
Temperature-Responsive Magnetic Nanoparticles for Bioanalysis of Lysozyme in Urine Samples


Abstract
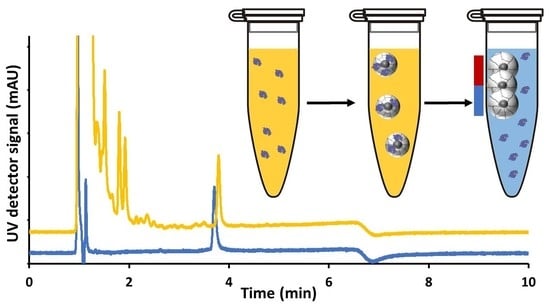
:
1. Introduction
2. Materials and Methods
2.1. Materials
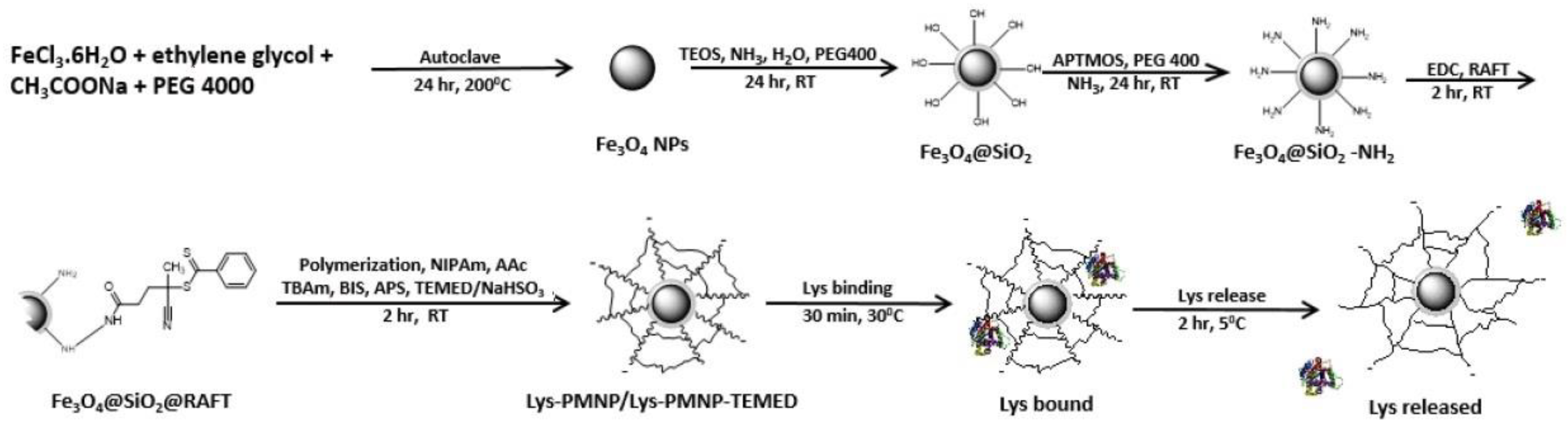
2.2. Synthesis of Lys-PMNPs
2.2.1. Synthesis of Magnetite NPs
2.2.2. Synthesis of Fe3O4@SiO2 Nanospheres
2.2.3. Synthesis of Amine-Modified Fe3O4 NPs (Fe3O4@SiO2-NH2)
2.2.4. Coupling of the RAFT Agent
2.2.5. Preparation of the Polymer Shell
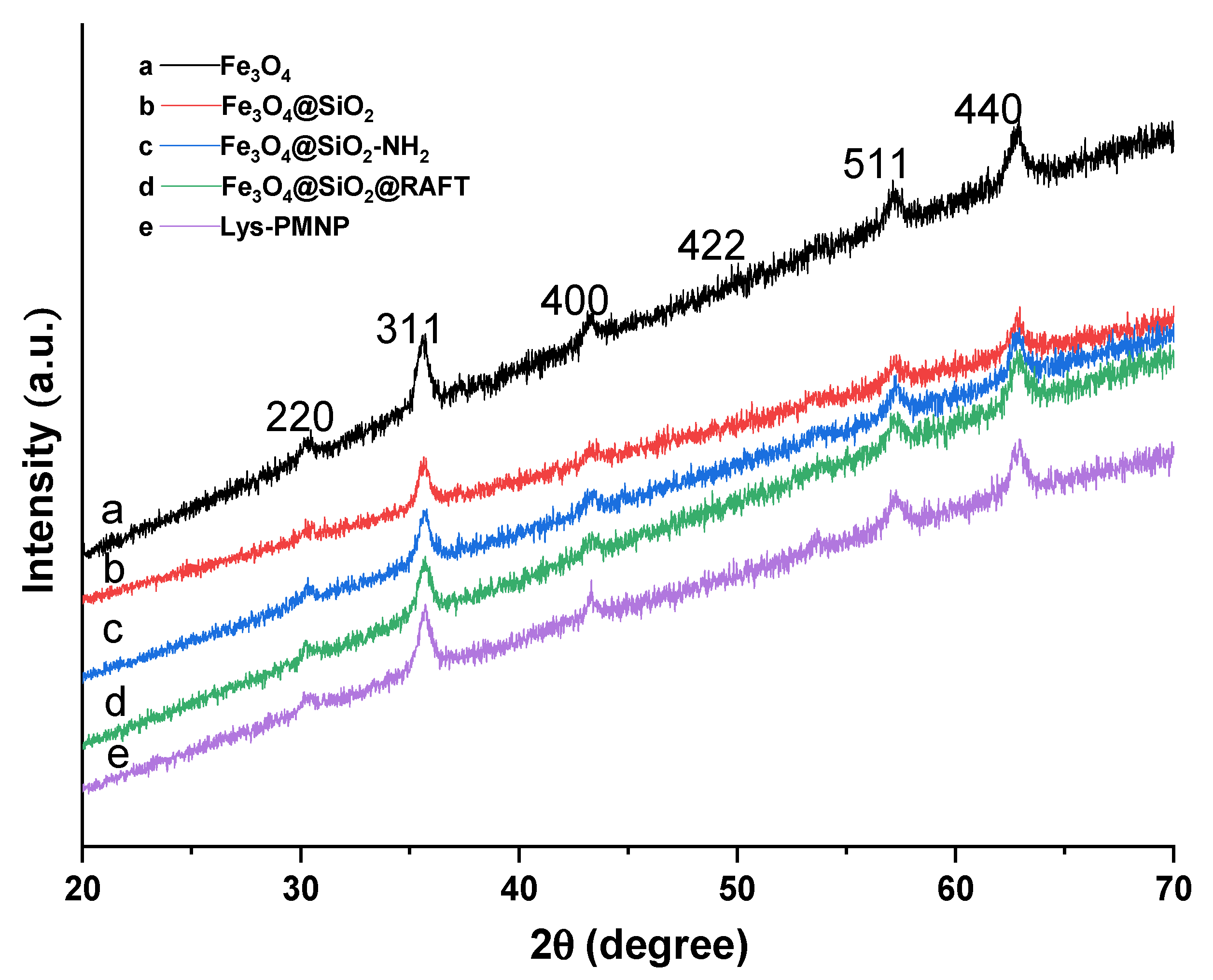
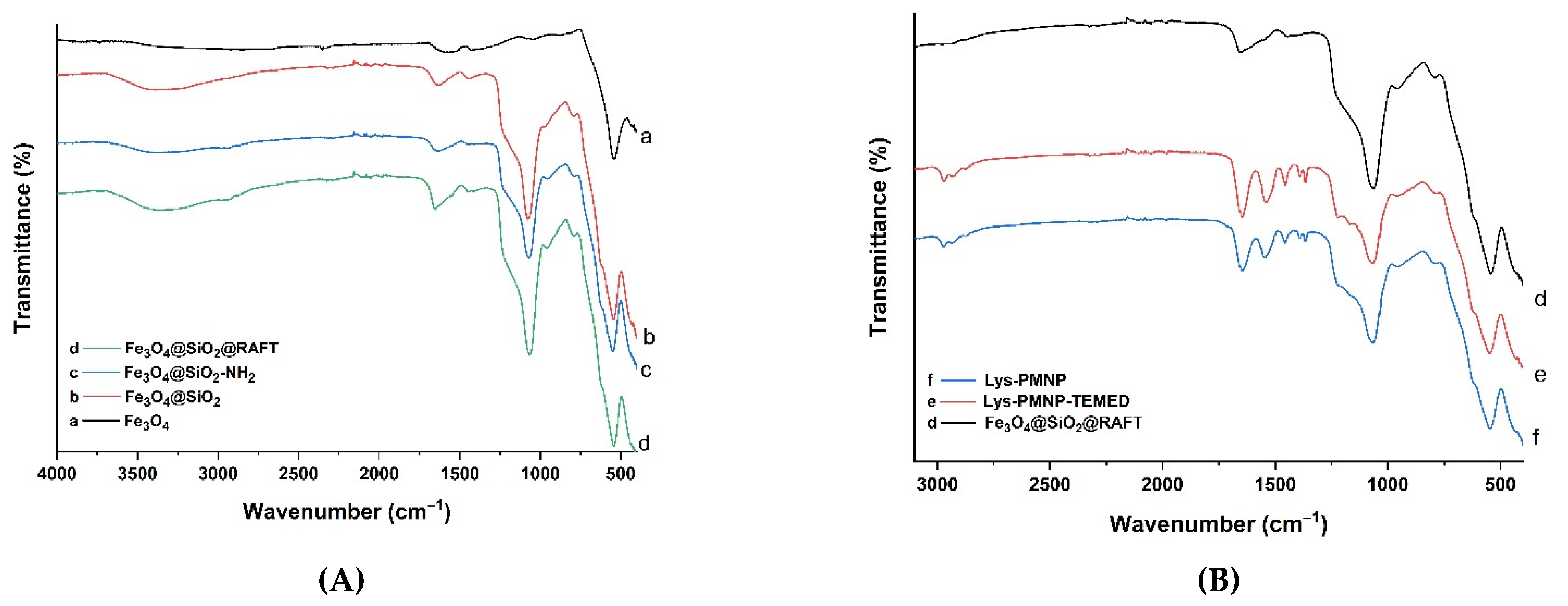
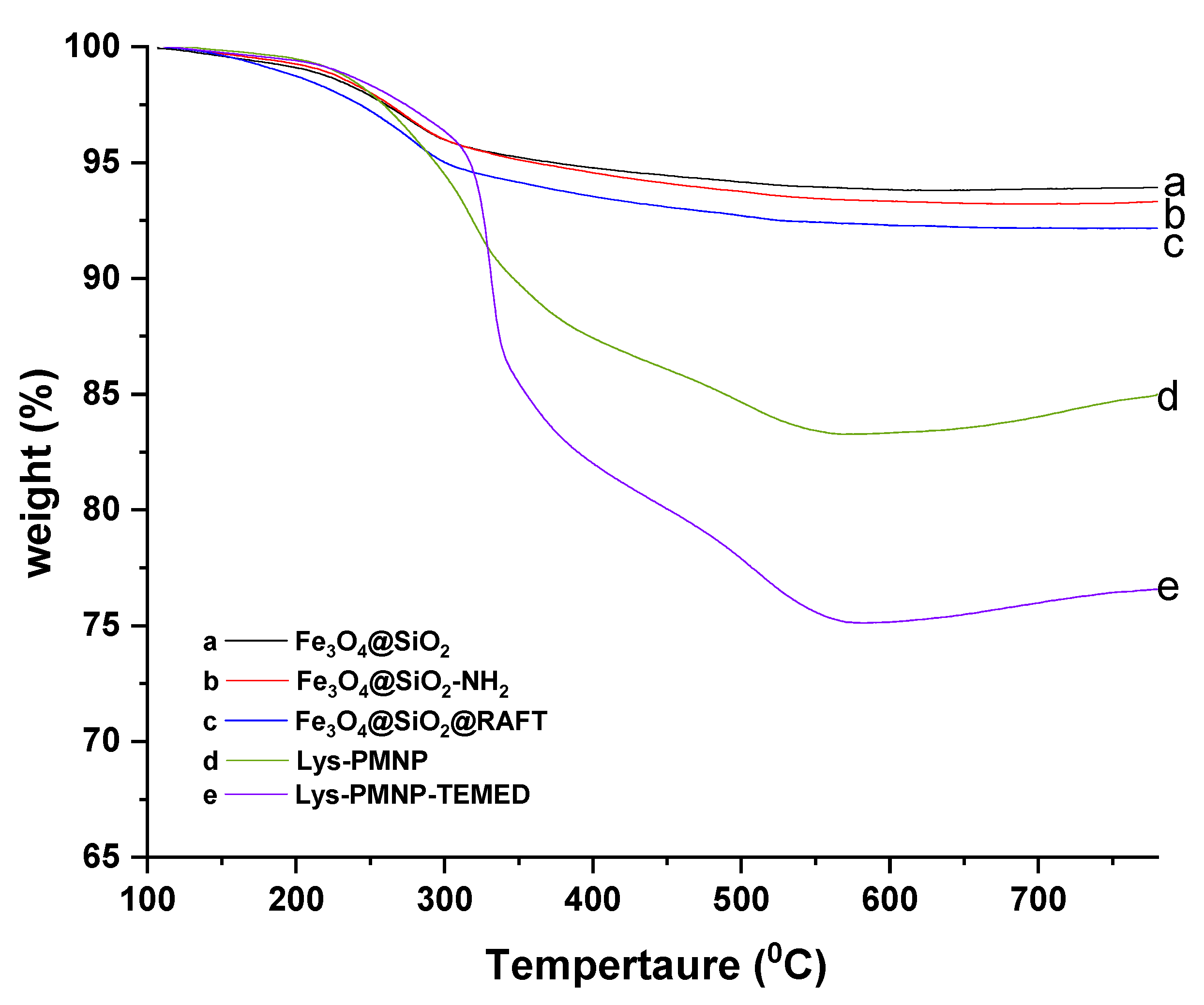
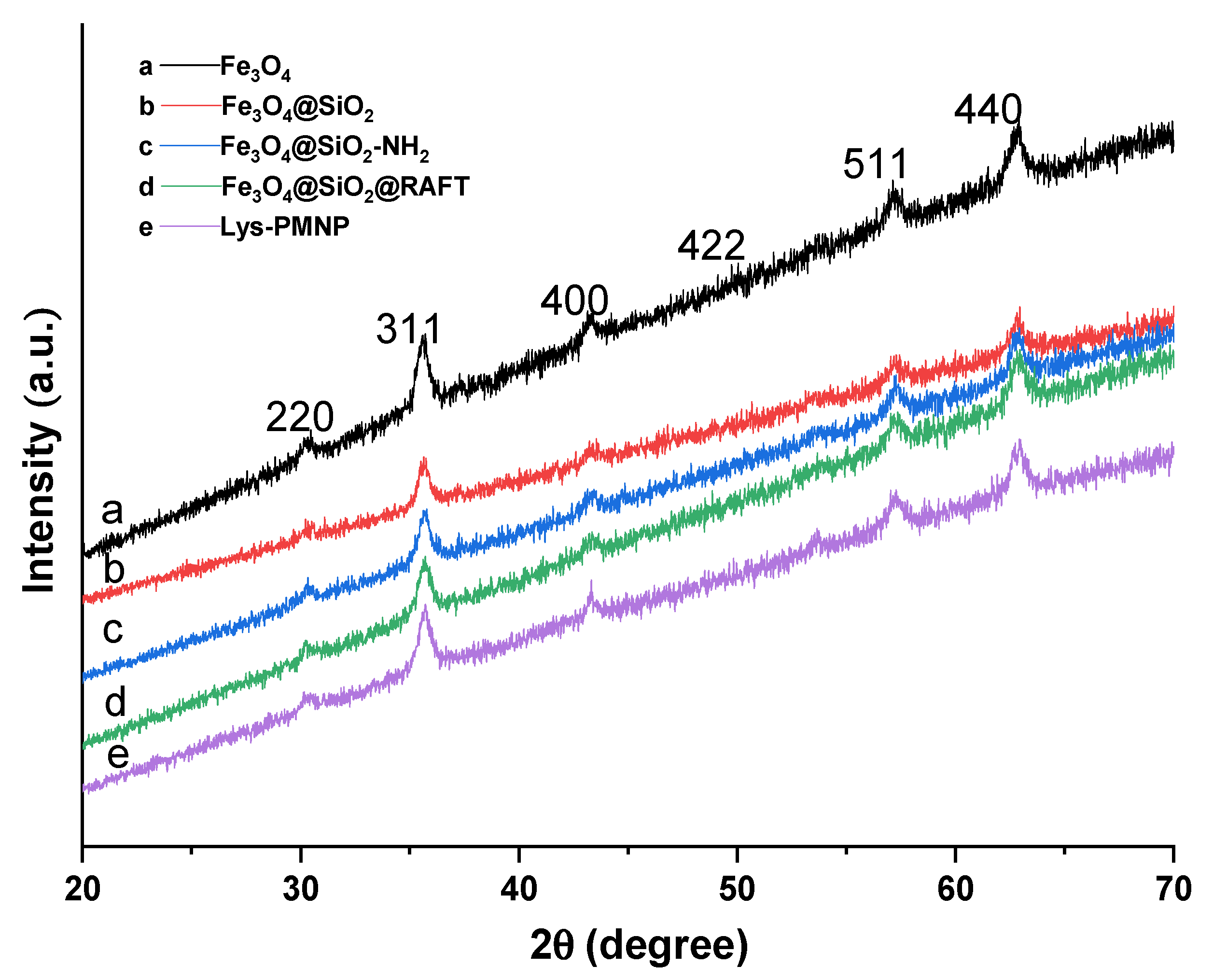
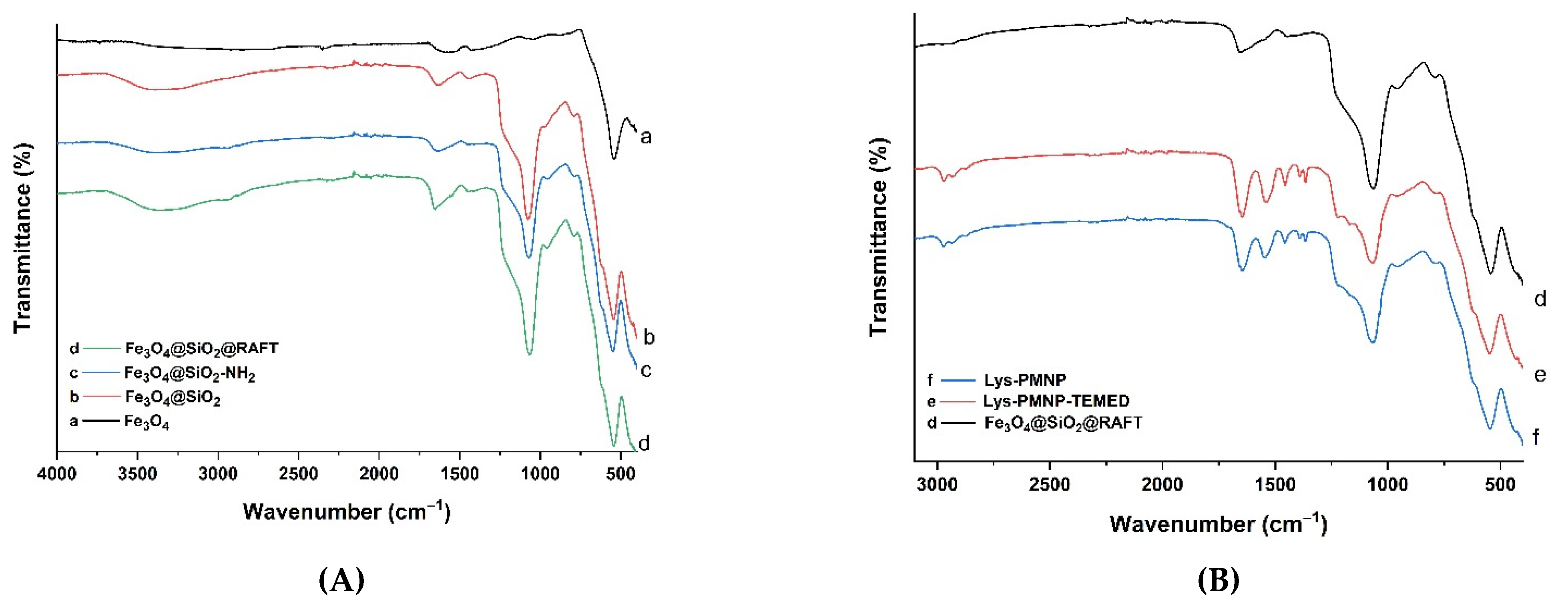
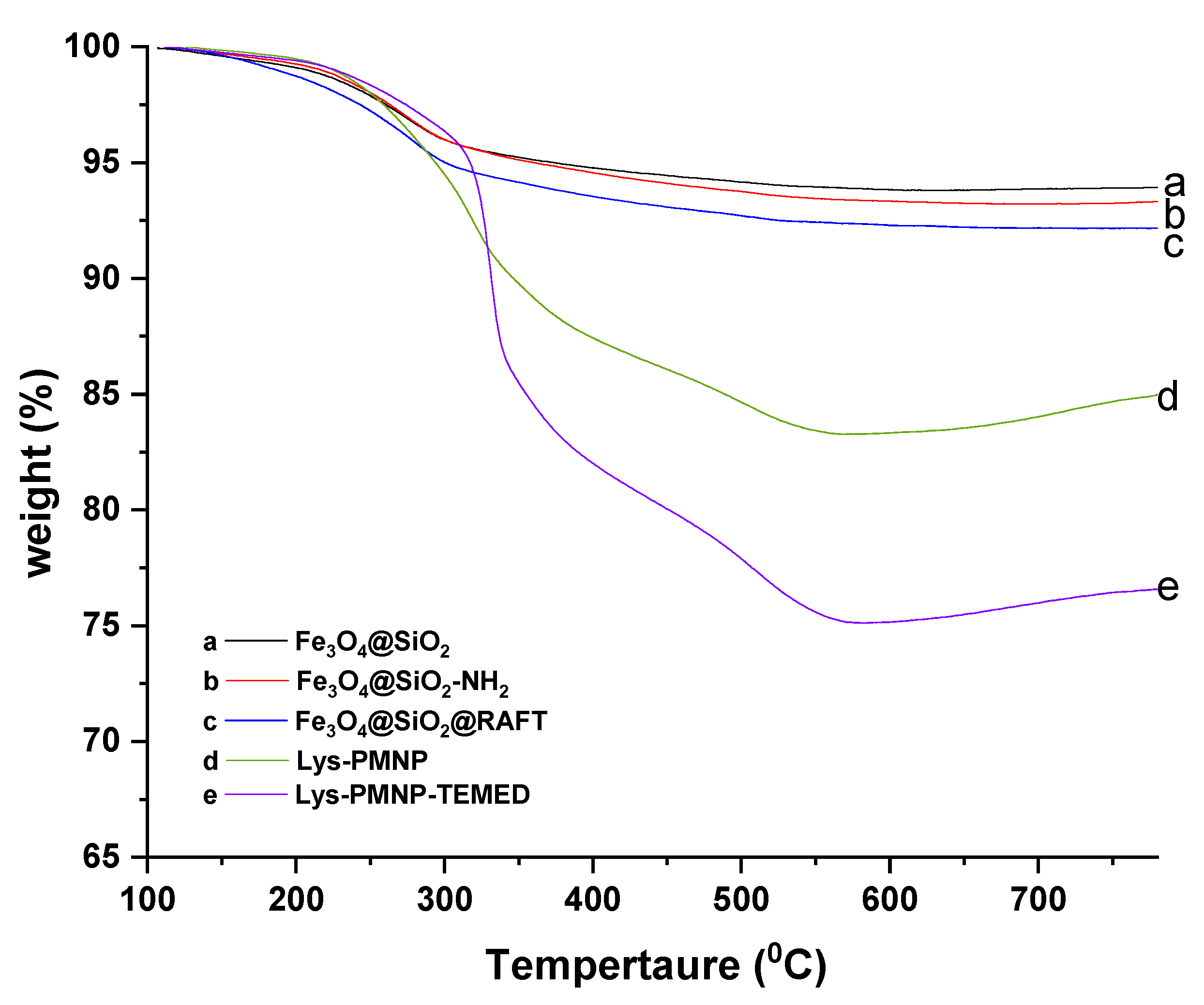
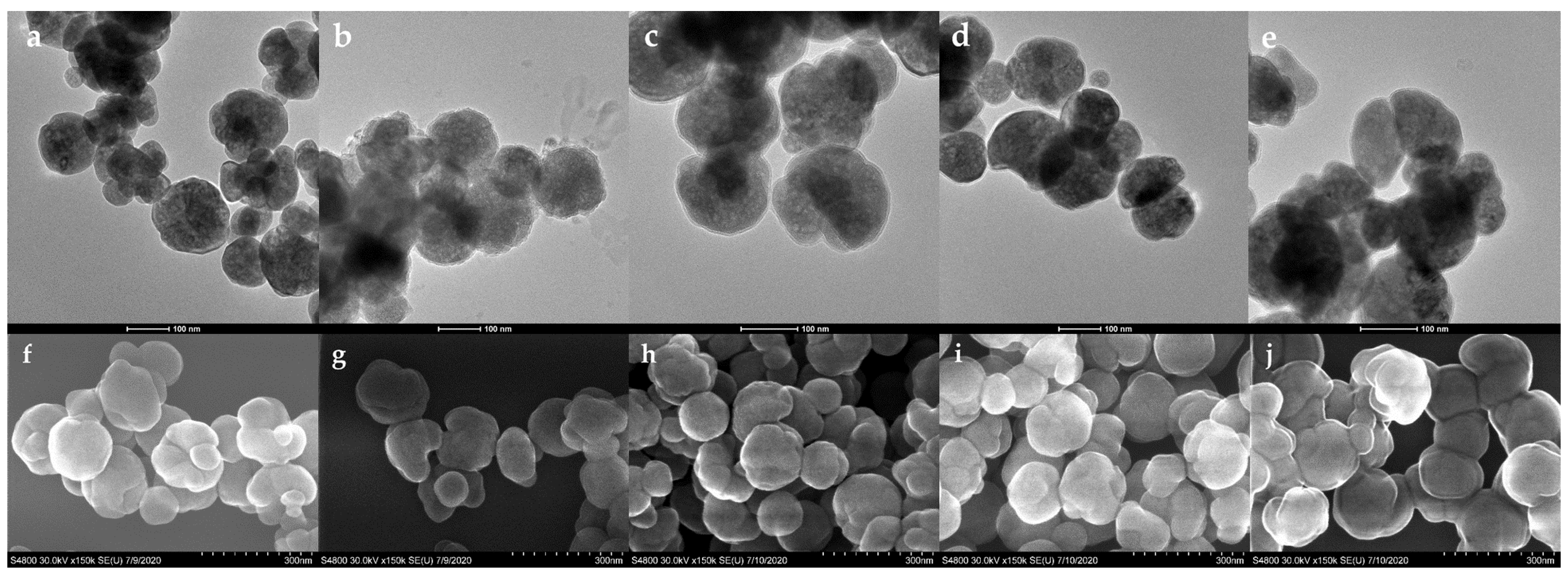
2.3. Characterization
2.4. Equilibrium Binding Measurement of Lys-PMNPs in Buffer
