
Hot Electrons in TiO2–Noble Metal Nano-Heterojunctions: Fundamental Science and Applications in Photocatalysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. What Are Hot Electrons and Why Are They Important?
1.2. Plasmonic Hot Electron Photocatalysis Using TiO2–Noble Metal Nanostructures
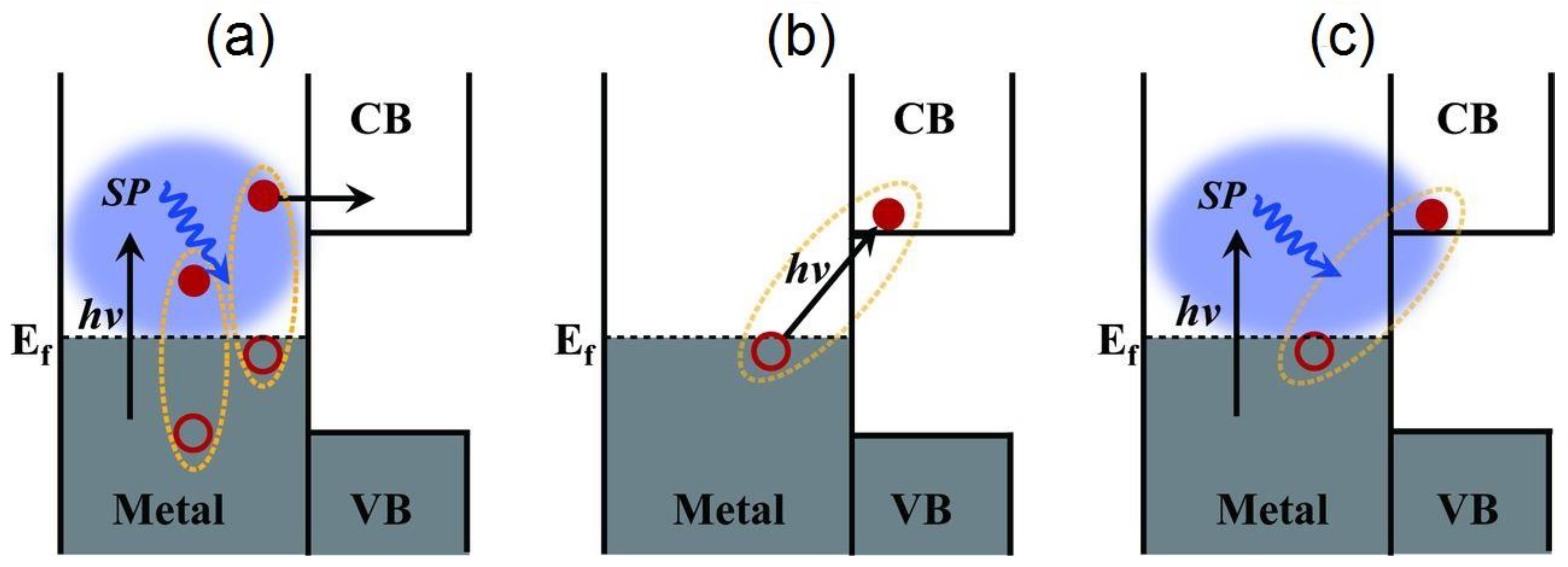
2. Digging Deeper into Hot Electrons
2.1. Surface Plasmons
2.2. Sequential Mechanism of Hot Electron Relaxation
2.3. Alternative Mechanisms of Hot Electron Relaxation
3. Probing Hot Electrons
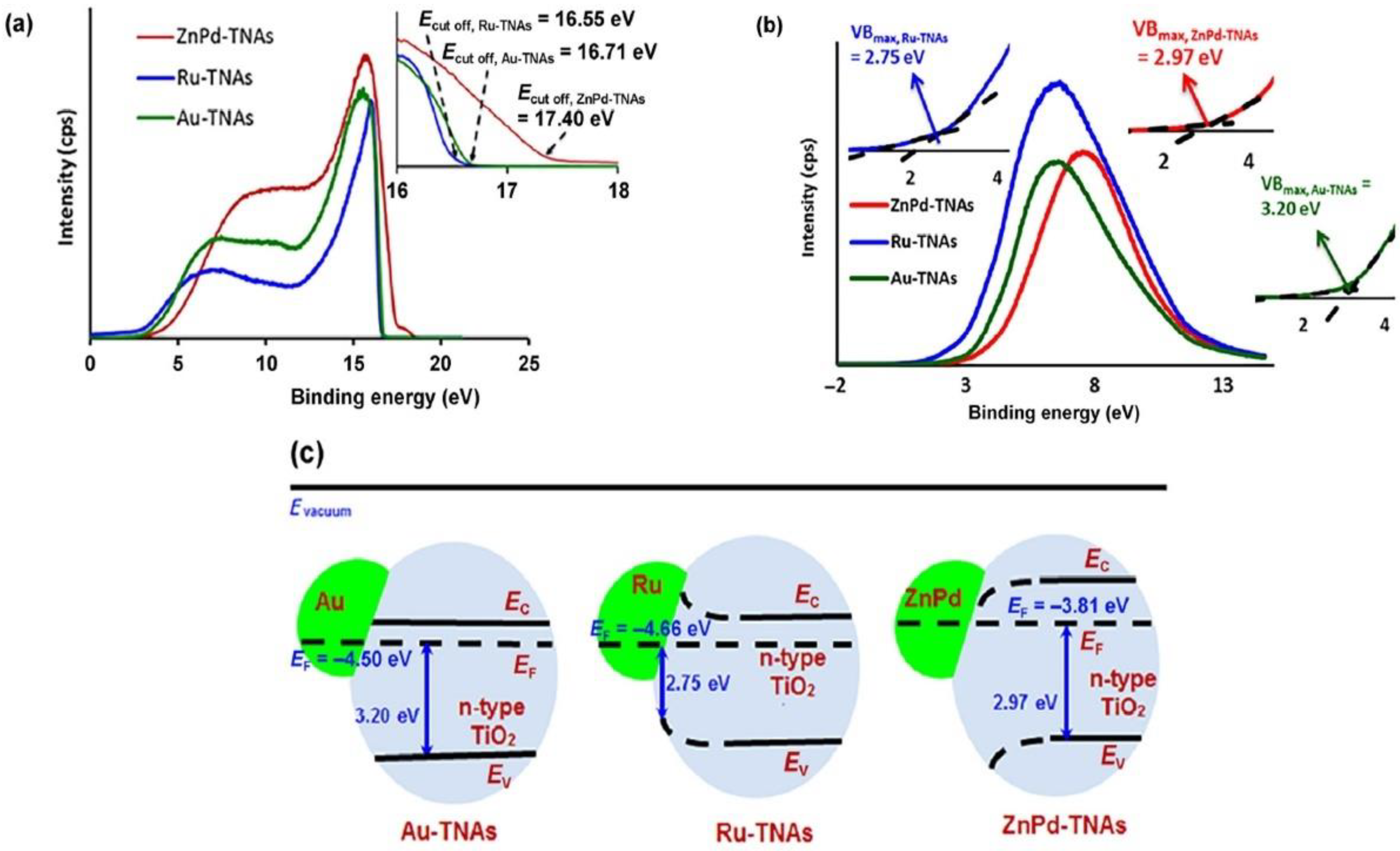
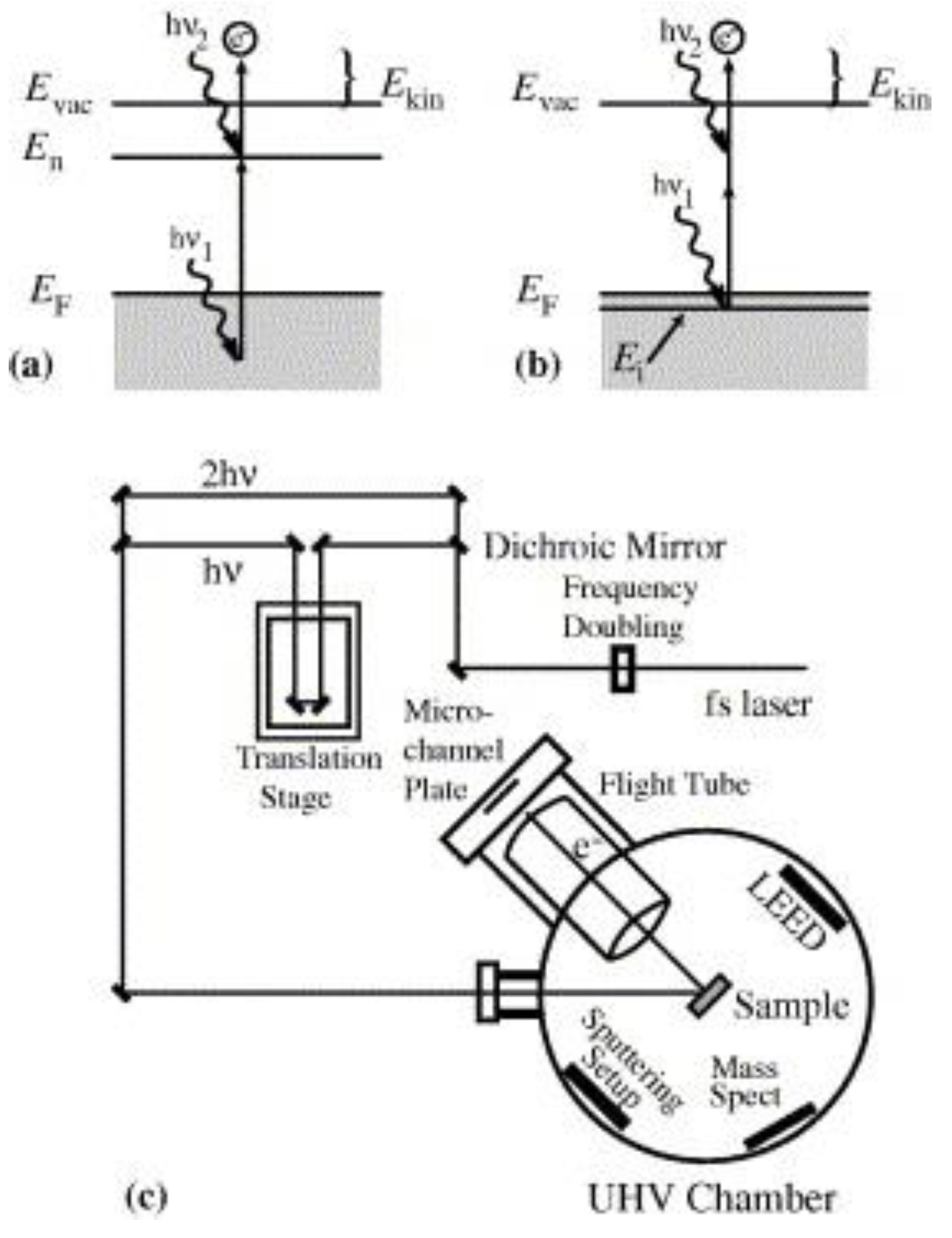
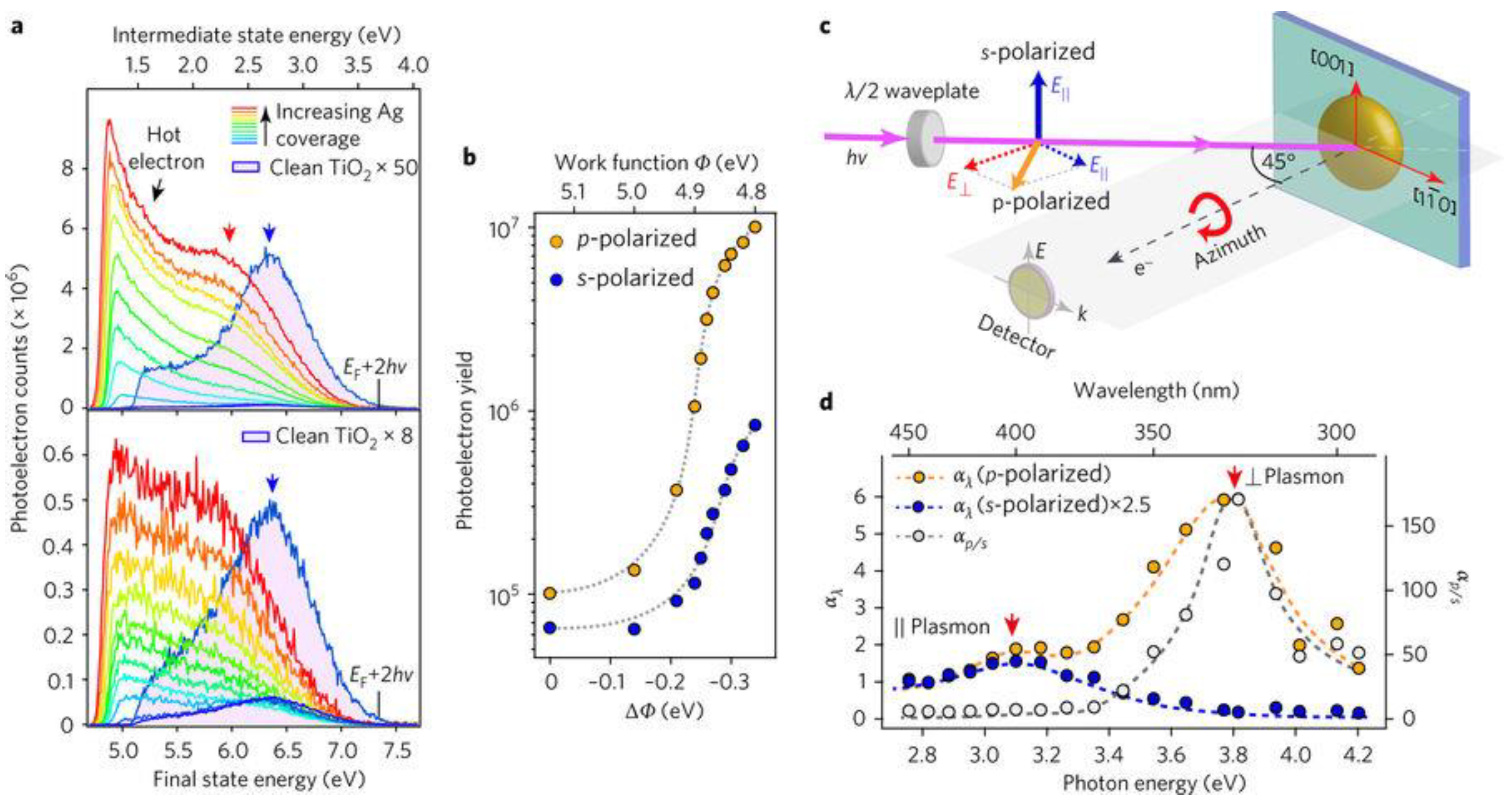
3.1. Photoemission Spectroscopy
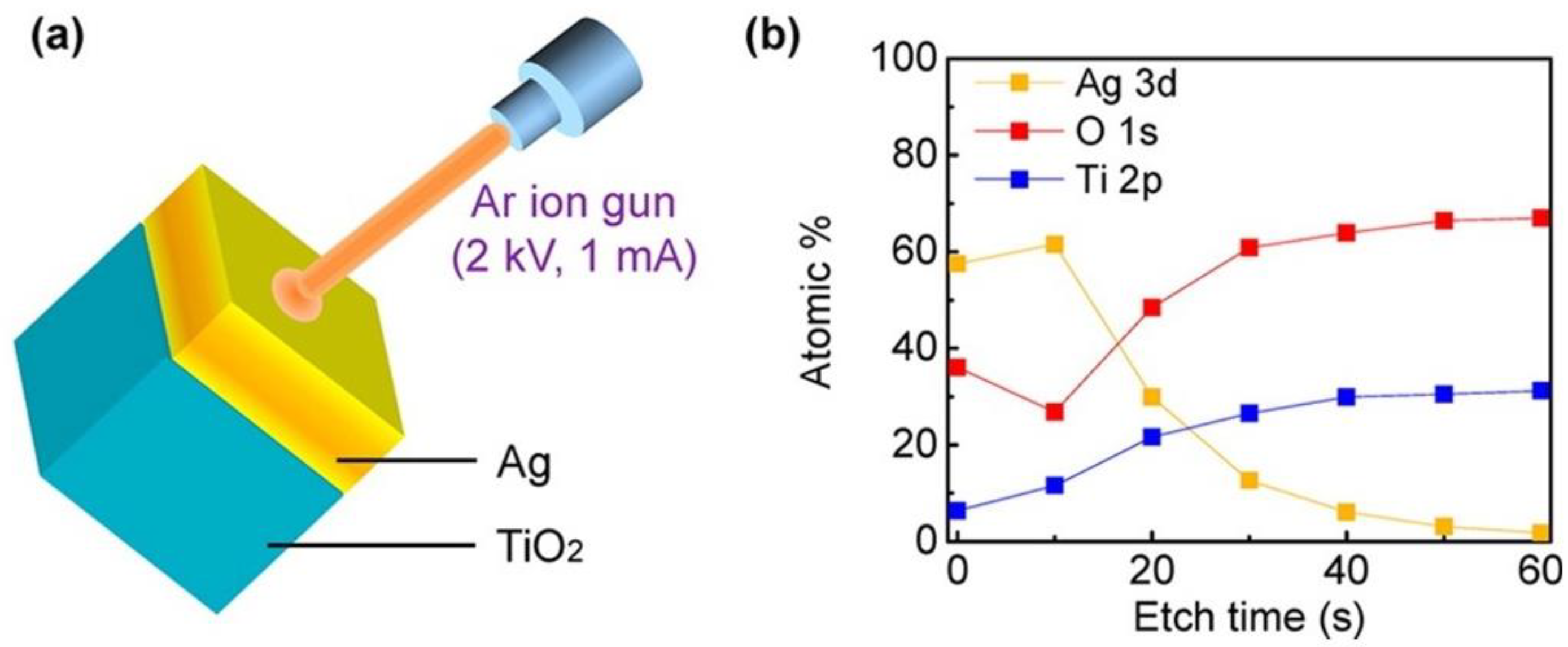
