Online 2006, 43, 43–5297. [Google Scholar] [CrossRef]
- Tejirian, A.; Xu, F. Inhibition of Cellulase-Catalyzed Lignocellulosic Hydrolysis by Iron and Oxidative Metal Ions and Complexes. Appl. Environ. Microbiol. 2010, 76, 7673–7682. [Google Scholar] [CrossRef] [PubMed]
- Sammond, D.W.; Yarbrough, J.M.; Mansfield, E.; Bomble, Y.J.; Hobdey, S.E.; Decker, S.R.; Taylor, L.E.; Resch, M.G.; Bozell, J.J.; Himmel, M.E.; et al. Predicting Enzyme Adsorption to Lignin Films by Calculating Enzyme Surface Hydrophobicity. J. Biol. Chem. 2014, 289, 20960–20969. [Google Scholar] [CrossRef]
- Tu, M.; Pan, X.; Saddler, J.N. Adsorption of Cellulase on Cellulolytic Enzyme Lignin from Lodgepole Pine. J. Agric. Food Chem. 2009, 57, 7771–7778. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yang, B.; Gregg, D.; Saddler, J.N.; Mansfield, S.D. Cellulase Adsorption and an Evaluation of Enzyme Recycle During Hydrolysis of Steam-Exploded Softwood Residues. Appl. Biochem. Biotechnol. 2002, 98–100, 641–654. [Google Scholar] [CrossRef]
- Korde, J.M.; Kandasubramanian, B. Biocompatible alkyl cyanoacrylates and their derivatives as bio-adhesives. Biomater. Sci. 2018, 6, 1691–1711. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose Composites | LOI (%) (ASTM-D2863) | Flammability Tests UL-94 HB, Time to Burn Marked Area of Samples in Secs (Std. dev) (ASTM D635-03) |
|---|---|---|
| No fillers | 19 | 23.7 (±1.2) |
| 10 wt%-CaSiO3 | 20.7 | 36.3 (±2.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, S.; Castaing, R.; Cruz-Izquierdo, A.; Scott, J.L. Influence of Calcium Silicate and Hydrophobic Agent Coatings on Thermal, Water Barrier, Mechanical and Biodegradation Properties of Cellulose. Nanomaterials 2021, 11, 1488. https://doi.org/10.3390/nano11061488
Chandrasekaran S, Castaing R, Cruz-Izquierdo A, Scott JL. Influence of Calcium Silicate and Hydrophobic Agent Coatings on Thermal, Water Barrier, Mechanical and Biodegradation Properties of Cellulose. Nanomaterials. 2021; 11(6):1488. https://doi.org/10.3390/nano11061488
Chicago/Turabian StyleChandrasekaran, Saravanan, Remi Castaing, Alvaro Cruz-Izquierdo, and Janet L. Scott. 2021. "Influence of Calcium Silicate and Hydrophobic Agent Coatings on Thermal, Water Barrier, Mechanical and Biodegradation Properties of Cellulose" Nanomaterials 11, no. 6: 1488. https://doi.org/10.3390/nano11061488
APA StyleChandrasekaran, S., Castaing, R., Cruz-Izquierdo, A., & Scott, J. L. (2021). Influence of Calcium Silicate and Hydrophobic Agent Coatings on Thermal, Water Barrier, Mechanical and Biodegradation Properties of Cellulose. Nanomaterials, 11(6), 1488. https://doi.org/10.3390/nano11061488