2.5. Release of Lysozyme from the Lys-PMNPs
2.6. Measurement of Selectivity
2.7. Binding and Release of Lys-PMNPs in Urine
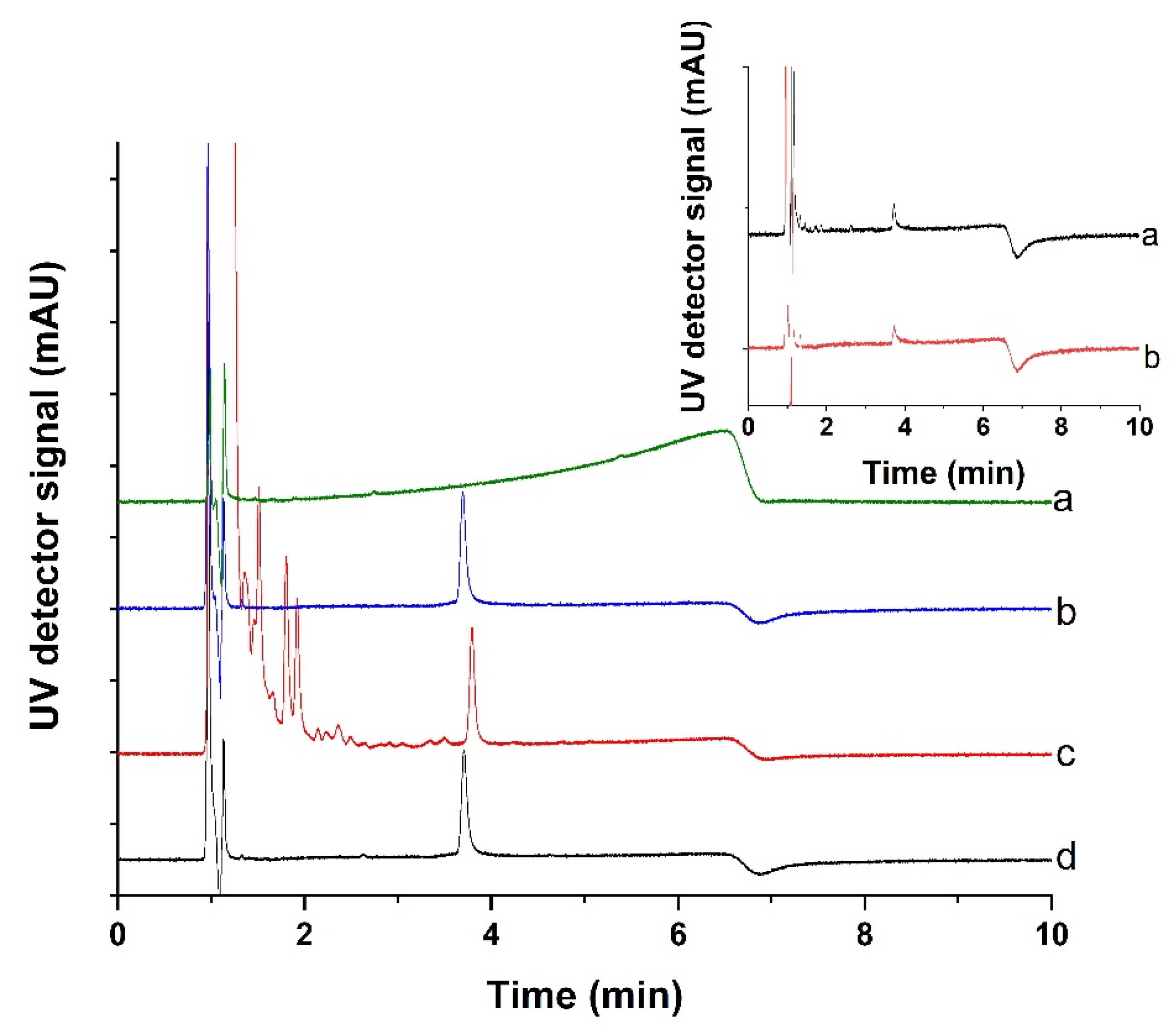
2.8. HPLC Analysis
2.9. Measurement of Urine Samples with Micrococcus lysodeikticus Assay
3. Results and Discussion
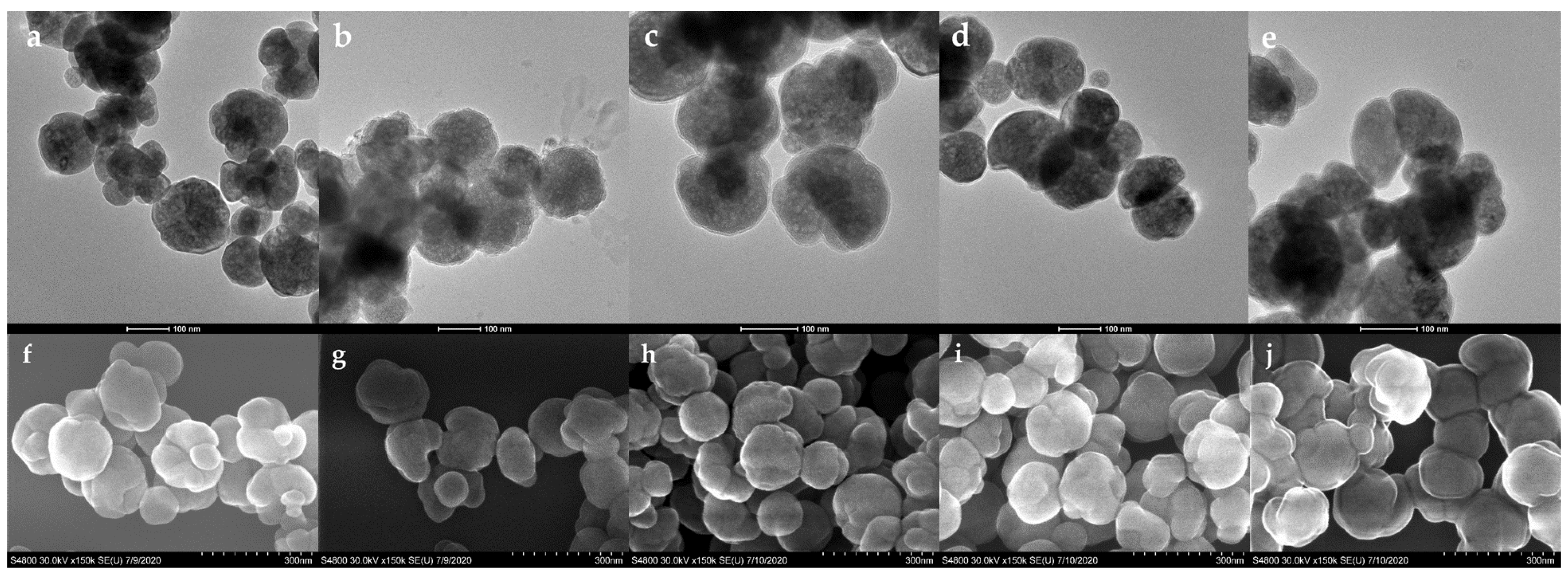
3.1. Preparation and Characterization of Poly(N-isopropylacrylamide-co-N-tert-butylacrylamide co-acrylic acid) Coated Magnetic Nanoparticles (Lys-PMNPs)
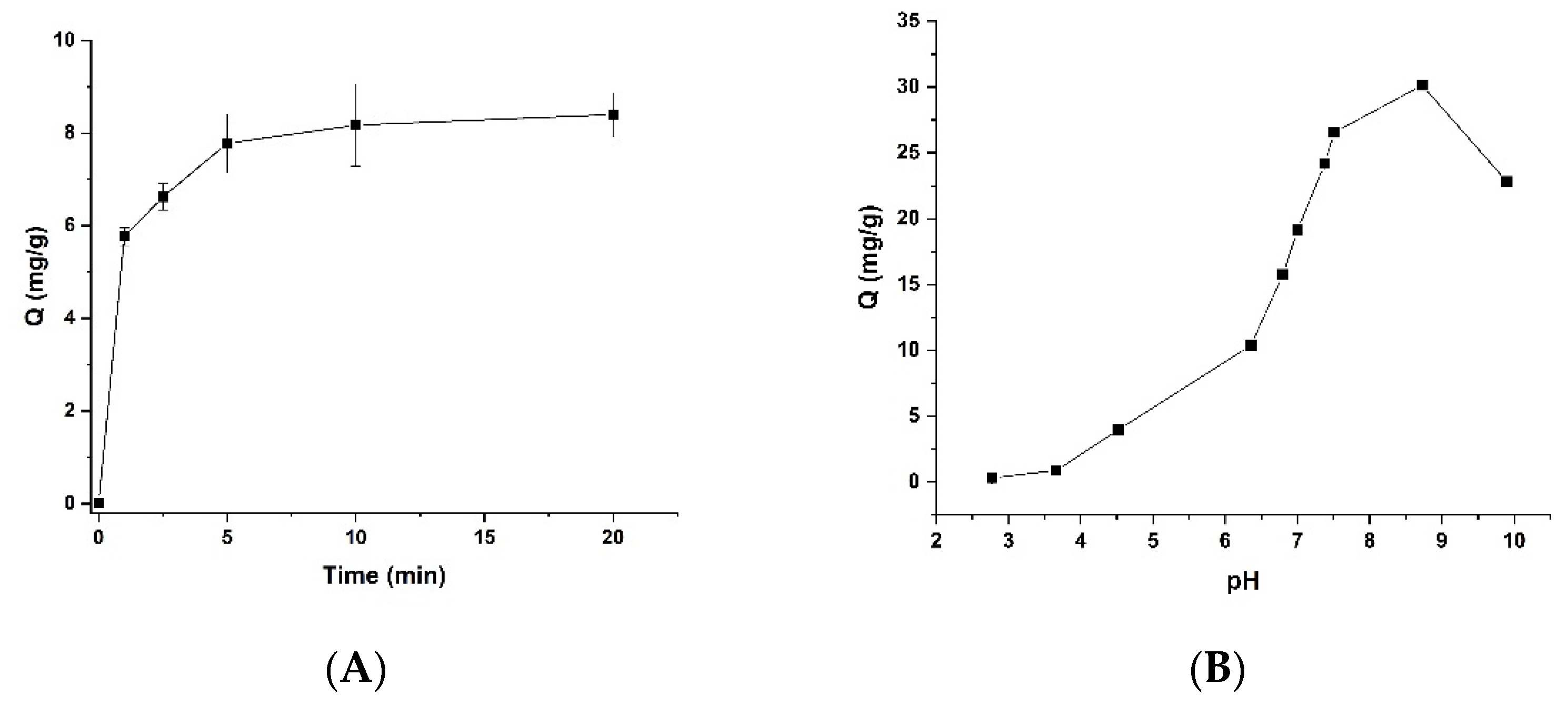
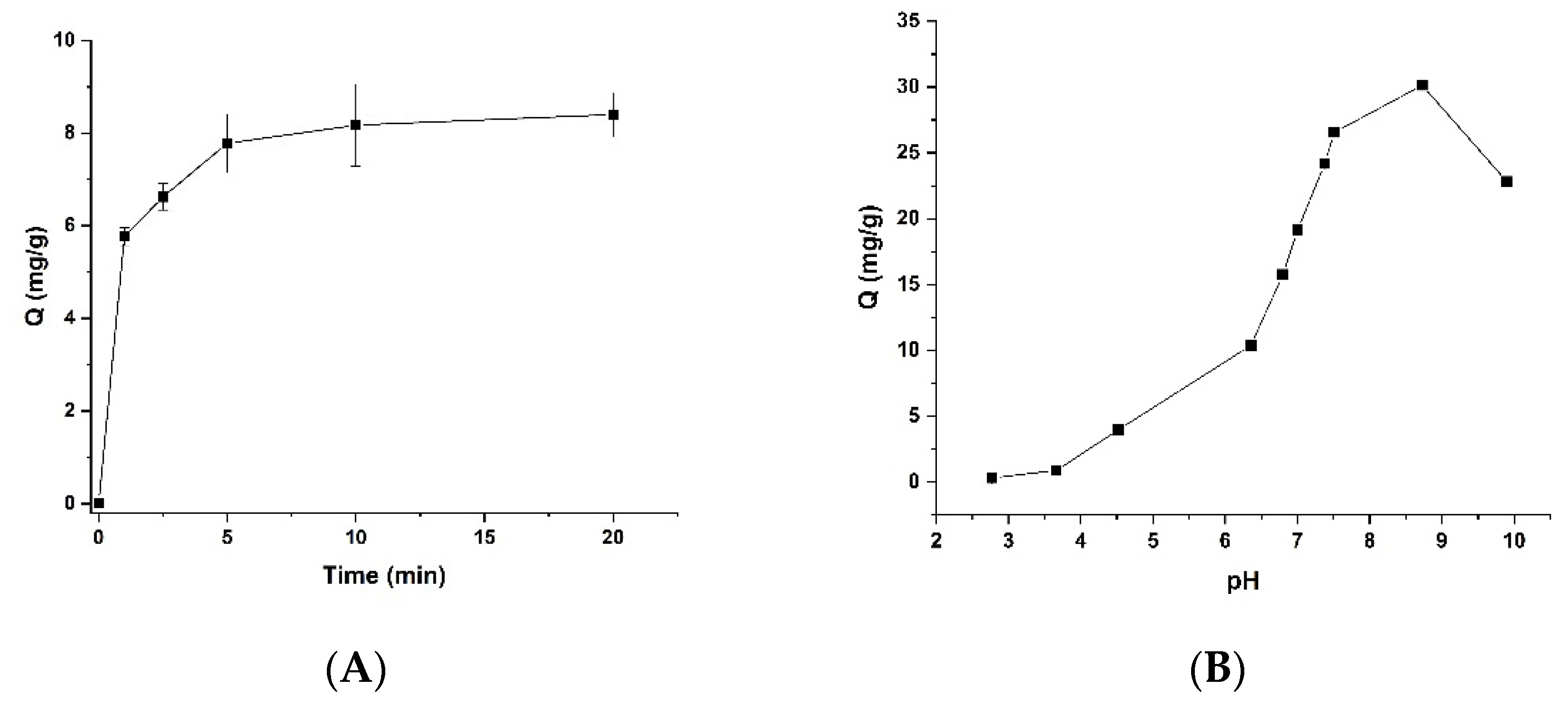
3.2. Effect of Time on the Binding of Lys-PMNPs
3.3. Effect of Buffer Ionic Strength and pH on the Protein Binding of Lys-PMNPs
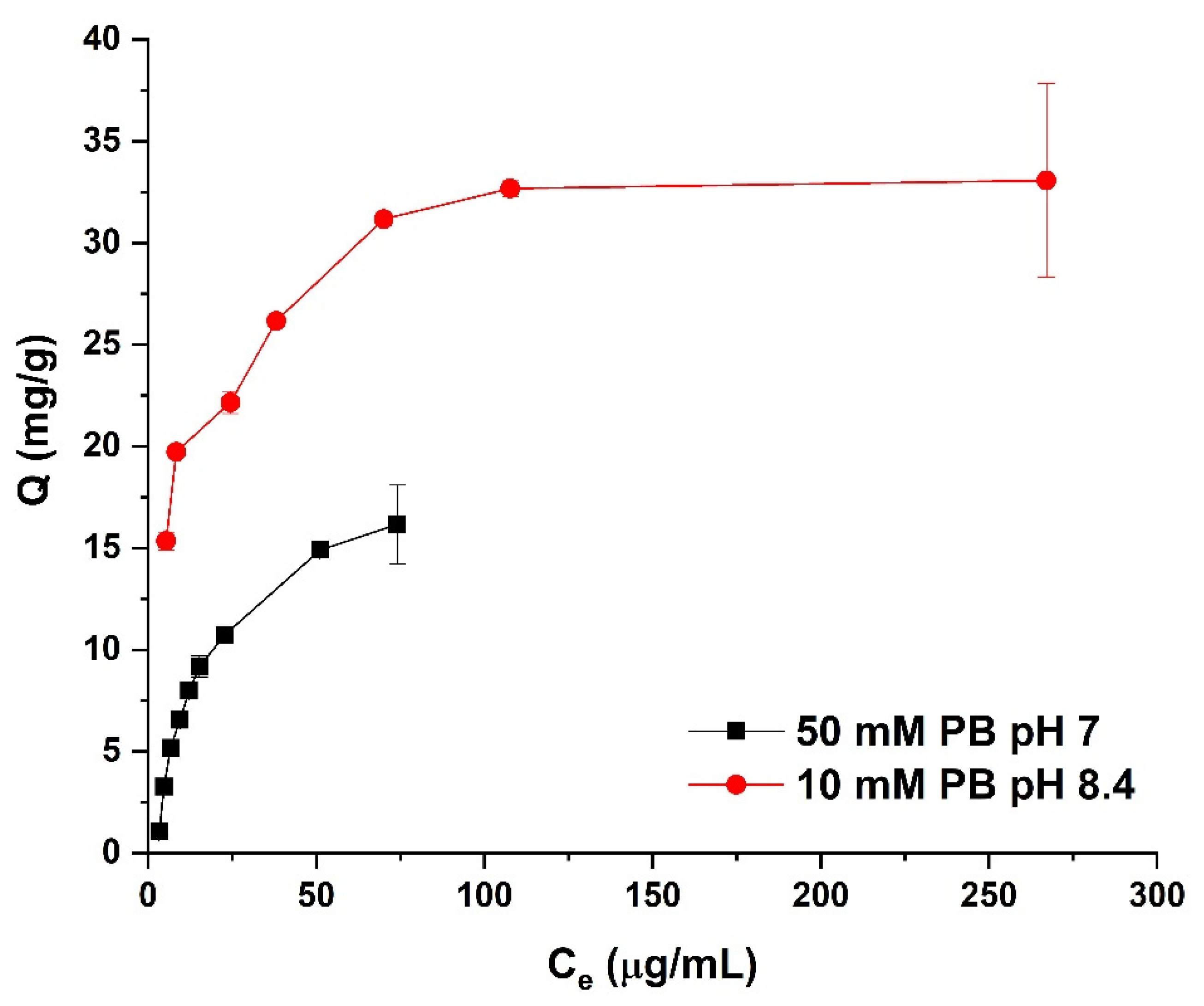
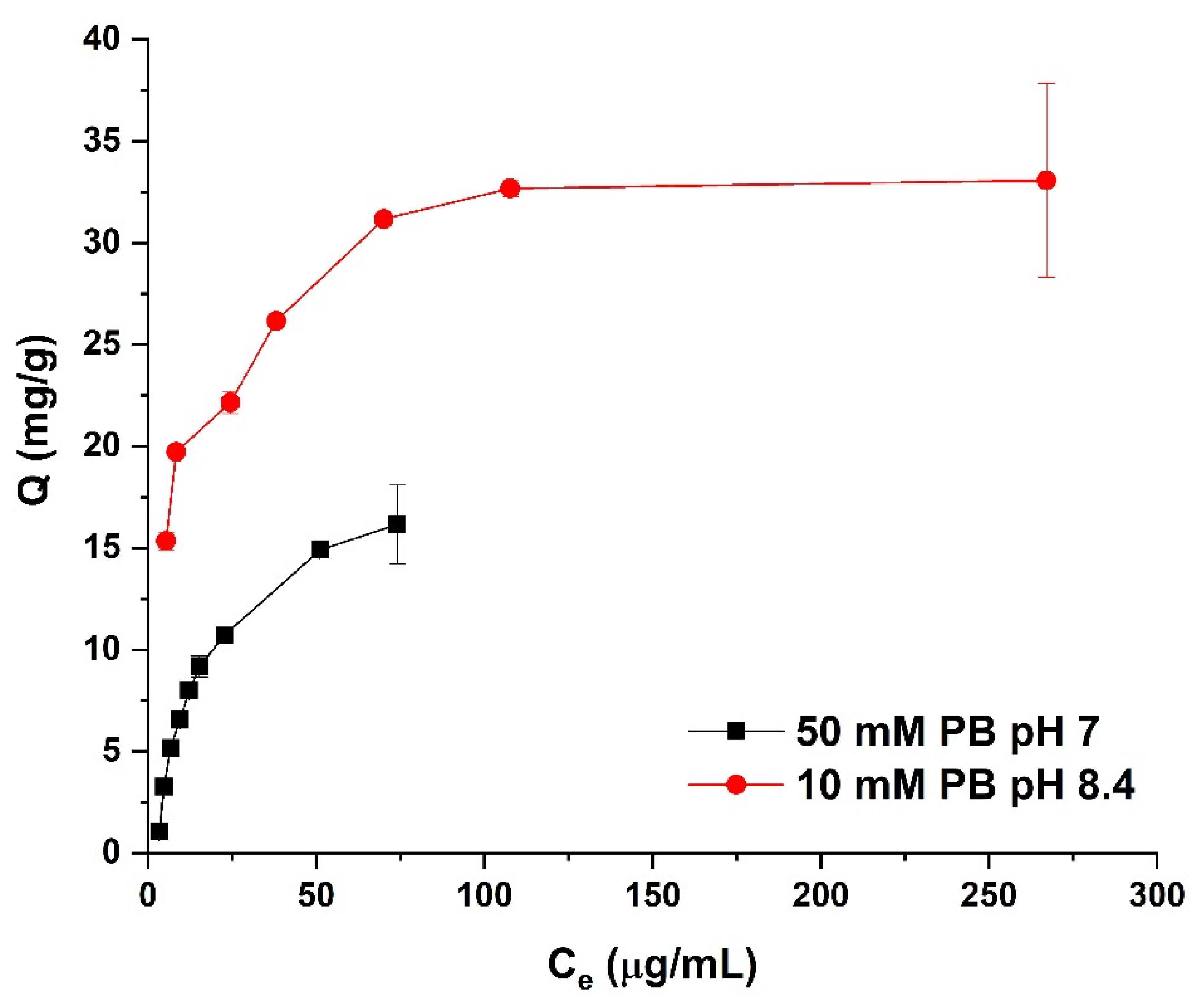
3.4. Adsorption Isotherms
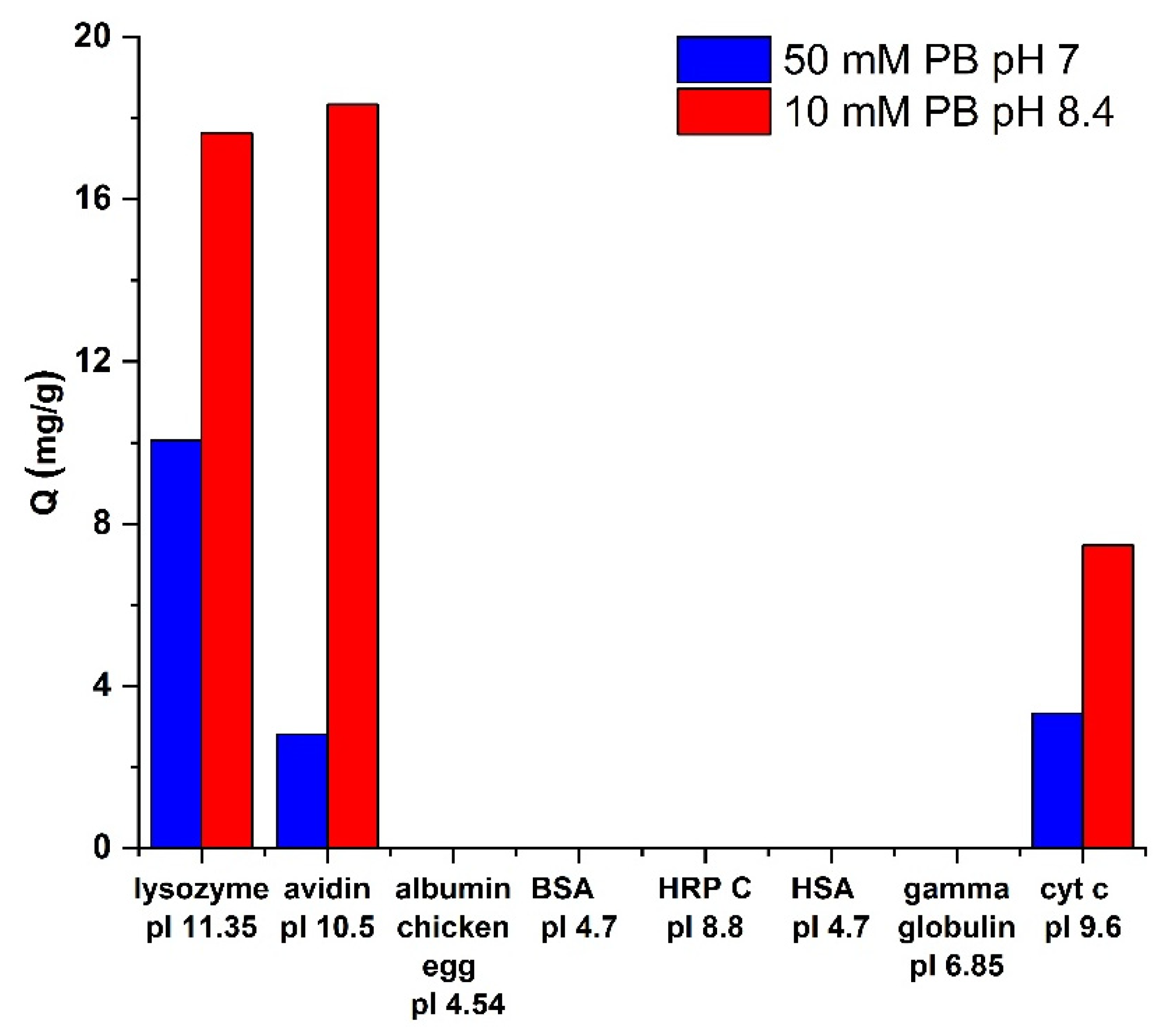
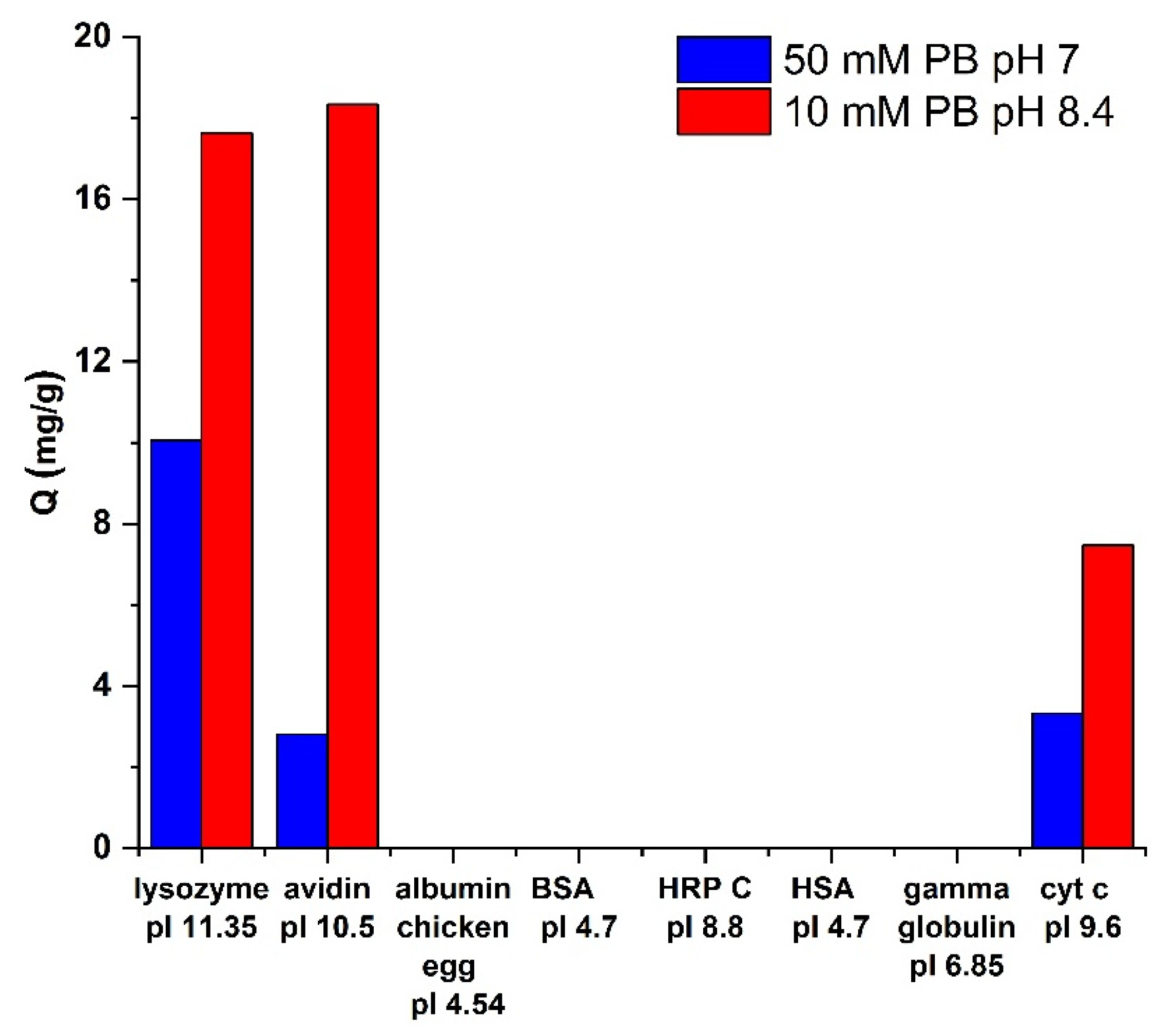