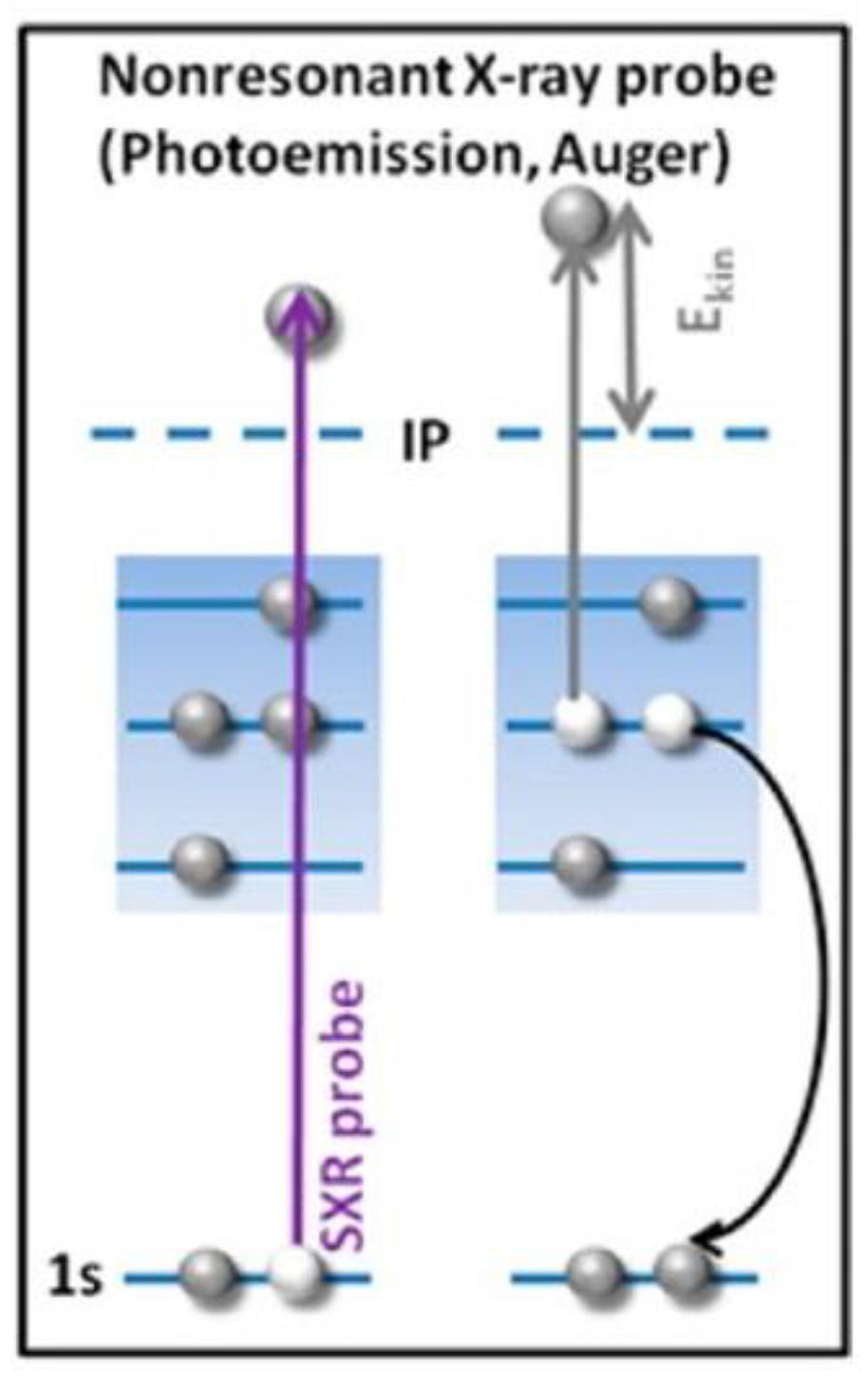
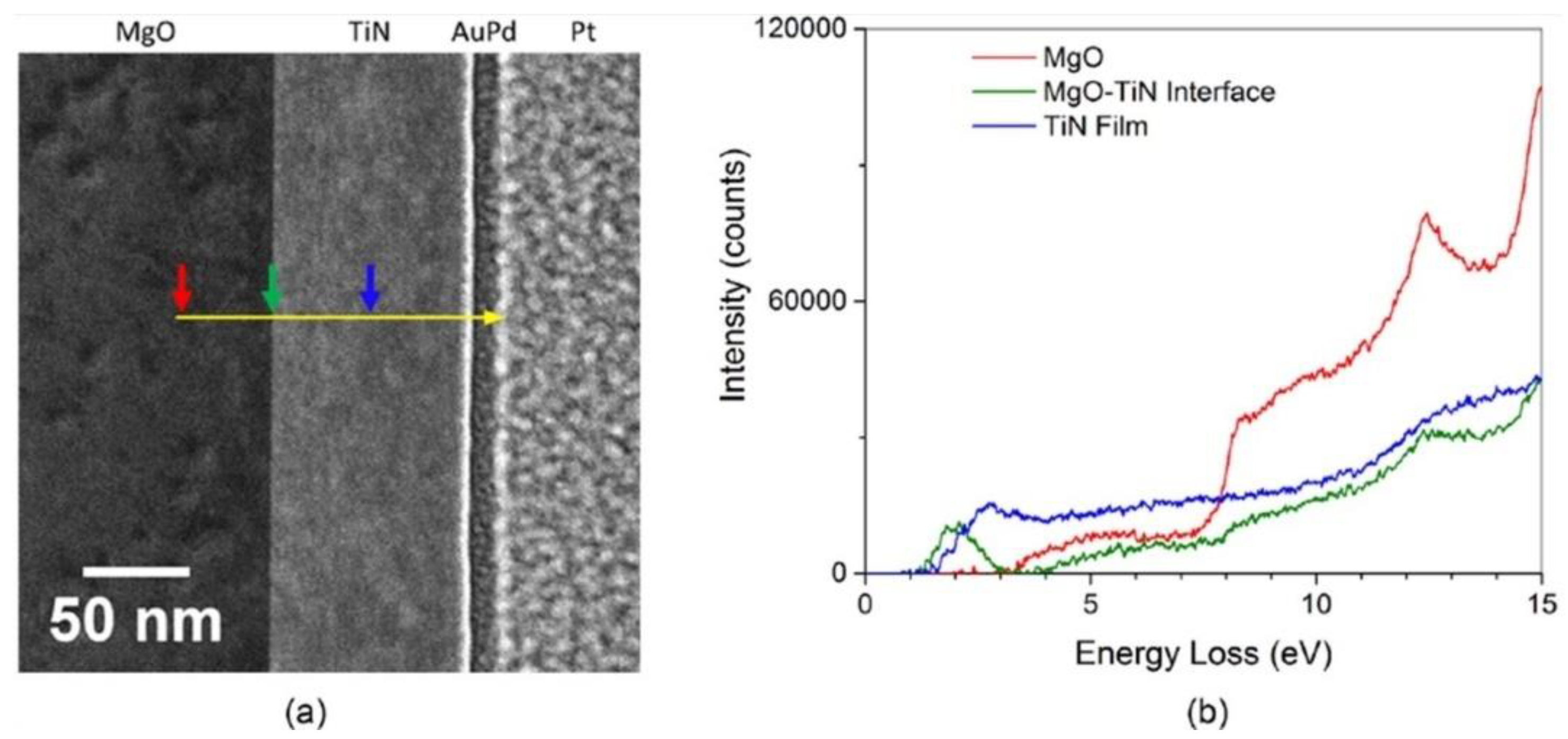
3.2. Auger and Electron Energy Loss Spectroscopy
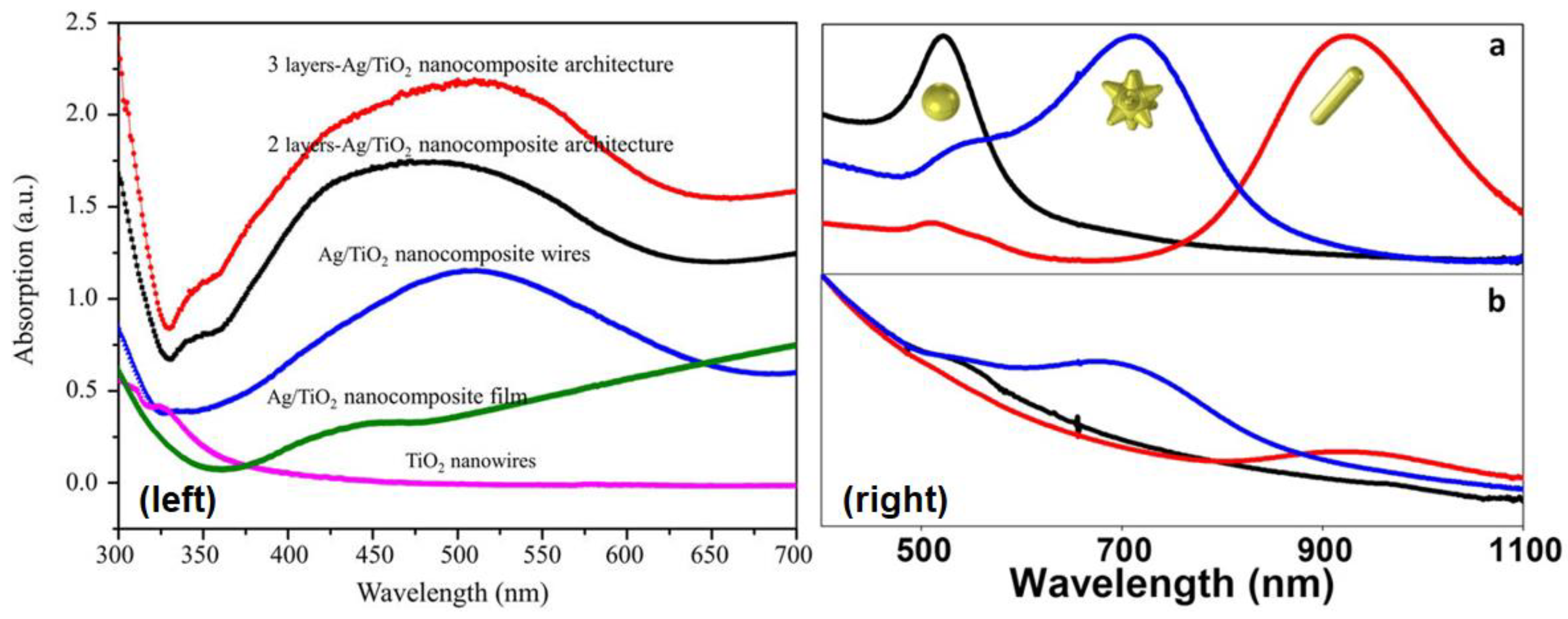
3.3. Absorption and Photoluminescence Spectroscopies
3.4. Raman Spectroscopy
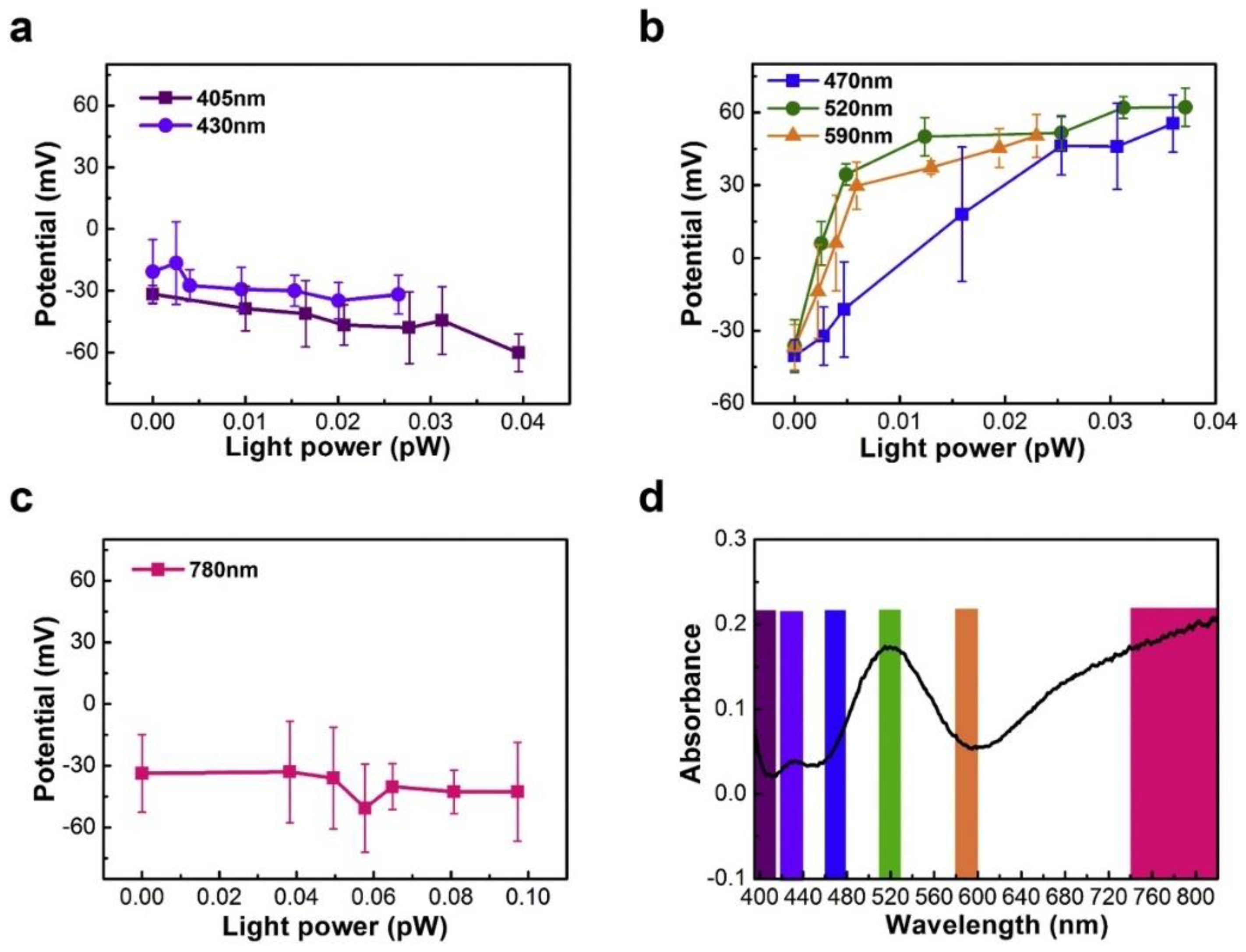
3.5. Kelvin Probe Force Microscopy
3.6. Other Prominent Methods
4. Exploiting Hot Electrons
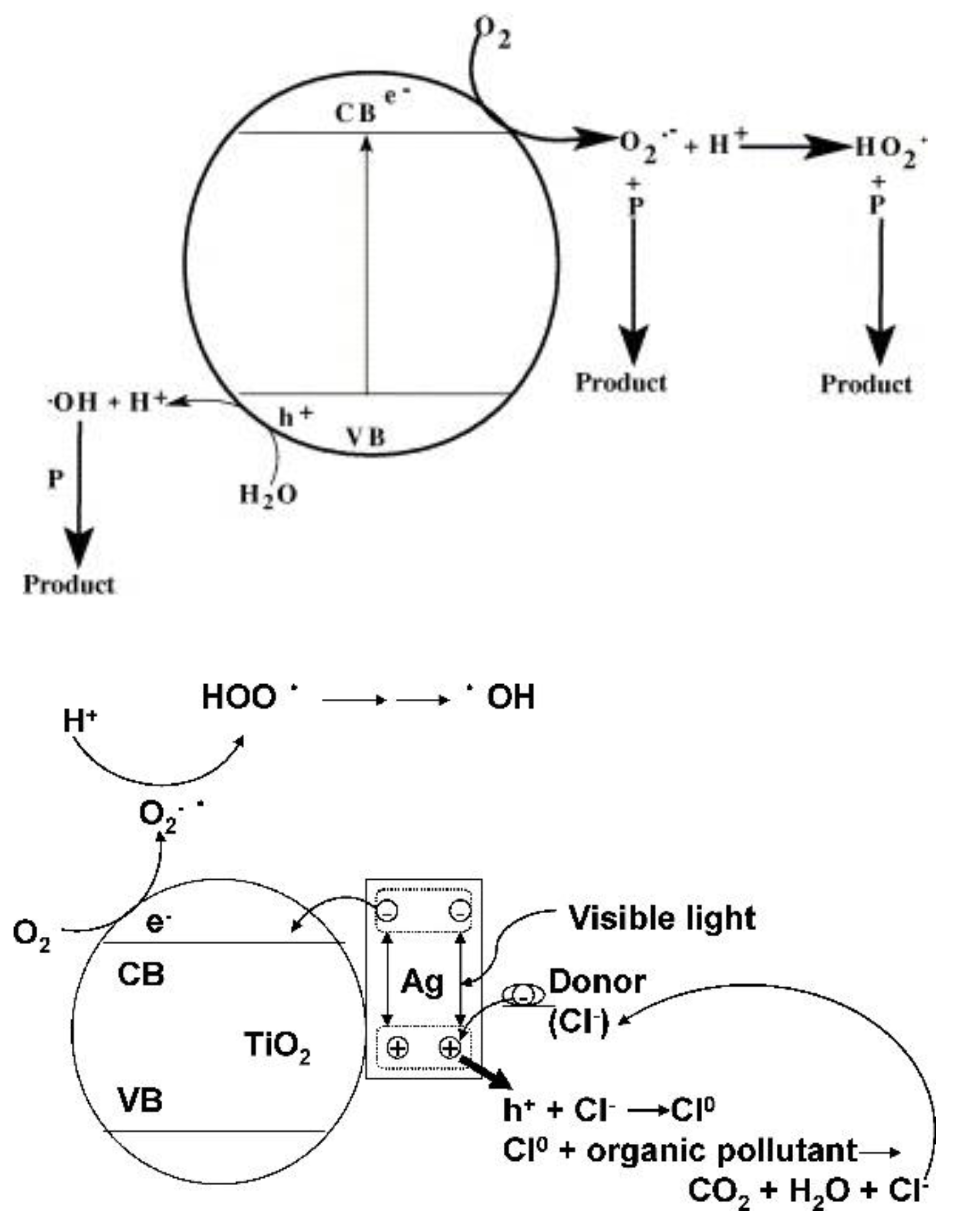
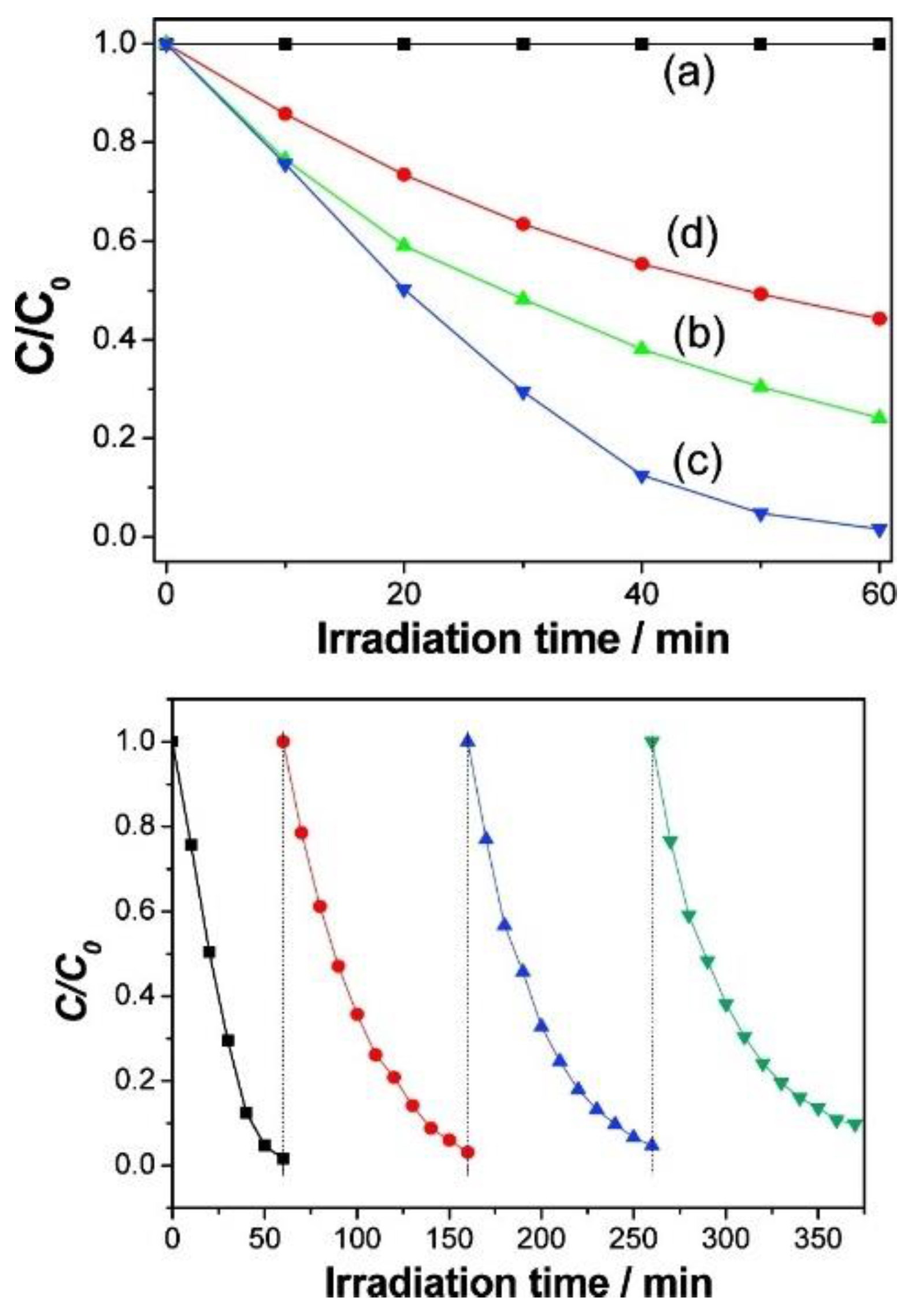
4.1. Photocatalytic Degradation/Aerobic Oxidation of Organic Compounds
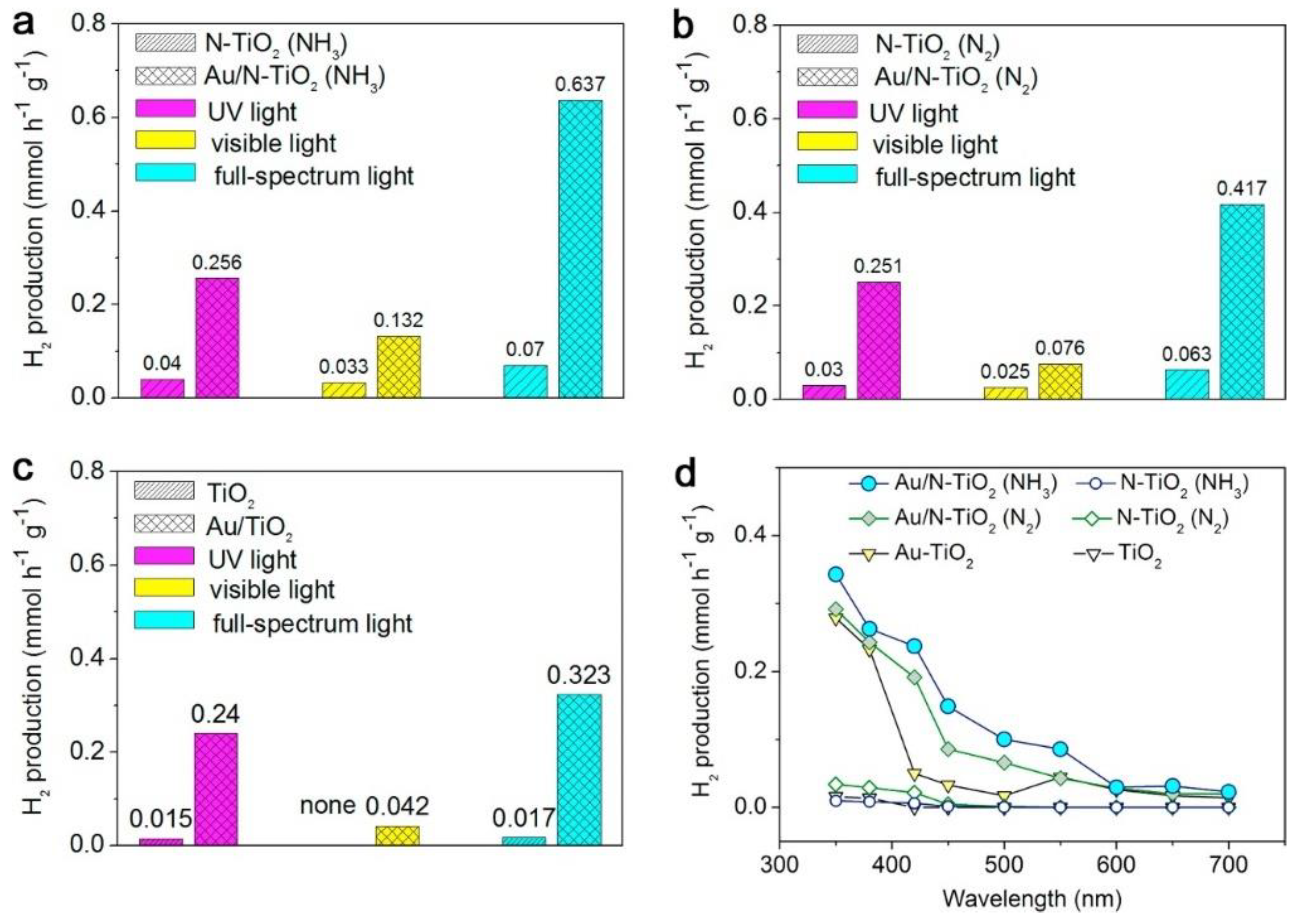
4.2. Photocatalytic CO2 Reduction and H2 Generation
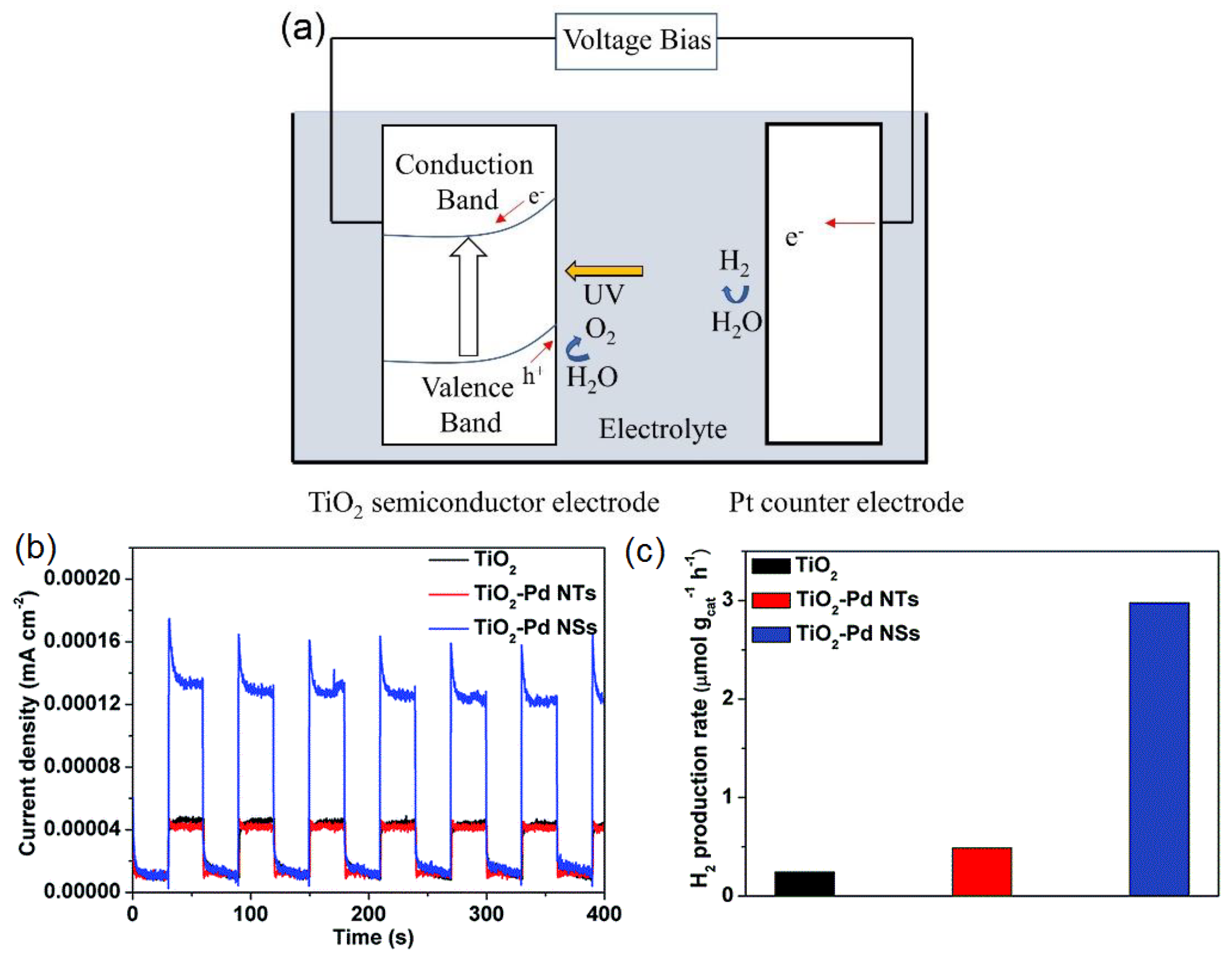
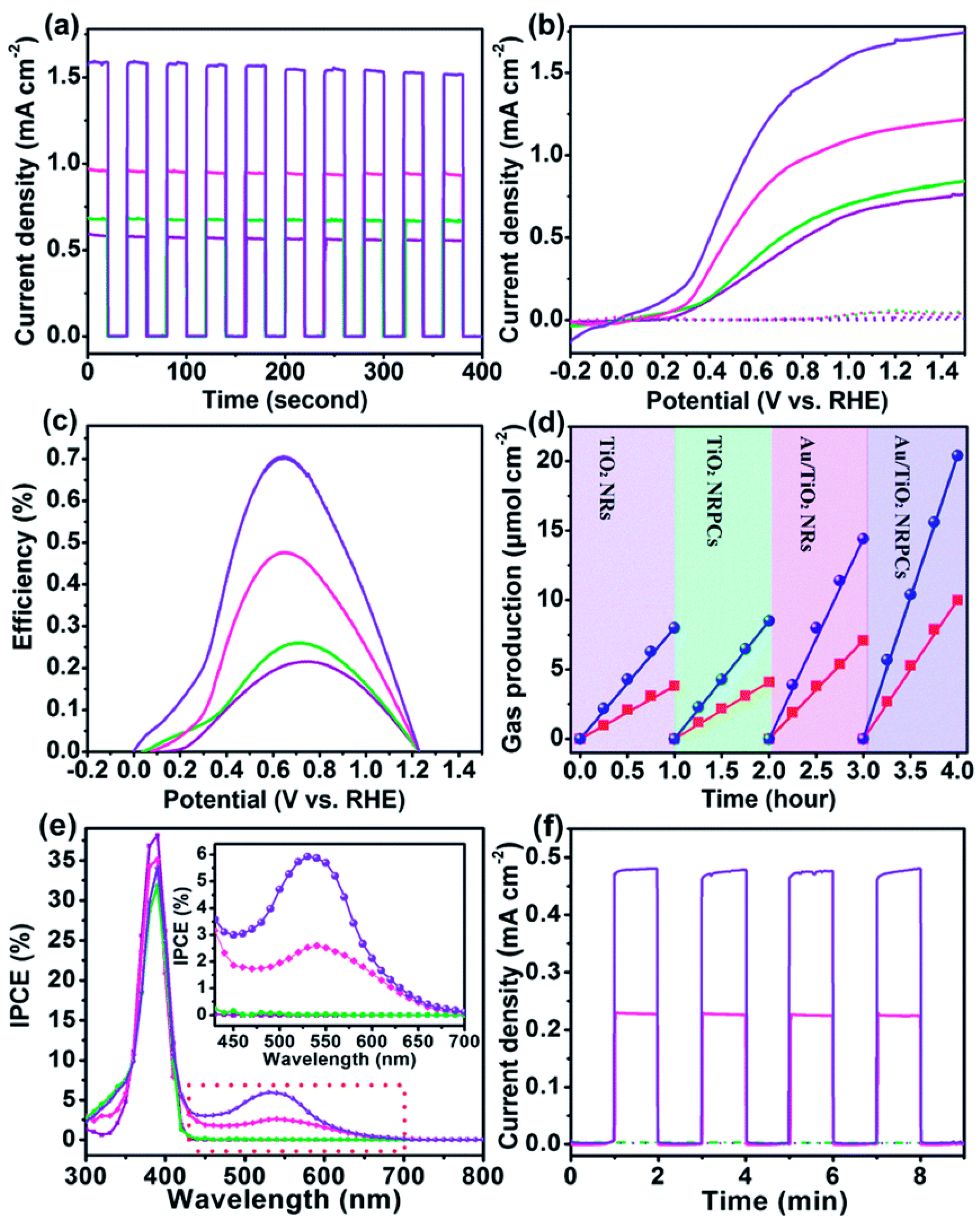
4.3. Photoelectrochemical Water Splitting
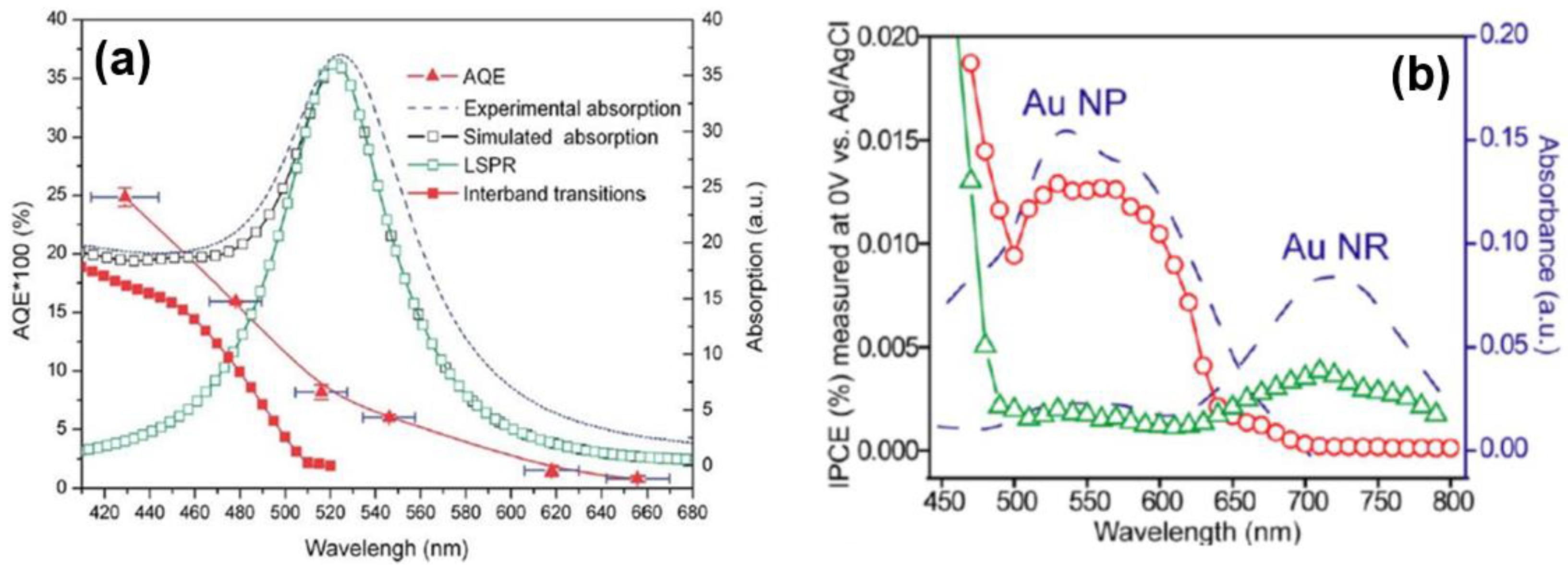
5. Mystery of the Action Spectrum: Reconciling Interband Transitions with Localized Surface Plasmon Resonances
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalyanasundaram, K.; Graetzel, M. Artificial photosynthesis: Biomimetic approaches to solar energy conversion and storage. Curr. Opin. Biotechnol. 2010, 21, 298–310. [Google Scholar] [CrossRef] [PubMed]
- BP. BP Statistical Review of World Energy; BP: London, UK, 2018. [Google Scholar]
- International Energy Agency. Key World Energy Statistics; International Energy Agency: Paris, France, 2018. [Google Scholar]
- U.S. Energy Information Administration. International Energy Outlook; U.S. Energy Information Administration: Washington, DC, USA, 2010.