3.5. Selectivity towards Different Proteins
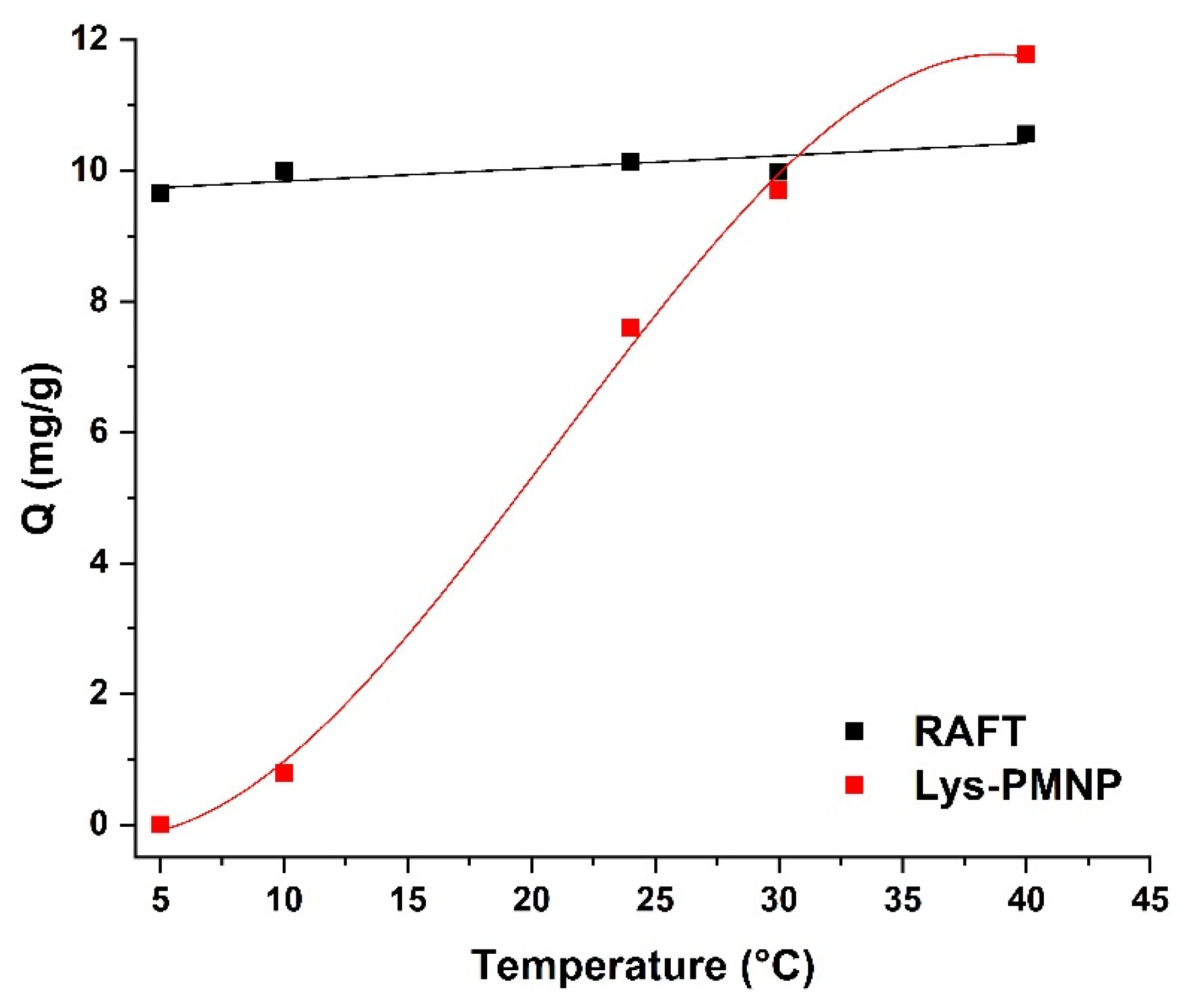
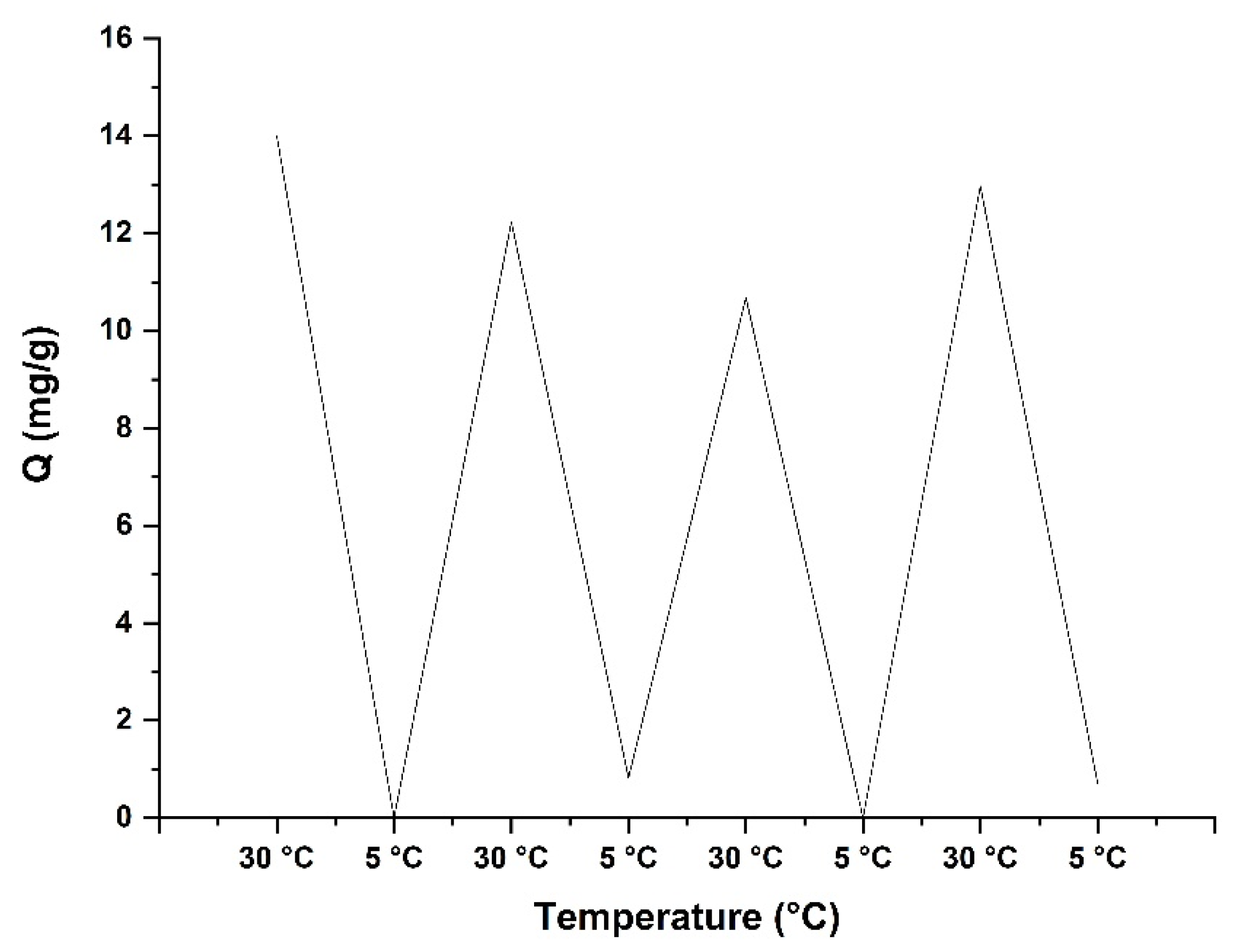
3.6. Thermally Modulated Binding and Release of Lysozyme
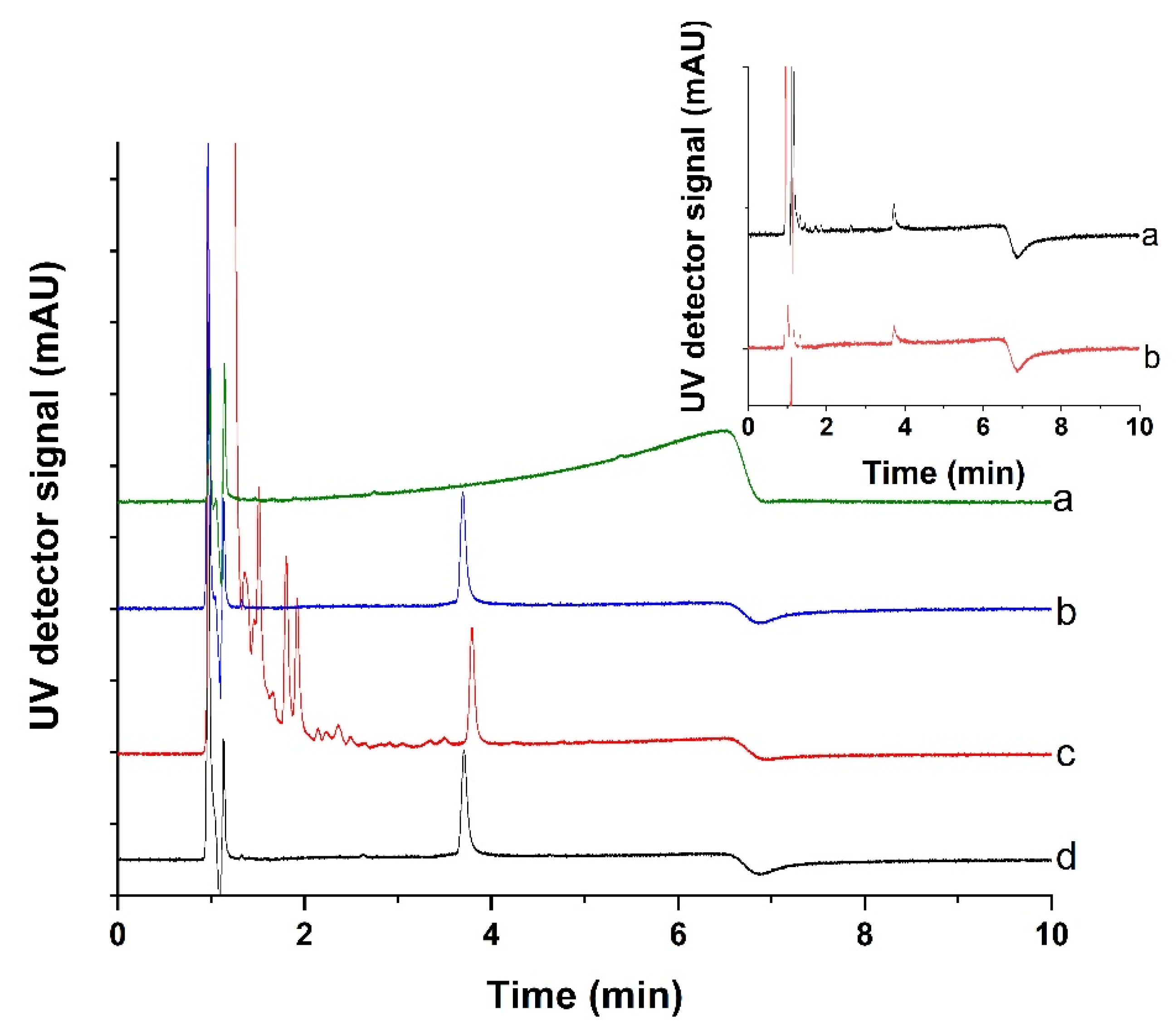
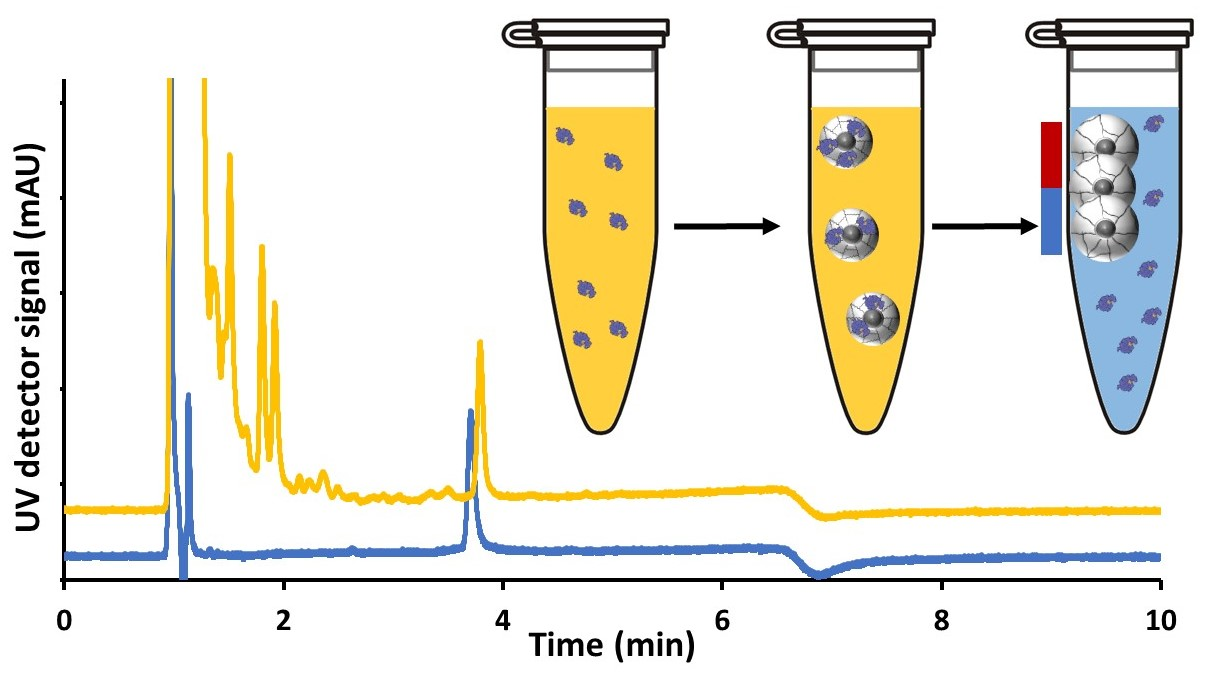
3.7. Development of a Sample Cleanup Procedure for the Measurement of Lysozyme in Clinical Urine Samples
3.8. Method Validation
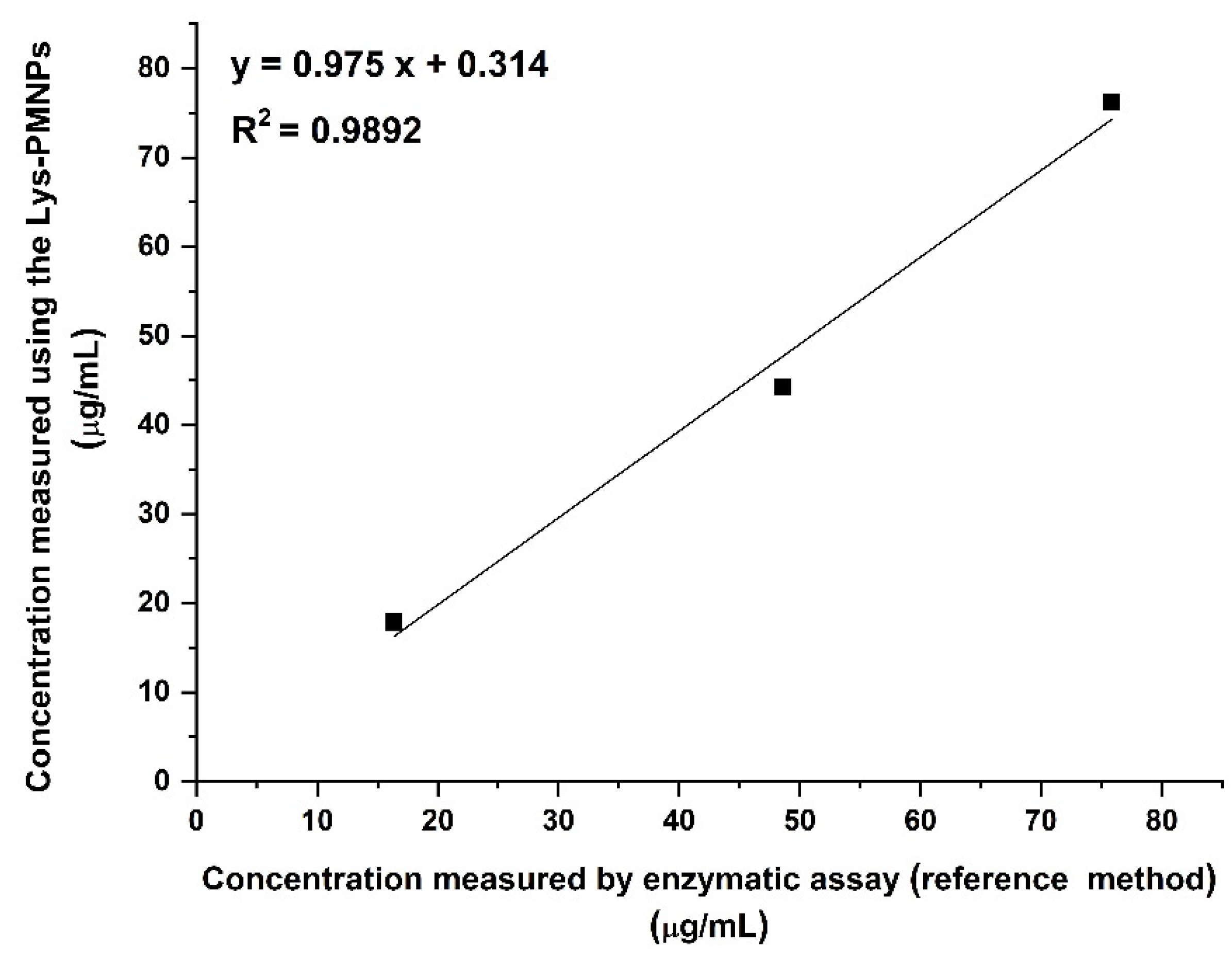
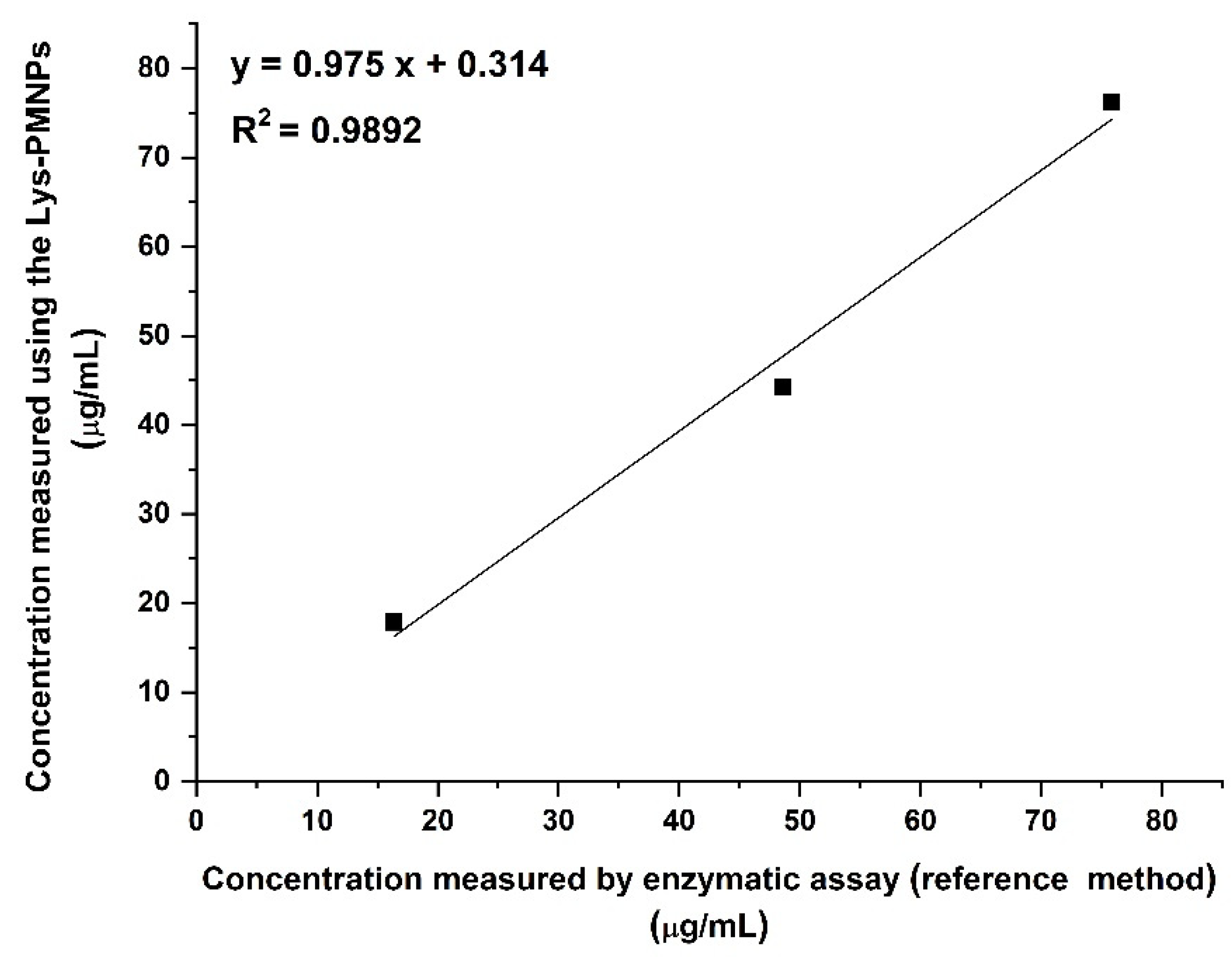
3.9. Comparison to a Reference Method by the Measurement of Unknown Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tibbals, H.F. Medical Nanotechnology and Nanomedicine; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Astruc, D. Introduction: Nanoparticles in Catalysis. Chem. Rev. 2020, 120, 461–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkin, A.; Krstic, P.; Wells, J. (Eds.) Nanotechnology