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward solar fuels: Photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-N. Comparison of CO2 Photoreduction Systems: A Review. Aerosol Air Qual. Res. 2014. [Google Scholar] [CrossRef]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar Energy Supply and Storage for the Legacy and Nonlegacy Worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L. Overview: Capturing the Sun for Energy Production. AMBIO 2012, 41, 103–107. [Google Scholar] [CrossRef]
- Li, J.; Wu, N. Semiconductor-based photocatalysts and photoelectrochemical cells for solar fuel generation: A review. Catal. Sci. Technol. 2015, 5, 1360–1384. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Arunachalam, P.; Ramachandran, K.; Al-Mayouf, A.M.; Karuppuchamy, S. Recent advances in semiconductor metal oxides with enhanced methods for solar photocatalytic applications. J. Alloys Compd. 2020, 828, 15. [Google Scholar] [CrossRef]
- Zeng, S.; Kar, P.; Thakur, U.K.; Shankar, K. A review on photocatalytic CO2 reduction using perovskite oxide nanomaterials. Nanotechnology 2018, 29, 052001. [Google Scholar] [CrossRef]
- Kumar, P.; Thakur, U.K.; Alam, K.; Kar, P.; Kisslinger, R.; Zeng, S.; Patel, S.; Shankar, K. Arrays of TiO2 nanorods embedded with fluorine doped carbon nitride quantum dots (CNFQDs) for visible light driven water splitting. Carbon 2018, 137, 174–187. [Google Scholar] [CrossRef]
- Enesca, A.; Isac, L. Tandem Structures Semiconductors Based on TiO2_SnO2 and ZnO_SnO2 for Photocatalytic Organic Pollutant Removal. Nanomaterials 2021, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Yao, L.; Deng, L.B.; Bowen, C.; Zhang, Y.; Chen, S.M.; Lin, Z.Q.; Peng, F.; Zhang, P.X. Recent advances in metal sulfides: From controlled fabrication to electrocatalytic, photocatalytic and photoelectrochemical water splitting and beyond. Chem. Soc. Rev. 2019, 48, 4178–4280. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Farsinezhad, S.; Zhang, X.; Shankar, K. Anodic Cu2S and CuS nanorod and nanowall arrays: Preparation, properties and application in CO2 photoreduction. Nanoscale 2014, 6, 14305–14318. [Google Scholar] [CrossRef]
- Varadhan, P.; Fu, H.C.; Priante, D.; Retamal, J.R.D.; Zhao, C.; Ebaid, M.; Ng, T.K.; Ajia, I.; Mitra, S.; Roqan, I.S.; et al. Surface Passivation of GaN Nanowires for Enhanced Photoelectrochemical Water-Splitting. Nano Lett. 2017, 17, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Ebaid, M.; Priante, D.; Liu, G.Y.; Zhao, C.; Alias, M.S.; Buttner, U.; Ng, T.K.; Isimjan, T.T.; Idriss, H.; Ooi, B.S. Unbiased photocatalytic hydrogen generation from pure water on stable Ir-treated In0.33Ga0.67N nanorods. Nano Energy 2017, 37, 158–167. [Google Scholar] [CrossRef]
- Wang, S.Y.; Hai, X.; Ding, X.; Chang, K.; Xiang, Y.G.; Meng, X.G.; Yang, Z.X.; Chen, H.; Ye, J.H. Light-Switchable Oxygen Vacancies in Ultrafine Bi5O7Br Nanotubes for Boosting Solar-Driven Nitrogen Fixation in Pure Water. Adv. Mater. 2017, 29, 7. [Google Scholar] [CrossRef]
- Alam, K.M.; Kumar, P.; Kar, P.; Thakur, U.K.; Zeng, S.; Cui, K.; Shankar, K. Enhanced charge separation in g-C3N4–BiOI heterostructures for visible light driven photoelectrochemical water splitting. Nanoscale Adv. 2019, 1, 1460–1471. [Google Scholar] [CrossRef]
- Farsinezhad, S.; Shanavas, T.; Mahdi, N.; Askar, A.M.; Kar, P.; Sharma, H.; Shankar, K. Core–shell titanium dioxide–titanium nitride nanotube arrays with near-infrared plasmon resonances. Nanotechnology 2018, 29, 154006. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Reddy, C.V.; Nadagouda, M.N.; Shetti, N.P.; Jaesool, S.; Aminabhavi, T.M. Polymeric graphitic carbon nitride (g-C3N4)-based semiconducting nanostructured materials: Synthesis methods, properties and photocatalytic applications. J. Environ. Manag. 2019, 238, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Shi, J.; Xu, Z.Z.; Zhang, B.L.; Liu, H.L.; Lin, Y.L.; Gao, F.L.; Li, S.T.; Li, G.Q. InGaN Nanorods Decorated with Au Nanoparticles for Enhanced Water Splitting Based on Surface Plasmon Resonance Effects. Nanomaterials 2020, 10, 912. [Google Scholar] [CrossRef] [PubMed]
- Dudita, M.; Bogatu, C.; Enesca, A.; Duta, A. The influence of the additives composition and concentration on the properties of SnOx thin films used in photocatalysis. Mater. Lett. 2011, 65, 2185–2189. [Google Scholar] [CrossRef]
- Mouchaal, Y.; Enesca, A.; Mihoreanu, C.; Khelil, A.; Duta, A. Tuning the opto-electrical properties of SnO2 thin films by Ag+1 and In+3 co-doping. Mater. Sci. Eng. B 2015, 199, 22–29. [Google Scholar] [CrossRef]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 Photocatalysis: A Historical Overview and Future Prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Ollis, D. Photocatalytic purification and remediation of contaminated air and water. Comptes Rendus l’Académie Sci. Ser. IIC Chem. 2000, 3, 405–411. [Google Scholar] [CrossRef]
- Herrmann, J.-M. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129. [Google Scholar] [CrossRef]
- Indrakanti, V.P.; Kubicki, J.D.; Schobert, H.H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: Current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2009, 2, 745–758. [Google Scholar] [CrossRef]
- Wang, M.; Ye, M.; Iocozzia, J.; Lin, C.; Lin, Z. Plasmon-Mediated Solar Energy Conversion via Photocatalysis in Noble Metal/Semiconductor Composites. Adv. Sci. (Weinh.) 2016, 3, 1600024. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, I.F.; Homsi, M.S.; Geonmonond, R.S.; Rocha, G.; Peng, Y.K.; Silva, I.F.; Quiroz, J.; Camargo, P.H.C. Hot Electrons, Hot Holes, or Both? Tandem Synthesis of Imines Driven by the Plasmonic Excitation in Au/CeO2Nanorods. Nanomaterials 2020, 10, 1530. [Google Scholar] [CrossRef] [PubMed]
- Hamans, R.F.; Kamarudheen, R.; Baldi, A. Single Particle Approaches to Plasmon-Driven Catalysis. Nanomaterials 2020, 10, 2377. [Google Scholar] [CrossRef]
- Tran, V.T.; Nguyen, H.Q.; Kim, Y.M.; Ok, G.; Lee, J. Photonic-Plasmonic Nanostructures for Solar Energy Utilization and Emerging Biosensors. Nanomaterials 2020, 10, 2248. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.S.; Li, J.; Li, J.G. Metal/Semiconductor Nanocomposites for Photocatalysis: Fundamentals, Structures, Applications and Properties. Nanomaterials 2019, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Abed, J.; Rajput, N.S.; El Moutaouakil, A.; Jouiad, M. Recent Advances in the Design of Plasmonic Au/TiO2 Nanostructures for Enhanced Photocatalytic Water Splitting. Nanomaterials 2020, 10, 2260. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, M.; Yamamoto, K.; Ohta, T.; Inoue, T.; Mizoguchi, K.; Nakaya, M.; Eguchi, T.; Nakajima, A. Confined Hot Electron Relaxation at the Molecular Heterointerface of the Size-Selected Plasmonic Noble Metal Nanocluster and Layered C60. ACS Nano 2021, 15, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Choi, J.; Kang, M.; Lee, H.; Ihee, H.; Park, J.Y. Relaxation Dynamics of Enhanced Hot-Electron Flow on Perovskite-Coupled Plasmonic Silver Schottky Nanodiodes. J. Phys. Chem. C 2021, 125, 2575–2582. [Google Scholar] [CrossRef]
- Hattori, Y.; Meng, J.; Zheng, K.; Meier de Andrade, A.; Kullgren, J.; Broqvist, P.; Nordlander, P.; Sá, J. Phonon-Assisted Hot Carrier Generation in Plasmonic Semiconductor Systems. Nano Lett. 2021, 21, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Cullen, D.A.; McBride, J.R.; Lian, T. Harvesting Sub-Bandgap IR Photons by Photothermionic Hot Electron Transfer in a Plasmonic p–n Junction. Nano Lett. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wu, S.; Dong, J.; Wang, R.; Wang, J.; Zhang, J.; Zhong, S.; Bai, S. Interfacial facet engineering on the Schottky barrier between plasmonic Au and TiO2 in boosting the photocatalytic CO2 reduction under ultraviolet and visible light irradiation. Chem. Eng. J. (Lausanne) 2021, 404, 127145. [Google Scholar] [CrossRef]
- Manuel, A.P.; Kirkey, A.; Mahdi, N.; Shankar, K. Plexcitonics—fundamental principles and optoelectronic applications. J. Mater. Chem. C 2019, 7, 1821–1853. [Google Scholar] [CrossRef]
- Zeng, S.; Vahidzadeh, E.; VanEssen, C.G.; Kar, P.; Kisslinger, R.; Goswami, A.; Zhang, Y.; Mahdi, N.; Riddell, S.; Kobryn, A.E.; et al. Optical control of selectivity of high rate CO2 photoreduction via interband- or hot electron Z-scheme reaction pathways in Au-TiO2 plasmonic photonic crystal photocatalyst. Appl. Catal. B-Environ. 2020, 267, 118644. [Google Scholar] [CrossRef]
- Ridley, B.K. Hot electrons in semiconductors. Sci. Prog. 1933 1986, 70, 425–459. [Google Scholar]
- Keyling, R.; Schöne, W.-D.; Ekardt, W. Comparison of the lifetime of excited electrons in noble metals. Phys. Rev. B 2000, 61, 1670–1673. [Google Scholar] [CrossRef]
- Mukherjee, S.; Libisch, F.; Large, N.; Neumann, O.; Brown, L.V.; Cheng, J.; Lassiter, J.B.; Carter, E.A.; Nordlander, P.; Halas, N.J. Hot Electrons Do the Impossible: Plasmon-Induced Dissociation of H2 on Au. Nano Lett. 2013, 13, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Brongersma, M.L.; Halas, N.J.; Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 2015, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Manuel, A.P.; Barya, P.; Riddell, S.; Zeng, S.; Alam, K.M.; Shankar, K. Plasmonic photocatalysis and SERS sensing using ellipsometrically modeled Ag nanoisland substrates. Nanotechnology 2020, 31, 365301. [Google Scholar] [CrossRef] [PubMed]
- Karaballi, R.A.; Esfahani Monfared, Y.; Dasog, M. Photothermal Transduction Efficiencies of Plasmonic Group 4 Metal Nitride Nanocrystals. Langmuir 2020, 36, 5058–5064. [Google Scholar] [CrossRef] [PubMed]
- Rej, S.; Mascaretti, L.; Santiago, E.Y.; Tomanec, O.; Kment, Š.; Wang, Z.; Zbořil, R.; Fornasiero, P.; Govorov, A.O.; Naldoni, A. Determining Plasmonic Hot Electrons and Photothermal Effects during H2 Evolution with TiN–Pt Nanohybrids. ACS Catal. 2020, 10, 5261–5271. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37. [Google Scholar] [CrossRef] [PubMed]
- Moskovits, M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. [Google Scholar] [CrossRef]
- Frischkorn, C.; Wolf, M. Femtochemistry at Metal Surfaces: Nonadiabatic Reaction Dynamics. Chem. Rev. 2006, 106, 4207–4233. [Google Scholar] [CrossRef] [PubMed]
- Buntin, S.A.; Richter, L.J.; Cavanagh, R.R.; King, D.S. Optically Driven Surface Reactions: Evidence for the Role of Hot Electrons. Phys. Rev. Lett. 1988, 61, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Bonn, M.; Funk, S.; Hess, C.; Denzler, D.N.; Stampfl, C.; Scheffler, M.; Wolf, M.; Ertl, G. Phonon- Versus Electron-Mediated Desorption and Oxidation of CO on Ru(0001). Science 1999, 285, 1042. [Google Scholar] [CrossRef]
- Kao, F.J.; Busch, D.G.; Gomes da Costa, D.; Ho, W. Femtosecond versus nanosecond surface photochemistry: O2+CO on Pt(111) at 80 K. Phys. Rev. Lett. 1993, 70, 4098–4101. [Google Scholar] [CrossRef] [PubMed]
- Eustis, S.; El-Sayed, M.A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217. [Google Scholar] [CrossRef]
- Yu, X.; Liu, F.; Bi, J.; Wang, B.; Yang, S. Improving the plasmonic efficiency of the Au nanorod-semiconductor photocatalysis toward water reduction by constructing a unique hot-dog nanostructure. Nano Energy 2017, 33, 469–475. [Google Scholar] [CrossRef]
- Wu, L.; Tsunenari, N.; Nishi, H.; Sugawa, K.; Otsuki, J.; Tatsuma, T. Two-Dimensional Arrays of Au Halfshells with Different Sizes for Plasmon-Induced Charge Separation. ChemistrySelect 2017, 2, 3744–3749. [Google Scholar] [CrossRef]
- Wu, X.; Ming, T.; Wang, X.; Wang, P.; Wang, J.; Chen, J. High-Photoluminescence-Yield Gold Nanocubes: For Cell Imaging and Photothermal Therapy. ACS Nano 2010, 4, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Fujitsuka, M.; Majima, T. Pt–Au Triangular Nanoprisms with Strong Dipole Plasmon Resonance for Hydrogen Generation Studied by Single-Particle Spectroscopy. ACS Nano 2016, 10, 6299–6305. [Google Scholar] [CrossRef] [PubMed]
- Hartland, G.V.; Besteiro, L.V.; Johns, P.; Govorov, A.O. What’s so Hot about Electrons in Metal Nanoparticles? ACS Energy Lett. 2017, 2, 1641–1653. [Google Scholar] [CrossRef]
- Haegel, N. Relaxation semiconductors: In theory and in practice. Appl. Phys. A 1991, 53, 1–7. [Google Scholar] [CrossRef]
- Zarifi, M.H.; Mohammadpour, A.; Farsinezhad, S.; Wiltshire, B.D.; Nosrati, M.; Askar, A.M.; Daneshmand, M.; Shankar, K. Time-Resolved Microwave Photoconductivity (TRMC) Using Planar Microwave Resonators: Application to the Study of Long-Lived Charge Pairs in Photoexcited Titania Nanotube Arrays. J. Phys. Chem. C 2015, 119, 14358–14365. [Google Scholar] [CrossRef]
- DuChene, J.S.; Sweeny, B.C.; Johnston-Peck, A.C.; Su, D.; Stach, E.A.; Wei, W.D. Prolonged Hot Electron Dynamics in Plasmonic-Metal/Semiconductor Heterostructures with Implications for Solar Photocatalysis. Angew. Chem. Int. Ed. 2014, 53, 7887–7891. [Google Scholar] [CrossRef]
- Sundararaman, R.; Narang, P.; Jermyn, A.S.; Goddard Iii, W.A.; Atwater, H.A. Theoretical predictions for hot-carrier generation from surface plasmon decay. Nat. Commun. 2014, 5, 5788. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Sundararaman, R.; Narang, P.; Goddard, W.A.; Atwater, H.A. Nonradiative Plasmon Decay and Hot Carrier Dynamics: Effects of Phonons, Surfaces, and Geometry. ACS Nano 2016, 10, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Hazra, A.; Das, S.; Kanungo, J.; Sarkar, C.K.; Basu, S. Studies on a resistive gas sensor based on sol–gel grown nanocrystalline p-TiO2 thin film for fast hydrogen detection. Sens. Actuators B Chem. 2013, 183, 87–95. [Google Scholar] [CrossRef]
- Kar, P.; Zhang, Y.; Farsinezhad, S.; Mohammadpour, A.; Wiltshire, B.D.; Sharma, H.; Shankar, K. Rutile phase n-and p-type anodic titania nanotube arrays with square-shaped pore morphologies. Chem. Commun. 2015, 51, 7816–7819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, S.; Su, X.; Xu, J.; Nie, G.; Zhang, Y.; He, Y.; Yu, S. Synthesis and characterization of Ag-loaded p-type TiO2 for adsorption and photocatalytic degradation of tetrabromobisphenol A. Water Environ. Res. 2020, 92, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei Akbari, M.; Hai, Z.; Wei, Z.; Hu, J.; Zhuiykov, S. Wafer-scale two-dimensional Au-TiO2 bilayer films for photocatalytic degradation of Palmitic acid under UV and visible light illumination. Mater. Res. Bull. 2017, 95, 380–391. [Google Scholar] [CrossRef]
- Jellison, G.E.; Boatner, L.A.; Budai, J.D.; Jeong, B.S.; Norton, D.P. Spectroscopic ellipsometry of thin film and bulk anatase (TiO2). J. Appl. Phys. 2003, 93, 9537–9541. [Google Scholar] [CrossRef]
- Takahashi, M.; Tsukigi, K.; Uchino, T.; Yoko, T. Enhanced photocurrent in thin film TiO2 electrodes prepared by sol–gel method. Thin Solid Films 2001, 388, 231–236. [Google Scholar] [CrossRef]
- Salvador, P. Hole diffusion length in n-TiO2 single crystals and sintered electrodes: Photoelectrochemical determination and comparative analysis. J. Appl. Phys. 1984, 55, 2977–2985. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Sun, P.; Lu, S.; Wang, L.; Wang, C.; Liu, Y. Photoelectrochemical Water Splitting with Rutile TiO2 Nanowires Array: Synergistic Effect of Hydrogen Treatment and Surface Modification with Anatase Nanoparticles. Electrochim. Acta 2014, 130, 290–295. [Google Scholar] [CrossRef]
- Mohammadpour, A.; Kar, P.; Wiltshire, B.D.; Askar, A.M.; Shankar, K. Electron transport, trapping and recombination in anodic TiO2 nanotube arrays. Curr. Nanosci. 2015, 11, 593–614. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 Reduction with H2O on TiO2 Nanocrystals: Comparison of Anatase, Rutile, and Brookite Polymorphs and Exploration of Surface Chemistry. ACS Catal. 2012, 2, 1817–1828. [Google Scholar] [CrossRef]
- Pan, J.; Wu, X.; Wang, L.; Liu, G.; Lu, G.Q.; Cheng, H.-M. Synthesis of anatase TiO2 rods with dominant reactive {010} facets for the photoreduction of CO2 to CH4 and use in dye-sensitized solar cells. Chem. Commun. 2011, 47, 8361–8363. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, L.; Andino, J.M.; Li, Y. Bicrystalline TiO2 with controllable anatase-brookite phase content for enhanced CO2 photoreduction to fuels. J. Mater. Chem. A 2013, 1, 8209–8216. [Google Scholar] [CrossRef]
- Anpo, M.; Nakaya, H.; Kodama, S.; Kubokawa, Y.; Domen, K.; Onishi, T. Photocatalysis over binary metal oxides. Enhancement of the photocatalytic activity of titanium dioxide in titanium-silicon oxides. J. Phys. Chem. 1986, 90, 1633–1636. [Google Scholar] [CrossRef]
- Wang, W.-N.; Park, J.; Biswas, P. Rapid synthesis of nanostructured Cu-TiO2-SiO2 composites for CO2 photoreduction by evaporation driven self-assembly. Catal. Sci. Technol. 2011, 1, 593–600. [Google Scholar] [CrossRef]
- Kongkanand, A.; Tvrdy, K.; Takechi, K.; Kuno, M.; Kamat, P.V. Quantum Dot Solar Cells. Tuning Photoresponse through Size and Shape Control of CdSe−TiO2 Architecture. J. Am. Chem. Soc. 2008, 130, 4007–4015. [Google Scholar] [CrossRef] [PubMed]
- Prabakar, K.; Minkyu, S.; Inyoung, S.; Heeje, K. CdSe quantum dots co-sensitized TiO 2 photoelectrodes: Particle size dependent properties. J. Phys. D Appl. Phys. 2010, 43, 012002. [Google Scholar] [CrossRef]
- Sachs, M.; Pastor, E.; Kafizas, A.; Durrant, J.R. Evaluation of Surface State Mediated Charge Recombination in Anatase and Rutile TiO2. J. Phys. Chem. Lett. 2016, 7, 3742–3746. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bian, Z.; Zhu, J.; Huo, Y.; Li, H.; Lu, Y. Mesoporous Au/TiO2 Nanocomposites with Enhanced Photocatalytic Activity. J. Am. Chem. Soc. 2007, 129, 4538–4539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Bao, J.; Liang, L.; Xie, Y.; Wu, H.B.; Lou, X.W. Ordered macroporous BiVO4 architectures with controllable dual porosity for efficient solar water splitting. Angew. Chem. Int. Ed. 2013, 52, 8579–8583. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tatsuma, T. Mechanisms and applications of plasmon-induced charge separation at TiO2 films loaded with gold nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637. [Google Scholar] [CrossRef] [PubMed]
- Radzig, M.; Koksharova, O.; Khmel, I.; Ivanov, V.; Yorov, K.; Kiwi, J.; Rtimi, S.; Tastekova, E.; Aybush, A.; Nadtochenko, V. Femtosecond Spectroscopy of Au Hot-Electron Injection into TiO2: Evidence for Au/TiO2 Plasmon Photocatalysis by Bactericidal Au Ions and Related Phenomena. Nanomaterials 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Negrin-Montecelo, Y.; Testa-Anta, M.; Marin-Caba, L.; Perez-Lorenzo, M.; Salgueirino, V.; Correa-Duarte, M.A.; Comesana-Hermo, M. Titanate Nanowires as One-Dimensional Hot Spot Generators for Broadband Au-TiO2 Photocatalysis. Nanomaterials 2019, 9, 990. [Google Scholar] [CrossRef] [PubMed]
- Linh, V.T.N.; Xiao, X.F.; Jung, H.S.; Giannini, V.; Maier, S.A.; Kim, D.H.; Lee, Y.I.; Park, S.G. Compact Integration of TiO2 Nanoparticles into the Cross-Points of 3D Vertically Stacked Ag Nanowires for Plasmon-Enhanced Photocatalysis. Nanomaterials 2019, 9, 468. [Google Scholar] [CrossRef]
- White, T.P.; Catchpole, K.R. Plasmon-enhanced internal photoemission for photovoltaics: Theoretical efficiency limits. Appl. Phys. Lett. 2012, 101, 073905. [Google Scholar] [CrossRef]
- Yu, H.; Peng, Y.; Yang, Y.; Li, Z.-Y. Plasmon-enhanced light–matter interactions and applications. NPJ Comput. Mater. 2019, 5, 45. [Google Scholar] [CrossRef]
- Zorić, I.; Zäch, M.; Kasemo, B.; Langhammer, C. Gold, Platinum, and Aluminum Nanodisk Plasmons: Material Independence, Subradiance, and Damping Mechanisms. ACS Nano 2011, 5, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Vahidzadeh, E.; Zeng, S.; Manuel, A.P.; Riddell, S.; Kumar, P.; Alam, K.M.; Shankar, K. Asymmetric Multipole Plasmon-Mediated Catalysis Shifts the Product Selectivity of CO2 Photoreduction toward C2+ Products. ACS Appl. Mater. Inter. 2021, 13, 7248–7258. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; El-Sayed, M.A. Gold and Silver Nanoparticles in Sensing and Imaging: Sensitivity of Plasmon Response to Size, Shape, and Metal Composition. J. Phys. Chem. B 2006, 110, 19220–19225. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Wang, Z.L.; El-Sayed, M.A. Alloy Formation of Gold−Silver Nanoparticles and the Dependence of the Plasmon Absorption on Their Composition. J. Phys. Chem. B 1999, 103, 3529–3533. [Google Scholar] [CrossRef]
- Jiang, T.; Jia, C.; Zhang, L.; He, S.; Sang, Y.; Li, H.; Li, Y.; Xu, X.; Liu, H. Gold and gold-palladium alloy nanoparticles on heterostructured TiO2 nanobelts as plasmonic photocatalysts for benzyl alcohol oxidation. Nanoscale 2015, 7, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Wiley, B.J.; Im, S.H.; Li, Z.-Y.; McLellan, J.; Siekkinen, A.; Xia, Y. Maneuvering the Surface Plasmon Resonance of Silver Nanostructures through Shape-Controlled Synthesis. J. Phys. Chem. B 2006, 110, 15666–15675. [Google Scholar] [CrossRef] [PubMed]
- Bonatti, L.; Gil, G.; Giovannini, T.; Corni, S.; Cappelli, C. Plasmonic Resonances of Metal Nanoparticles: Atomistic vs. Continuum Approaches. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Gutiérrez, Y.; Brown, A.S.; Moreno, F.; Losurdo, M. Plasmonics beyond noble metals: Exploiting phase and compositional changes for manipulating plasmonic performance. J. Appl. Phys. 2020, 128, 080901. [Google Scholar] [CrossRef]
- Zhao, Y.; Burda, C. Development of plasmonic semiconductor nanomaterials with copper chalcogenides for a future with sustainable energy materials. Energy Environ. Sci. 2012, 5, 5564–5576. [Google Scholar] [CrossRef]
- Naik, G.V.; Schroeder, J.L.; Ni, X.; Kildishev, A.V.; Sands, T.D.; Boltasseva, A. Titanium nitride as a plasmonic material for visible and near-infrared wavelengths. Opt. Mater. Express 2012, 2, 478–489. [Google Scholar] [CrossRef]
- Karaballi, R.A.; Humagain, G.; Fleischman, B.R.; Dasog, M. Synthesis of Plasmonic Group-4 Nitride Nanocrystals by Solid-State Metathesis. Angew. Chem. 2019, 131, 3179–3182. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, I.H.; Qian, W.; El-Sayed, M.A. Cancer Cell Imaging and Photothermal Therapy in the Near-Infrared Region by Using Gold Nanorods. J. Am. Chem. Soc. 2006, 128, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.R.; Stafford, R.J.; Bankson, J.A.; Sershen, S.R.; Rivera, B.; Price, R.E.; Hazle, J.D.; Halas, N.J.; West, J.L. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc. Natl. Acad. Sci. USA 2003, 100, 13549–13554. [Google Scholar] [CrossRef] [PubMed]
- Laura, B.C.; Lissett, R.B.; Germaine, A.; Tse-Kuan, Y.; Rachel, S.; Yi, L.; Rebekah, A.D. Immunoconjugated gold nanoshell-mediated photothermal ablation of trastuzumab-resistant breast cancer cells. Breast Cancer Res. Treat. 2011, 125, 27–34. [Google Scholar] [CrossRef]
- Zhou, W.; Dridi, M.; Suh, J.Y.; Kim, C.H.; Co, D.T.; Wasielewski, M.R.; Schatz, G.C.; Odom, T.W. Lasing action in strongly coupled plasmonic nanocavity arrays. Nat. Nanotechnol. 2013, 8, 506. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-J.; Kim, J.; Chen, H.-Y.; Wu, C.; Dabidian, N.; Sanders, C.E.; Wang, C.-Y.; Lu, M.-Y.; Li, B.-H.; Qiu, X.; et al. Plasmonic Nanolaser Using Epitaxially Grown Silver Film. Science 2012, 337, 450. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.K.; Huang, X.; El-Sayed, I.H.; El-Sayed, M.A. Noble Metals on the Nanoscale: Optical and Photothermal Properties and Some Applications in Imaging, Sensing, Biology, and Medicine. Acc. Chem. Res. 2008, 41, 1578–1586. [Google Scholar] [CrossRef]
- Loo, C.; Lin, A.; Hirsch, L.; Lee, M.-H.; Barton, J.; Halas, N.; West, J.; Drezek, R. Nanoshell-Enabled Photonics-Based Imaging and Therapy of Cancer. Technol. Cancer Res. Treat. 2004, 3, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Moskovits, M. Surface-enhanced Raman spectroscopy: A brief retrospective. J. Raman Spectrosc. 2005, 36, 485–496. [Google Scholar] [CrossRef]
- Marimuthu, A.; Christopher, P.; Linic, S. Design of Plasmonic Platforms for Selective Molecular Sensing Based on Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2012, 116, 9824–9829. [Google Scholar] [CrossRef]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, G.; Yu, J.; Fan, W. Surface plasmon resonance-mediated photocatalysis by noble metal-based composites under visible light. J. Mater. Chem. 2012, 22, 21337–21354. [Google Scholar] [CrossRef]
- Atwater, H.A.; Polman, A. Plasmonics for improved photovoltaic devices. Nat. Mater. 2010, 9, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, X.; Wu, Y.; Zhang, X.; Ren, X. Plasmon-Enhanced Light Absorption in GaAs Nanowire Array Solar Cells. Nanoscale Res. Lett. 2015, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Boerigter, C.; Aslam, U.; Linic, S. Mechanism of Charge Transfer from Plasmonic Nanostructures to Chemically Attached Materials. ACS Nano 2016, 10, 6108–6115. [Google Scholar] [CrossRef]
- Alexei, D.S.; Gregory, N.G.t.; Roman, S. Hot-electron effect in superconductors and its applications for radiation sensors. Supercond. Sci. Technol. 2002, 15, R1. [Google Scholar]
- Le Ru, E.C.; Etchegoin, P.G. Principles of Surface-Enhanced Raman Spectroscopy. [Electronic Resource]: And Related Plasmonic Effects, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Jana, J.; Ganguly, M.; Pal, T. Enlightening surface plasmon resonance effect of metal nanoparticles for practical spectroscopic application. RSC Adv. 2016, 6, 86174–86211. [Google Scholar] [CrossRef]
- Wark, A.W.; Lee, H.J.; Corn, R.M. Long-Range Surface Plasmon Resonance Imaging for Bioaffinity Sensors. Anal. Chem. 2005, 77, 3904–3907. [Google Scholar] [CrossRef]
- Stewart, M.E.; Anderton, C.R.; Thompson, L.B.; Maria, J.; Gray, S.K.; Rogers, J.A.; Nuzzo, R.G. Nanostructured Plasmonic Sensors. Chem. Rev. 2008, 108, 494–521. [Google Scholar] [CrossRef]
- Kale, M.J.; Avanesian, T.; Christopher, P. Direct Photocatalysis by Plasmonic Nanostructures. ACS Catal. 2013, 4, 116–128. [Google Scholar] [CrossRef]
- Sönnichsen, C.; Franzl, T.; Wilk, T.; von Plessen, G.; Feldmann, J.; Wilson, O.; Mulvaney, P. Drastic Reduction of Plasmon Damping in Gold Nanorods. Phys. Rev. Lett. 2002, 88, 077402. [Google Scholar] [CrossRef]
- Lamprecht, B.; Leitner, A.; Aussenegg, F.R. SHG studies of plasmon dephasing in nanoparticles. Appl. Phys. B 1999, 68, 419–423. [Google Scholar] [CrossRef]
- Tsu, R. Landau Damping and Dispersion of Phonon, Plasmon, and Photon Waves in Polar Semiconductors. Phys. Rev. 1967, 164, 380–383. [Google Scholar] [CrossRef]