for Elelctronics, Photonics and Renewable Energy; Springer: New York, NY, USA, 2010. [Google Scholar]
- de Dios, A.S.; Díaz-García, M.E. Multifunctional nanoparticles: Analytical prospects. Anal. Chim. Acta 2010, 666, 1–22. [Google Scholar] [CrossRef]
- Moghaddam, A.Z.; Bameri, A.E.; Ganjali, M.R.; Alexovič, M.; Jazi, M.E.; Tabani, H. A low-voltage electro-membrane extraction for quantification of imatinib and sunitinib in biological fluids. Bioanalysis 2021, 13, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Kim, M.; Abelmann, L. Magnetic microgels and nanogels: Physical mechanisms and biomedical applications. Bioeng. Transl. Med. 2020, 6, e10190. [Google Scholar] [CrossRef]
- Ahmadi, M.; Elmongy, H.; Madrakian, T.; Abdel-Rehim, M. Nanomaterials as sorbents for sample preparation in bioanalysis: A review. Anal. Chim. Acta 2017, 958, 1–21. [Google Scholar] [CrossRef]
- He, J.; Huang, M.; Wang, D.; Zhang, Z.; Li, G. Magnetic separation techniques in sample preparation for biological analysis: A review. J. Pharm. Biomed. Anal. 2014, 101, 84–101. [Google Scholar] [CrossRef]
- Guo, P.-F.; Wang, X.-M.; Chen, X.-W.; Yang, T.; Chen, M.-L.; Wang, J.-H. Nanostructures serve as adsorbents for the selective separation/enrichment of proteins. TrAC Trends Anal. Chem. 2019, 120, 115650. [Google Scholar] [CrossRef]
- Wu, W.; He, Q.; Jiang, C. Magnetic Iron Oxide Nanoparticles: Synthesis and Surface Functionalization Strategies. Nanoscale Res. Lett. 2008, 3, 397–415. [Google Scholar] [CrossRef] [Green Version]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Lv, Y.; Tan, T.; Svec, F. Molecular imprinting of proteins in polymers attached to the surface of nanomaterials for selective recognition of biomacromolecules. Biotechnol. Adv. 2013, 31, 1172–1186. [Google Scholar] [CrossRef]