- Perner, M.; Klar, T.; Grosse, S.; Lemmer, U.; von Plessen, G.; Spirkl, W.; Feldmann, J. Homogeneous line widths of surface plasmons in gold nanoparticles measured by femtosecond pump-and-probe and near-field optical spectroscopy. J. Lumin. 1998, 76–77, 181–184. [Google Scholar] [CrossRef]
- Voisin, C.; Christofilos, D.; Del Fatti, N.; Vallée, F.; Prével, B.; Cottancin, E.; Lermé, J.; Pellarin, M.; Broyer, M. Size-Dependent Electron-Electron Interactions in Metal Nanoparticles. Phys. Rev. Lett. 2000, 85, 2200–2203. [Google Scholar] [CrossRef] [PubMed]
- Voisin, C.; Del Fatti, N.; Christofilos, D.; Vallée, F. Ultrafast Electron Dynamics and Optical Nonlinearities in Metal Nanoparticles. J. Phys. Chem. B 2001, 105, 2264–2280. [Google Scholar] [CrossRef]
- Ratchford, D.C.; Dunkelberger, A.D.; Owrutsky, J.C.; Pehrsson, P.E.; Vurgaftman, I. Quantification of Efficient Plasmonic Hot-Electron Injection in Gold Nanoparticle-TiO2 Films. Nano Lett. 2017, 17, 6047–6055. [Google Scholar] [CrossRef]
- Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast Plasmon-Induced Electron Transfer from Gold Nanodots into TiO2 Nanoparticles. J. Am. Chem. Soc. 2007, 129, 14852–14853. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Furube, A.; Hara, K.; Katoh, R.; Tachiya, M. Ultrafast plasmon induced electron injection mechanism in gold–TiO2 nanoparticle system. J. Photochem. Photobiol. C Photochem. Rev. 2013, 15, 21–30. [Google Scholar] [CrossRef]
- Du, L.; Furube, A.; Yamamoto, K.; Hara, K.; Katoh, R.; Tachiya, M. Plasmon-Induced Charge Separation and Recombination Dynamics in Gold−TiO2 Nanoparticle Systems: Dependence on TiO2 Particle Size. J. Phys. Chem. C 2009, 113, 6454–6462. [Google Scholar] [CrossRef]
- Wu, K.; Chen, J.; McBride, J.R.; Lian, T. Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition. Science 2015, 349, 632. [Google Scholar] [CrossRef]
- Nosaka, Y.; Takahashi, S.; Sakamoto, H.; Nosaka, A.Y. Reaction Mechanism of Cu(II)-Grafted Visible-Light Responsive TiO2 and WO3 Photocatalysts Studied by Means of ESR Spectroscopy and Chemiluminescence Photometry. J. Phys. Chem. C 2011, 115, 21283–21290. [Google Scholar] [CrossRef]
- Nogawa, T.; Isobe, T.; Matsushita, S.; Nakajima, A. Preparation and visible-light photocatalytic activity of Au- and Cu-modified TiO2 powders. Mater. Lett. 2012, 82, 174–177. [Google Scholar] [CrossRef]
- Zhang, X.; Han, F.; Shi, B.; Farsinezhad, S.; Dechaine, G.P.; Shankar, K. Photocatalytic Conversion of Diluted CO2 into Light Hydrocarbons Using Periodically Modulated Multiwalled Nanotube Arrays. Angew. Chem. 2012, 124, 12904–12907. [Google Scholar] [CrossRef]
- Farsinezhad, S.; Sharma, H.; Shankar, K. Interfacial band alignment for photocatalytic charge separation in TiO2 nanotube arrays coated with CuPt nanoparticles. Phys. Chem. Chem. Phys. 2015, 17, 29723–29733. [Google Scholar] [CrossRef] [PubMed]
- Rtimi, S.; Pulgarin, C.; Sanjines, R.; Kiwi, J. Accelerated self-cleaning by Cu promoted semiconductor binary-oxides under low intensity sunlight irradiation. Appl. Catal. B-Environ. 2016, 180, 648–655. [Google Scholar] [CrossRef]
- Bernareggi, M.; Dozzi, M.; Bettini, L.; Ferretti, A.; Chiarello, G.; Selli, E. Flame-Made Cu/TiO2 and Cu-Pt/TiO2 Photocatalysts for Hydrogen Production. Catalysts 2017, 7, 301. [Google Scholar] [CrossRef]
- Kar, P.; Zhang, Y.; Mahdi, N.; Thakur, U.K.; Wiltshire, B.D.; Kisslinger, R.; Shankar, K. Heterojunctions of mixed phase TiO2 nanotubes with Cu, CuPt, and Pt nanoparticles: Interfacial band alignment and visible light photoelectrochemical activity. Nanotechnology 2017, 29, 014002. [Google Scholar] [CrossRef] [PubMed]
- Asbury, J.B.; Hao, E.; Wang, Y.; Ghosh, H.N.; Lian, T. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semiconductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105, 4545–4557. [Google Scholar] [CrossRef]
- Anderson, N.A.; Lian, T. Ultrafast Electron Transfer at the Molecule-Semiconductor Nanoparticle Interface. Annu. Rev. Phys. Chem. 2004, 56, 491–519. [Google Scholar] [CrossRef] [PubMed]
- Boerigter, C.; Campana, R.; Morabito, M.; Linic, S. Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis. Nat. Commun. 2016, 7, 10545. [Google Scholar] [CrossRef] [PubMed]
- Foerster, B.; Kaefer, K.; Celiksoy, S.; Sönnichsen, C.; Joplin, A.; Link, S. Chemical Interface Damping Depends on Electrons Reaching the Surface. ACS Nano 2017, 11, 2886–2893. [Google Scholar] [CrossRef]
- Hou, W.; Hung, W.H.; Pavaskar, P.; Goeppert, A.; Aykol, M.; Cronin, S.B. Photocatalytic Conversion of CO2 to Hydrocarbon Fuels via Plasmon-Enhanced Absorption and Metallic Interband Transitions. ACS Catal. 2011, 1, 929–936. [Google Scholar] [CrossRef]
- Deng, X.Q.; Zhu, B.; Li, X.S.; Liu, J.L.; Zhu, X.B.; Zhu, A.M. Visible-light photocatalytic oxidation of CO over plasmonic Au/TiO2: Unusual features of oxygen plasma activation. Appl. Catal. B-Environ. 2016, 188, 48–55. [Google Scholar] [CrossRef]
- Subramanian, A.; Pan, Z.H.; Li, H.F.; Zhou, L.S.; Li, W.F.; Qiu, Y.C.; Xu, Y.J.; Hou, Y.; Muzi, C.; Zhang, Y.G. Synergistic promotion of photoelectrochemical water splitting efficiency of TiO2 nanorods using metal-semiconducting nanoparticles. Appl. Surf. Sci. 2017, 420, 631–637. [Google Scholar] [CrossRef]
- Lang, Q.Q.; Chen, Y.H.; Huang, T.L.; Yang, L.N.; Zhong, S.X.; Wu, L.J.; Chen, J.R.; Bai, S. Graphene “bridge” in transferring hot electrons from plasmonic Ag nanocubes to TiO2 nanosheets for enhanced visible light photocatalytic hydrogen evolution. Appl. Catal. B-Environ. 2018, 220, 182–190. [Google Scholar] [CrossRef]
- Lee, S.Y.; Tsalu, P.V.; Kim, G.W.; Seo, M.J.; Hong, J.W.; Ha, J.W. Tuning Chemical Interface Damping: Interfacial Electronic Effects of Adsorbate Molecules and Sharp Tips of Single Gold Bipyramids. Nano Lett. 2019, 19, 2568–2574. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Yasumoto, N.; Imai, J.; Sakamoto, H.; Tanaka, S.; Ichikawa, S.; Ohtani, B.; Hirai, T. Quantum tunneling injection of hot electrons in Au/TiO2 plasmonic photocatalysts. Nanoscale 2017, 9, 8349–8361. [Google Scholar] [CrossRef]
- Valenti, M.; Venugopal, A.; Schmidt-Ott, A.; Smith, W.A.; Tordera, D.; Jonsson, M.P.; Biskos, G. Hot Carrier Generation and Extraction of Plasmonic Alloy Nanoparticles. ACS Photonics 2017, 4, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Niemantsverdriet, J.W. Spectroscopy in Catalysis: An Introduction, 3rd ed.; Wiley-VCH: Weinheim, Germamy; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Lee, H.; Lee, Y.K.; Hwang, E.; Park, J.Y. Enhanced Surface Plasmon Effect of Ag/TiO2 Nanodiodes on Internal Photoemission. J. Phys. Chem. C 2014, 118, 5650–5656. [Google Scholar] [CrossRef]
- Kar, P.; Farsinezhad, S.; Mahdi, N.; Zhang, Y.; Obuekwe, U.; Sharma, H.; Shen, J.; Semagina, N.; Shankar, K. Enhanced CH4 yield by photocatalytic CO2 reduction using TiO2 nanotube arrays grafted with Au, Ru, and ZnPd nanoparticles. Nano Res. 2016, 9, 3478–3493. [Google Scholar] [CrossRef]
- Ayas, S.; Cupallari, A.; Dana, A. Probing hot-electron effects in wide area plasmonic surfaces using X-ray photoelectron spectroscopy. Appl. Phys. Lett. 2014, 105, 221608. [Google Scholar] [CrossRef]
- Gessner, O.; Gühr, M. Monitoring Ultrafast Chemical Dynamics by Time-Domain X-ray Photo- and Auger-Electron Spectroscopy. Acc. Chem. Res. 2016, 49, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, P.; Adisak, B.; Ouyang, S.; Umezawa, N.; Ye, J.; Kodiyath, R.; Tanabe, T.; Ramesh, G.V.; Ueda, S.; et al. Gold photosensitized SrTiO3 for visible-light water oxidation induced by Au interband transitions. J. Mater. Chem. A 2014, 2. [Google Scholar] [CrossRef]
- Szymanski, P.; Garrett-Roe, S.; Harris, C.B. Time- and angle-resolved two-photon photoemission studies of electron localization and solvation at interfaces. Prog. Surf. Sci. 2005, 78, 1–39. [Google Scholar] [CrossRef]
- Bauer, M.; Marienfeld, A.; Aeschlimann, M. Hot electron lifetimes in metals probed by time-resolved two-photon photoemission. Prog. Surf. Sci. 2015, 90, 319–376. [Google Scholar] [CrossRef]
- Tan, S.; Argondizzo, A.; Ren, J.; Liu, L.; Zhao, J.; Petek, H. Plasmonic coupling at a metal/semiconductor interface. Nat. Photonics 2017, 11, 806–812. [Google Scholar] [CrossRef]
- Lüth, H. Solid Surfaces, Interfaces and Thin Films, 6th ed.; Springer: Cham, Switzerland; New York, NY, USA, 2015. [Google Scholar]
- Ertl, G.; Küppers, J. Low Energy Electrons and Surface Chemistry, 2nd ed.; VCH: Weinheim, Germany; Deerfield Beach, FL, USA, 1985. [Google Scholar]
- Briggs, D.; Seah, M.P. Practical Surface Analysis, 2nd ed.; Wiley: Chichester, UK; New York, NY, USA, 1990. [Google Scholar]
- Carlson, T.A. Photoelectron and Auger Spectroscopy; Plenum Press: New York, NY, USA, 1975. [Google Scholar]
- Oura, K.; Katayama, M.; Zotov, A.V.; Lifshits, V.G.; Saranin, A.A. Surface Science: An Introduction; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Berenyi, Z.; Aszalós-Kiss, B.; Csik, A.; Tóth, J.; Kövér, L.; Varga, D. Separation of extrinsic and intrinsic plasmon excitations in Ge KLL Auger spectra. ATOMKI Annu. Rep. 2002, 33, 31. [Google Scholar]
- Herzing, A.A.; Guler, U.; Boltasseva, A.; Shalaev, V.; Zhou, X.; Norris, T.B. Electron energy loss spectroscopy of plasmon resonances in titanium nitride thin films. Appl. Phys. Lett. 2016, 108. [Google Scholar] [CrossRef]
- Kawata, S.; Inouye, Y.; Verma, P. Plasmonics for near-field nano-imaging and superlensing. Nat. Photonics 2009, 3, 388. [Google Scholar] [CrossRef]
- Colliex, C.; Kociak, M.; Stéphan, O. Electron Energy Loss Spectroscopy imaging of surface plasmons at the nanometer scale. Ultramicroscopy 2016, 162, A1–A24. [Google Scholar] [CrossRef]
- Forcherio, G.T.; DeJarnette, D.; Benamara, M.; Roper, D.K. Electron Energy Loss Spectroscopy of Surface Plasmon Resonances on Aberrant Gold Nanostructures. J. Phys. Chem. C 2016, 120, 24950–24956. [Google Scholar] [CrossRef]
- Steiner, P.; Reiter, F.J.; Höchst, H.; Hüfner, S. The KLL Auger spectra of Na and Mg metal and their plasmon structure. Phys. Status Solidi B 1978, 90, 45–51. [Google Scholar] [CrossRef]
- Stolterfoht, N.; Bremer, J.H.; Hoffmann, V.; Rösler, M.; Baragiola, R.A. Auger transitions and plasmon decay produced by hollow atoms at an Al(111) surface. Nuclear Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2002, 193, 523–529. [Google Scholar] [CrossRef]
- Boulenouar, K.; Bouslama, M.H.; Mokadem, A.; Vizzini, S.; Lounis, Z.; Abdellaoui, A.; Reguad, B.; Bedrouni, M.; Hamaida, K.; Guenouna, T.; et al. Auger Electron Spectroscopy, Electron Energy Loss Spectroscopy, UV Photoelectron Spectroscopy, and Photoluminescence Characterization of In2O3 Associated to the Theoretical Calculations Based on the Generalized Gradient Approximation and Modified Becke Johnson. J. Phys. Chem. C 2017, 121, 8345–8352. [Google Scholar] [CrossRef]
- Klar, T.; Perner, M.; Grosse, S.; von Plessen, G.; Spirkl, W.; Feldmann, J. Surface-Plasmon Resonances in Single Metallic Nanoparticles. Phys. Rev. Lett. 1998, 80, 4249–4252. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Park, S.H.; Hwang, S.H.; Jeon, S.; Hwang, B.; Jung, J.Y.; Lee, J.; Jeong, J.H. Three-dimensional plasmonic Ag/TiO2 nanocomposite architectures on flexible substrates for visible-light photocatalytic activity. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Sousa-Castillo, A.; Comesaña-Hermo, M.; Rodríguez-González, B.; Pérez-Lorenzo, M.; Wang, Z.; Kong, X.-T.; Govorov, A.O.; Correa-Duarte, M.A. Boosting Hot Electron-Driven Photocatalysis through Anisotropic Plasmonic Nanoparticles with Hot Spots in Au–TiO2 Nanoarchitectures. J. Phys. Chem. C 2016, 120, 11690–11699. [Google Scholar] [CrossRef]
- Mubeen, S.; Lee, J.; Liu, D.; Stucky, G.D.; Moskovits, M. Panchromatic photoproduction of H2 with surface plasmons. Nano Lett. 2015, 15, 2132–2136. [Google Scholar] [CrossRef] [PubMed]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence: Principles and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Kühn, S.; Mori, G.; Agio, M.; Sandoghdar, V. Modification of single molecule fluorescence close to a nanostructure: Radiation pattern, spontaneous emission and quenching. Mol. Phys. 2008, 106, 893–908. [Google Scholar] [CrossRef]
- Zhang, Y.; Aslan, K.; Previte, M.J.R.; Geddes, C.D. Metal-enhanced fluorescence: Surface plasmons can radiate a fluorophore’s structured emission. Appl. Phys. Lett. 2007, 90, 053107. [Google Scholar] [CrossRef]
- Razgoniaeva, N.; Lambright, S.; Sharma, N.; Acharya, A.; Khon, E.; Moroz, P.; Razgoniaev, A.; Ostrowski, A.; Zamkov, M. Exciton Generation in Semiconductor Nanocrystals via the Near-Field Plasmon Energy Transfer. J. Phys. Chem. C 2015, 119, 15562–15571. [Google Scholar] [CrossRef]
- Fofang, N.T.; Park, T.-H.; Neumann, O.; Mirin, N.A.; Nordlander, P.; Halas, N.J. Plexcitonic Nanoparticles: Plasmon−Exciton Coupling in Nanoshell−J-Aggregate Complexes. Nano Lett. 2008, 8, 3481–3487. [Google Scholar] [CrossRef]
- Soganci, I.M.; Nizamoglu, S.; Mutlugun, E.; Akin, O.; Demir, H.V. Localized plasmon-engineered spontaneous emission of CdSe/ZnS nanocrystals closely-packed in the proximity of Ag nanoisland films for controlling emission linewidth, peak, and intensity. Optics Express 2007, 15, 14289–14298. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.Y.; Choi, K.C.; Ahn, C.W. Surface plasmon-enhanced spontaneous emission rate in an organic light-emitting device structure: Cathode structure for plasmonic application. Appl. Phys. Lett. 2009, 94, 173301. [Google Scholar] [CrossRef]
- Choulis, S.A.; Mathai, M.K.; Choong, V.-E. Influence of metallic nanoparticles on the performance of organic electrophosphorescence devices. Appl. Phys. Lett. 2006, 88, 213503. [Google Scholar] [CrossRef]
- Dostálek, J.; Knoll, W. Biosensors based on surface plasmon-enhanced fluorescence spectroscopy (Review). Biointerphases 2008, 3, FD12–FD22. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, H.-Y.; Wang, H.; Gao, B.-R.; Hao, Y.-w.; Jin, Y.; Chen, Q.-D.; Sun, H.-B. Surface Plasmon Enhanced Fluorescence of Dye Molecules on Metal Grating Films. J. Phys. Chem. C 2011, 115, 12636–12642. [Google Scholar] [CrossRef]
- Lin, K.-Q.; Yi, J.; Hu, S.; Sun, J.-J.; Zheng, J.-T.; Wang, X.; Ren, B. Intraband Hot-Electron Photoluminescence from Single Silver Nanorods. ACS Photonics 2016, 3, 1248–1255. [Google Scholar] [CrossRef]
- Shahbazyan, T.V. Theory of Plasmon-Enhanced Metal Photoluminescence. Nano Lett. 2013, 13, 194–198. [Google Scholar] [CrossRef]
- Najmaei, S.; Mlayah, A.; Arbouet, A.; Girard, C.; Léotin, J.; Lou, J. Plasmonic Pumping of Excitonic Photoluminescence in Hybrid MoS2–Au Nanostructures. ACS Nano 2014, 8, 12682–12689. [Google Scholar] [CrossRef] [PubMed]
- Dulkeith, E.; Niedereichholz, T.; Klar, T.A.; Feldmann, J.; von Plessen, G.; Gittins, D.I.; Mayya, K.S.; Caruso, F. Plasmon emission in photoexcited gold nanoparticles. Phys. Rev. B 2004, 70, 205424. [Google Scholar] [CrossRef]
- Mohamed, M.B.; Volkov, V.; Link, S.; El-Sayed, M.A. The ‘lightning’ gold nanorods: Fluorescence enhancement of over a million compared to the gold metal. Chem. Phys. Lett. 2000, 317, 517–523. [Google Scholar] [CrossRef]
- Paul, K.K.; Giri, P.K. Role of Surface Plasmons and Hot Electrons on the Multi-Step Photocatalytic Decay by Defect Enriched Ag@TiO2 Nanorods under Visible Light. J. Phys. Chem. C 2017, 121, 20016–20030. [Google Scholar] [CrossRef]
- Farsinezhad, S.; Banerjee, S.P.; Bangalore Rajeeva, B.; Wiltshire, B.D.; Sharma, H.; Sura, A.; Mohammadpour, A.; Kar, P.; Fedosejevs, R.; Shankar, K. Reduced Ensemble Plasmon Line Widths and Enhanced Two-Photon Luminescence in Anodically Formed High Surface Area Au-TiO2 3D Nanocomposites. ACS Appl. Mater. Inter. 2017, 9, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, C.; El Harfouch, Y.; Palleau, E.; Eloi, F.; Douillard, L.; Charra, F.; Fiorini-Debuisschert, C.; Marguet, S. Two-Photon Luminescence of Single Colloidal Gold Nanorods: Revealing the Origin of Plasmon Relaxation in Small Nanocrystals. J. Phys. Chem. C 2016, 120, 23136–23143. [Google Scholar] [CrossRef]
- Wang, D.S.; Lin, C.W.; Hsu, F.Y. Surface plasmon effects on two photon luminescence of Gold nanorods. Opt. Express 2009, 17, 11350–11359. [Google Scholar] [CrossRef] [PubMed]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73. [Google Scholar] [CrossRef]
- Haug, T.; Klemm, P.; Bange, S.; Lupton, J.M. Hot-Electron Intraband Luminescence from Single Hot Spots in Noble-Metal Nanoparticle Films. Phys. Rev. Lett. 2015, 115, 067403. [Google Scholar] [CrossRef] [PubMed]
- Imura, K.; Okamoto, H. Properties of Photoluminescence from Single Gold Nanorods Induced by Near-Field Two-Photon Excitation. J. Phys. Chem. C 2009, 113, 11756–11759. [Google Scholar] [CrossRef]
- Siddiquee, A.M.; Taylor, A.B.; Syed, S.; Chon, J.W.M.; Lim, G.H.; Lim, B. Measurement of Plasmon-Mediated Two-Photon Luminescence Action Cross Sections of Single Gold Bipyramids, Dumbbells, and Hemispherically Capped Cylindrical Nanorods. J. Phys. Chem. C 2015, 119, 28536–28543. [Google Scholar] [CrossRef]
- Shank, C.V.; Zakharchenya, B.P. Spectroscopy of Nonequilibrium Electrons and Phonons; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Gardiner, D.J.; Graves, P.R. Practical Raman Spectroscopy; Springer: Berlin/Heidelberg, Germany, 1989. [Google Scholar]
- Ember, K.; Hoeve, M.; McAughtrie, S.; Bergholt, M.S.; Dwyer, B.; Stevens, M.M.; Faulds, K.; Forbes, S.; Campbell, C. Raman Spectroscopy and Regenerative Medicine: A Review. NPJ Regen. Med. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.K.; Gopinath, C.S. Bimetallic and Plasmonic Ag-Au on TiO2 for Solar Water Splitting: An Active Nanocomposite for Entire Visible-Light-Region Absorption. ChemCatChem 2016, 8, 3294–3311. [Google Scholar] [CrossRef]
- Prezgot, D.; Ianoul, A. Probing the Anisotropy of SERS Enhancement with Spatially Separated Plasmonic Modes in Strongly Coupled Silver Nanocubes on a Dielectric Substrate. J. Phys. Chem. C 2015, 119, 3293–3301. [Google Scholar] [CrossRef]
- Jailaubekov, A.E.; Willard, A.P.; Tritsch, J.R.; Chan, W.-L.; Sai, N.; Gearba, R.; Kaake, L.G.; Williams, K.J.; Leung, K.; Rossky, P.J.; et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic photovoltaics. Nat. Mater. 2013, 12, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, Y.; Moser, J.E.; Grätzel, M.; Klug, D.R.; Durrant, J.R. Subpicosecond Interfacial Charge Separation in Dye-Sensitized Nanocrystalline Titanium Dioxide Films. J. Phys. Chem. 1996, 100, 20056–20062. [Google Scholar] [CrossRef]
- Melitz, W.; Shen, J.; Kummel, A.C.; Lee, S. Kelvin probe force microscopy and its application. Surf. Sci. Rep. 2011, 66, 1–27. [Google Scholar] [CrossRef]
- Adhikari, N.; Dubey, A.; Khatiwada, D.; Mitul, A.F.; Wang, Q.; Venkatesan, S.; Iefanova, A.; Zai, J.; Qian, X.; Kumar, M.; et al. Interfacial Study To Suppress Charge Carrier Recombination for High Efficiency Perovskite Solar Cells. ACS Appl. Mater. Inter. 2015, 7, 26445–26454. [Google Scholar] [CrossRef]
- Siddiki, M.K.; Venkatesan, S.; Galipeau, D.; Qiao, Q. Kelvin probe force microscopic imaging of the energy barrier and energetically favorable offset of interfaces in double-junction organic solar cells. ACS Appl. Mater. Inter. 2013, 5, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Jo, J.-S.; Park, J.H.; Lee, S.W.; Jang, J.-W. A hot-electron-triggered catalytic oxidation reaction of plasmonic silver nanoparticles evidenced by surface potential mapping. J. Mater. Chem. A 2018, 6, 20939–20946. [Google Scholar] [CrossRef]
- Kang, Z.; Si, H.; Shi, M.; Xu, C.; Fan, W.; Ma, S.; Kausar, A.; Liao, Q.; Zhang, Z.; Zhang, Y. Kelvin probe force microscopy for perovskite solar cells. Sci. China Mater. 2019, 62, 776–789. [Google Scholar] [CrossRef]
- Chen, R.; Fan, F.; Dittrich, T.; Li, C. Imaging photogenerated charge carriers on surfaces and interfaces of photocatalysts with surface photovoltage microscopy. Chem. Soc. Rev. 2018, 47, 8238–8262. [Google Scholar] [CrossRef]
- Lee, H.; Lee, W.; Lee, J.H.; Yoon, D.S. Surface Potential Analysis of Nanoscale Biomaterials and Devices Using Kelvin Probe Force Microscopy. J. Nanomater. 2016, 2016, 4209130. [Google Scholar] [CrossRef]
- Rosenwaks, Y.; Shikler, R.; Glatzel, T.; Sadewasser, S. Kelvin probe force microscopy of semiconductor surface defects. Phys. Rev. B 2004, 70, 085320. [Google Scholar] [CrossRef]
- Jian, A.; Feng, K.; Jia, H.; Zhang, Q.; Sang, S.; Zhang, X. Quantitative investigation of plasmonic hot-electron injection by KPFM. Appl. Surf. Sci. 2019, 492, 644–650. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, S.W.; Oh, T.; Petrosko, S.H.; Mirkin, C.A.; Jang, J.-W. Direct Observation of Plasmon-Induced Interfacial Charge Separation in Metal/Semiconductor Hybrid Nanostructures by Measuring Surface Potentials. Nano Lett. 2018, 18, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-B.; Sun, X.-J.; Jia, Y.-P.; Stockman, M.I.; Paudel, H.P.; Song, H.; Jiang, H.; Li, Z.-M. Direct observation of localized surface plasmon field enhancement by Kelvin probe force microscopy. Light Sci. Appl. 2017, 6, e17038. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-H.; Wu, J.-J.; Chou, M.M.C.; Chang, Y.-M.; Yoshimura, M. Charge Transfer in Au Nanoparticle–Nonpolar ZnO Photocatalysts Illustrated by Surface-Potential-Derived Three-Dimensional Band Diagram. J. Phys. Chem. C 2014, 118, 19814–19821. [Google Scholar] [CrossRef]
- Yoo, H.; Bae, C.; Yang, Y.; Lee, S.; Kim, M.; Kim, H.; Kim, Y.; Shin, H. Spatial Charge Separation in Asymmetric Structure of Au Nanoparticle on TiO2 Nanotube by Light-Induced Surface Potential Imaging. Nano Lett. 2014, 14, 4413–4417. [Google Scholar] [CrossRef] [PubMed]
- Sönnichsen, C.; Franzl, T.; Wilk, T.; Plessen, G.v.; Feldmann, J. Plasmon resonances in large noble-metal clusters. New J. Phys. 2002, 4, 93. [Google Scholar] [CrossRef]
- Novo, C.; Gomez, D.; Perez-Juste, J.; Zhang, Z.; Petrova, H.; Reismann, M.; Mulvaney, P.; Hartland, G.V. Contributions from radiation damping and surface scattering to the linewidth of the longitudinal plasmon band of gold nanorods: A single particle study. Phys. Chem. Chem. Phys. 2006, 8, 3540–3546. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Novo, C.; Funston, A.; Wang, H.; Staleva, H.; Zou, S.; Mulvaney, P.; Xia, Y.; Hartland, G.V. Dark-field microscopy studies of single metal nanoparticles: Understanding the factors that influence the linewidth of the localized surface plasmon resonance. J. Mater. Chem. 2008, 18, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Femtosecond dynamics of electronic excitations at metal surfaces. Surf. Sci. 1997, 377, 343–349. [Google Scholar] [CrossRef]
- Schmuttenmaer, C.A.; Aeschlimann, M.; Elsayed-Ali, H.E.; Miller, R.J.D.; Mantell, D.A.; Cao, J.; Gao, Y. Time-resolved two-photon photoemission from Cu(100): Energy dependence of electron relaxation. Phys. Rev. B 1994, 50, 8957–8960. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, M.; Bauer, M.; Pawlik, S. Competing nonradiative channels for hot electron induced surface photochemistry. Chem. Phys. 1996, 205, 127–141. [Google Scholar] [CrossRef]
- Ogawa, S.; Petek, H. Femtosecond dynamics of hot-electron relaxation in Cu(110) and Cu(100). Surf. Sci. 1996, 357–358, 585–594. [Google Scholar] [CrossRef]
- Mathias, S.; Wiesenmayer, M.; Deicke, F.; Ruffing, A.; Miaja-Avila, L.; Murnane, M.M.; Kapteyn, H.C.; Bauer, M.; Aeschlimann, M. Time and angle resolved photoemission spectroscopy using femtosecond visible and high-harmonic light. J. Phys. Conf. Ser. 2009, 148, 012042. [Google Scholar] [CrossRef]
- Harutyunyan, H.; Martinson, A.B.; Rosenmann, D.; Khorashad, L.K.; Besteiro, L.V.; Govorov, A.O.; Wiederrecht, G.P. Anomalous ultrafast dynamics of hot plasmonic electrons in nanostructures with hot spots. Nat. Nanotechnol. 2015, 10, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, R.; Hendry, E.; Shan, J.; Heinz, T.F.; Bonn, M. Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 2011, 83, 543–586. [Google Scholar] [CrossRef]
- Beard, M.C.; Turner, G.M.; Schmuttenmaer, C.A. Transient photoconductivity in GaAs as measured by time-resolved terahertz spectroscopy. Phys. Rev. B 2000, 62, 15764–15777. [Google Scholar] [CrossRef]
- Changhwan, L.; Ievgen, I.N.; Young Keun, L.; Changui, A.; Hyosun, L.; Seokwoo, J.; Jeong Young, P. Amplification of hot electron flow by the surface plasmon effect on metal–insulator–metal nanodiodes. Nanotechnology 2015, 26, 445201. [Google Scholar]
- Clavero, C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photonics 2014, 8, 95. [Google Scholar] [CrossRef]
- Ahn, W.; Ratchford, D.C.; Pehrsson, P.E.; Simpkins, B.S. Surface plasmon polariton-induced hot carrier generation for photocatalysis. Nanoscale 2017, 9, 3010–3022. [Google Scholar] [CrossRef]
- Kodiyath, R.; Manikandan, M.; Liu, L.; Ramesh, G.V.; Koyasu, S.; Miyauchi, M.; Sakuma, Y.; Tanabe, T.; Gunji, T.; Duy Dao, T.; et al. Visible-light photodecomposition of acetaldehyde by TiO2-coated gold nanocages: Plasmon-mediated hot electron transport via defect states. Chem. Commun. 2014, 50, 15553–15556. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.S.; Trafela, S.; Rozman, K.Z.; Kovac, J.; Djinovic, P.; Pintar, A. Determination of Schottky barrier height and enhanced photoelectron generation in novel plasmonic immobilized multisegmented (Au/TiO2) nanorod arrays (NRAs) suitable for solar energy conversion applications. J. Mater. Chem. C 2017, 5, 10509–10516. [Google Scholar] [CrossRef]
- Barad, H.-N.; Ginsburg, A.; Cohen, H.; Rietwyk, K.J.; Keller, D.A.; Tirosh, S.; Bouhadana, Y.; Anderson, A.Y.; Zaban, A. Hot Electron-Based Solid State TiO2|Ag Solar Cells. Adv. Mater. Interfaces 2016, 3, 1500789. [Google Scholar] [CrossRef]
- Fang, Y.; Jiao, Y.; Xiong, K.; Ogier, R.; Yang, Z.-J.; Gao, S.; Dahlin, A.B.; Käll, M. Plasmon Enhanced Internal Photoemission in Antenna-Spacer-Mirror Based Au/TiO2 Nanostructures. Nano Lett. 2015, 15, 4059–4065. [Google Scholar] [CrossRef]
- Chatterjee, D.; Dasgupta, S. Visible light induced photocatalytic degradation of organic pollutants. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 186–205. [Google Scholar] [CrossRef]
- Umar, M.; Aziz, H.A. Photocatalytic Degradation of Organic Pollutants in Water. In Organic Pollutants—Monitoring, Risk and Treatment; Rashed, M.N., Ed.; InTech: Rijeka, Croatia, 2013; Chapter 8. [Google Scholar]
- Yu, J.; Dai, G.; Huang, B. Fabrication and Characterization of Visible-Light-Driven Plasmonic Photocatalyst Ag/AgCl/TiO2 Nanotube Arrays. J. Phys. Chem. C 2009, 113, 16394–16401. [Google Scholar] [CrossRef]
- Linic, S.; Aslam, U.; Boerigter, C.; Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 2015, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, W.J.; Lee, S.-H.A.; Maeda, K.; Mallouk, T.E. Visible Light Water Splitting Using Dye-Sensitized Oxide Semiconductors. Acc. Chem. Res. 2009, 42, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Swierk, J.R.; Mallouk, T.E. Correction: Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 2017, 46, 559. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Long, M.; Zeng, H.; Cai, W.; Zhou, B.; Zhang, J.; Wu, Y.; Ding, D.; Wu, D. Preparation, characterization and visible-light activity of carbon modified TiO2 with two kinds of carbonaceous species. J. Mol. Catal. A Chem. 2009, 314, 35–41. [Google Scholar] [CrossRef]
- Hong, X.; Wang, Z.; Cai, W.; Lu, F.; Zhang, J.; Yang, Y.; Ma, N.; Liu, Y. Visible-Light-Activated Nanoparticle Photocatalyst of Iodine-Doped Titanium Dioxide. Chem. Mater. 2005, 17, 1548–1552. [Google Scholar] [CrossRef]
- Zhang, P.; Li, X.; Wu, X. Photocatalytic degradation of methyl orange by N-doped and Ag-loaded nano-TiO2 under visible light. Appl. Nanosci. 2016, 21, 775–782. [Google Scholar]
- Naldoni, A.; Riboni, F.; Marelli, M.; Bossola, F.; Ulisse, G.; Di Carlo, A.; Pis, I.; Nappini, S.; Malvestuto, M.; Dozzi, M.V.; et al. Influence of TiO2 electronic structure and strong metal-support interaction on plasmonic Au photocatalytic oxidations. Catal. Sci. Technol. 2016, 6, 3220–3229. [Google Scholar] [CrossRef]
- Liu, L.; Dao, T.D.; Kodiyath, R.; Kang, Q.; Abe, H.; Nagao, T.; Ye, J. Plasmonic Janus-Composite Photocatalyst Comprising Au and C–TiO2 for Enhanced Aerobic Oxidation over a Broad Visible-Light Range. Adv. Funct. Mater. 2014, 24, 7754–7762. [Google Scholar] [CrossRef]
- Wu, D.; Long, M. Visible light assisted photocatalytic degradation of methyl orange using Ag/N-TiO(2) photocatalysts. Water Sci. Technol. 2012, 65, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Paramasivam, I.; Macak, J.M.; Schmuki, P. Photocatalytic activity of TiO2 nanotube layers loaded with Ag and Au nanoparticles. Electrochem. Commun. 2008, 10, 71–75. [Google Scholar] [CrossRef]
- Tsukamoto, D.; Shiraishi, Y.; Sugano, Y.; Ichikawa, S.; Tanaka, S.; Hirai, T. Gold Nanoparticles Located at the Interface of Anatase/Rutile TiO2 Particles as Active Plasmonic Photocatalysts for Aerobic Oxidation. J. Am. Chem. Soc. 2012, 134, 6309–6315. [Google Scholar] [CrossRef]
- Bian, Z.; Tachikawa, T.; Zhang, P.; Fujitsuka, M.; Majima, T. Au/TiO2 Superstructure-Based Plasmonic Photocatalysts Exhibiting Efficient Charge Separation and Unprecedented Activity. J. Am. Chem. Soc. 2014, 136, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Jedsukontorn, T.; Saito, N.; Hunsom, M. Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials 2018, 8, 269. [Google Scholar] [CrossRef]
- Chou, J.B.; Li, X.-H.; Wang, Y.; Fenning, D.P.; Elfaer, A.; Viegas, J.; Jouiad, M.; Shao-Horn, Y.; Kim, S.-G. Surface plasmon assisted hot electron collection in wafer-scale metallic-semiconductor photonic crystals. Opt. Express 2016, 24, A1234–A1244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Y.; Lee, S.-T.; Yang, S.; Kang, Z. Coupling surface plasmon resonance of gold nanoparticles with slow-photon-effect of TiO2 photonic crystals for synergistically enhanced photoelectrochemical water splitting. Energy Environ. Sci. 2014, 7, 1409–1419. [Google Scholar] [CrossRef]
- Fan, W.; Leung, K.M. Recent Development of Plasmonic Resonance-Based Photocatalysis and Photovoltaics for Solar Utilization. Molecules 2016, 21, 180. [Google Scholar] [CrossRef]
- Boxi, S.S.; Paria, S. Visible light induced enhanced photocatalytic degradation of organic pollutants in aqueous media using Ag doped hollow TiO2 nanospheres. RSC Adv. 2015, 5, 37657–37668. [Google Scholar] [CrossRef]
- Chehadi, Z.; Alkees, N.; Bruyant, A.; Toufaily, J.; Girardon, J.-S.; Capron, M.; Dumeignil, F.; Hamieh, T.; Bachelot, R.; Jradi, S. Plasmonic enhanced photocatalytic activity of semiconductors for the degradation of organic pollutants under visible light. Mater. Sci. Semicond. Process. 2016, 42, 81–84. [Google Scholar] [CrossRef]
- Wang, L.; Wen, M.; Wang, W.; Momuinou, N.; Wang, Z.; Li, S. Photocatalytic degradation of organic pollutants using rGO supported TiO2-CdS composite under visible light irradiation. J. Alloys Compd. 2016, 683, 318–328. [Google Scholar] [CrossRef]
- Wang, S.; Hou, Y.; Wang, X. Development of a Stable MnCo2O4 Cocatalyst for Photocatalytic CO2 Reduction with Visible Light. ACS Appl. Mater. Inter. 2015, 7, 4327–4335. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Xie, S.; Fan, W.; Zhang, Q.; Wang, Y.; Deng, W.; Wang, Y. Photocatalytic Conversion of Carbon Dioxide with Water into Methane: Platinum and Copper(I) Oxide Co-catalysts with a Core–Shell Structure. Angew. Chem. Int. Ed. 2013, 52, 5776–5779. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar] [CrossRef] [PubMed]
- Marszewski, M.; Cao, S.; Yu, J.; Jaroniec, M. Semiconductor-based photocatalytic CO2 conversion. Mater. Horiz. 2015, 2, 261–278. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Wei, Y.; Zhao, Y.; Zhao, Z.; Duan, A.; Liu, J.; Pang, Y.; Li, J.; Jiang, G.; Wang, Y. AuPd/3DOM-TiO2 catalysts for photocatalytic reduction of CO2: High efficient separation of photogenerated charge carriers. Appl. Catal. B-Environ. 2017, 209, 228–239. [Google Scholar] [CrossRef]
- Wu, B.-H.; Liu, W.-T.; Chen, T.-Y.; Perng, T.-P.; Huang, J.-H.; Chen, L.-J. Plasmon-enhanced photocatalytic hydrogen production on Au/TiO2 hybrid nanocrystal arrays. Nano Energy 2016, 27, 412–419. [Google Scholar] [CrossRef]
- Jiang, R.; Li, B.; Fang, C.; Wang, J. Metal/Semiconductor Hybrid Nanostructures for Plasmon-Enhanced Applications. Adv. Mater. 2014, 26, 5274–5309. [Google Scholar] [CrossRef] [PubMed]
- Naik, G.V.; Shalaev, V.M.; Boltasseva, A. Alternative Plasmonic Materials: Beyond Gold and Silver. Adv. Mater. 2013, 25, 3264–3294. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, Z.; Jiang, W.; Yin, W.; Zhong, S.; Gong, P.; Qiao, R.; Li, Z.; Bai, S. Ultrathin nanosheets of palladium in boosting its cocatalyst role and plasmonic effect towards enhanced photocatalytic hydrogen evolution. RSC Adv. 2016, 6, 56800–56806. [Google Scholar] [CrossRef]
- Yoo, J.; Zazpe, R.; Cha, G.; Prikryl, J.; Hwang, I.; Macak, J.M.; Schmuki, P. Uniform ALD deposition of Pt nanoparticles within 1D anodic TiO2 nanotubes for photocatalytic H2 generation. Electrochem. Commun. 2018, 86, 6–11. [Google Scholar] [CrossRef]
- Ozkan, S.; Yoo, J.; Nguyen, N.T.; Mohajernia, S.; Zazpe, R.; Prikryl, J.; Macak, J.M.; Schmuki, P. Spaced TiO2 Nanotubes Enable Optimized Pt Atomic Layer Deposition for Efficient Photocatalytic H2 Generation. ChemistryOpen 2018, 7, 797–802. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of Au/TiO2 with Metal Cocatalysts Exhibiting Strong Surface Plasmon Resonance Effective for Photoinduced Hydrogen Formation under Irradiation of Visible Light. ACS Catal. 2013, 3, 79–85. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Cao, S.-W.; Xue, C. Au/Pt Nanoparticle-Decorated TiO2 Nanofibers with Plasmon-Enhanced Photocatalytic Activities for Solar-to-Fuel Conversion. J. Phys. Chem. C 2013, 117, 25939–25947. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Q.L.; Du, C.; Sun, S.S.; Steinkruger, J.D.; Zhou, C.; Yang, S.Y. Synergistic Effect of Dual Particle-Size AuNPs on TiO2 for Efficient Photocatalytic Hydrogen Evolution. Nanomaterials 2019, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Nahar, S.; Zain, M.F.M.; Kadhum, A.A.H.; Hasan, H.A.; Hasan, M.R. Advances in Photocatalytic CO2 Reduction with Water: A Review. Materials 2017, 10, 629. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, S.; Baldinozzi, G.; Friedrich, D.; Kressman, S.; Strub, H.; Artero, V.; Laberty-Robert, C. Mesoporous thin film WO3 photoanode for photoelectrochemical water splitting: A sol-gel dip coating approach. Sustain. Energy Fuels 2017, 1, 145–153. [Google Scholar] [CrossRef]
- Nishijima, Y.; Ueno, K.; Yokota, Y.; Murakoshi, K.; Misawa, H. Plasmon-Assisted Photocurrent Generation from Visible to Near-Infrared Wavelength Using a Au-Nanorods/TiO2 Electrode. J. Phys. Chem. Lett. 2010, 1, 2031–2036. [Google Scholar] [CrossRef]
- Wang, X.; Long, R.; Liu, D.; Yang, D.; Wang, C.; Xiong, Y. Enhanced full-spectrum water splitting by confining plasmonic Au nanoparticles in N-doped TiO2 bowl nanoarrays. Nano Energy 2016, 24, 87–93. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Hedhili, M.N.; Zhang, H.; Wang, P. Plasmonic gold nanocrystals coupled with photonic crystal seamlessly on TiO2 nanotube photoelectrodes for efficient visible light photoelectrochemical water splitting. Nano Lett. 2013, 13, 14–20. [Google Scholar] [CrossRef]
- Tian, Y.; Tatsuma, T. Plasmon-induced photoelectrochemistry at metal nanoparticles supported on nanoporous TiO2. Chem. Commun. 2004, 1810–1811. [Google Scholar] [CrossRef] [PubMed]
- Christopher, P.; Xin, H.; Linic, S. Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures. Nat. Chem. 2011, 3, 467. [Google Scholar] [CrossRef] [PubMed]
- Bora, T.; Zoepfl, D.; Dutta, J. Importance of Plasmonic Heating on Visible Light Driven Photocatalysis of Gold Nanoparticle Decorated Zinc Oxide Nanorods. Sci. Rep. 2016, 6, 26913. [Google Scholar] [CrossRef] [PubMed]
- Barman, T.; Hussain, A.A.; Sharma, B.; Pal, A.R. Plasmonic Hot Hole Generation by Interband Transition in Gold-Polyaniline. Sci. Rep. 2015, 5, 18276. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.-C.; Wang, G.; Chang, K.-D.; Ling, Y.; Lin, Y.-K.; Fitzmorris, B.C.; Liu, C.-M.; Lu, X.; Tong, Y.; Zhang, J.Z.; et al. Au Nanostructure-Decorated TiO2 Nanowires Exhibiting Photoactivity Across Entire UV-visible Region for Photoelectrochemical Water Splitting. Nano Lett. 2013, 13, 3817–3823. [Google Scholar] [CrossRef]
- Bernardi, M.; Mustafa, J.; Neaton, J.B.; Louie, S.G. Theory and computation of hot carriers generated by surface plasmon polaritons in noble metals. Nat. Commun. 2015, 6, 7044. [Google Scholar] [CrossRef] [PubMed]
- Manjavacas, A.; Liu, J.G.; Kulkarni, V.; Nordlander, P. Plasmon-Induced Hot Carriers in Metallic Nanoparticles. ACS Nano 2014, 8, 7630–7638. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, H.; Wu, L.; Png, C.E.; Bai, P. Interference-Induced Broadband Absorption Enhancement for Plasmonic-Metal@Semiconductor Microsphere as Visible Light Photocatalyst. ACS Catal. 2014, 4, 4269–4276. [Google Scholar] [CrossRef]
- Nagamitsu, M.; Awa, K.; Tada, H. Hydrogen peroxide synthesis from water and oxygen using a three-component nanohybrid photocatalyst consisting of Au particle-loaded rutile TiO2 and RuO2 with a heteroepitaxial junction. Chem. Commun. 2020, 56, 8190–8193. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.T.; Nicolas, A.G.; Edvinsson, T.; Meng, J.; Zheng, K.B.; Abdellah, M.; Sa, J. Molecular Linking Selectivity on Self-Assembled Metal-Semiconductor Nano-Hybrid Systems. Nanomaterials 2020, 10, 1378. [Google Scholar] [CrossRef] [PubMed]
- Naldoni, A.; Malara, F.; Boldrini, C.L.; Marelli, M.; Dal Santo, V.; Montini, T.; Beltram, A.; Romero-Ocaña, I.; Fornasiero, P.; Mróz, M.M.; et al. Hot Electron Collection on Brookite Nanorods Lateral Facets for Plasmon-Enhanced Water Oxidation. ACS Catal. 2017, 7, 1270–1278. [Google Scholar] [CrossRef]
- Lee, K.-S.; El-Sayed, M.A. Dependence of the Enhanced Optical Scattering Efficiency Relative to That of Absorption for Gold Metal Nanorods on Aspect Ratio, Size, End-Cap Shape, and Medium Refractive Index. J. Phys. Chem. B 2005, 109, 20331–20338. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.; Cadusch, J.J.; Dligatch, S.; Roberts, A.; Davis, T.J.; Mulvaney, P.; Gómez, D.E. Hot Carrier Extraction with Plasmonic Broadband Absorbers. ACS Nano 2016, 10, 4704–4711. [Google Scholar] [CrossRef] [PubMed]
- Pelayo Garcia de Arquer, F.; Mihi, A.; Konstantatos, G. Molecular interfaces for plasmonic hot electron photovoltaics. Nanoscale 2015, 7, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, H.; Park, J.Y. Tandem-structured, hot electron based photovoltaic cell with double Schottky barriers. Sci. Rep. 2014, 4, 4580. [Google Scholar] [CrossRef] [PubMed]



























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manuel, A.P.; Shankar, K. Hot Electrons in TiO2–Noble Metal Nano-Heterojunctions: Fundamental Science and Applications in Photocatalysis. Nanomaterials 2021, 11, 1249. https://doi.org/10.3390/nano11051249
Manuel AP, Shankar K. Hot Electrons in TiO2–Noble Metal Nano-Heterojunctions: Fundamental Science and Applications in Photocatalysis. Nanomaterials. 2021; 11(5):1249. https://doi.org/10.3390/nano11051249
Chicago/Turabian StyleManuel, Ajay P., and Karthik Shankar. 2021. "Hot Electrons in TiO2–Noble Metal Nano-Heterojunctions: Fundamental Science and Applications in Photocatalysis" Nanomaterials 11, no. 5: 1249. https://doi.org/10.3390/nano11051249
APA StyleManuel, A. P., & Shankar, K. (2021). Hot Electrons in TiO2–Noble Metal Nano-Heterojunctions: Fundamental Science and Applications in Photocatalysis. Nanomaterials, 11(5), 1249. https://doi.org/10.3390/nano11051249