- Hoshino, Y.; Lee, H.; Miura, Y. Interaction between synthetic particles and biomacromolecules: Fundamental study of nonspecific interaction and design of nanoparticles that recognize target molecules. Polym. J. 2014, 46, 537–545. [Google Scholar] [CrossRef]
- Dinc, M.; Esen, C.; Mizaikoff, B. Recent advances on core–shell magnetic molecularly imprinted polymers for biomacromolecules. TrAC Trends Anal. Chem. 2019, 114, 202–217. [Google Scholar] [CrossRef]
- Koch, S.J.; Renner, C.; Xie, X.; Schrader, T. Tuning Linear Copolymers into Protein-Specific Hosts. Angew. Chem. Int. Ed. 2006, 45, 6352–6355. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Lesel, B.K.; Yonamine, Y.; Beierle, J.M.; Hoshino, Y.; Shea, K.J. Temperature-Responsive “Catch and Release” of Proteins by using Multifunctional Polymer-Based Nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 2405–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Shea, K.J. Tuning the Protein Corona of Hydrogel Nanoparticles: The Synthesis of Abiotic Protein and Peptide Affinity Reagents. Acc. Chem. Res. 2016, 49, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Qiao, J.; Qi, L. Dual-functional polymer-modified magnetic nanoparticles for isolation of lysozyme. Anal. Chim. Acta 2018, 1035, 70–76. [Google Scholar] [CrossRef]
- Callewaert, L.; Michiels, C. Lysozymes in the animal kingdom. J. Biosci. 2010, 35, 127–160. [Google Scholar] [CrossRef] [PubMed]
- Hankiewicz, J.; Swierczek, E. Lysozyme in human body fluids. Clin. Chim. Acta 1974, 57, 205–209. [Google Scholar] [CrossRef]
- Pruzanski, W.; Saito, S.G. The Diagnostic Value of Lysozyme (Muramidase) Estimation in Biological Fluids. Am. J. Med. Sci. 1969, 258, 405–415. [Google Scholar] [CrossRef]
- Kovanyi, G.; Letnansky, K. Urine and blood serum muramidase (lysozyme) in patients with urogenital tumors. Eur. J. Cancer 1971, 7, 25–31. [Google Scholar] [CrossRef]
- Porstmann, B.; Jung, K.; Schmechta, H.; Evers, U.; Pergande, M.; Porstmann, T.; Kramm, H.-J.; Krause, H. Measurement of lysozyme in human body fluids: Comparison of various enzyme immunoassay techniques and their diagnostic application. Clin. Biochem. 1989, 22, 349–355. [Google Scholar] [CrossRef]
- Osserman, E.F.; Lawlor, D.P. Serum and urinary lysozyme (muramidase) in monocytic and monomyelocytic leukemia. J. Exp. Med. 1966, 124, 921–952. [Google Scholar] [CrossRef] [PubMed]
- Smolelis, A.N.; Hartsell, S.E. The Determination of Lysozyme. J. Bacteriol. 1949, 58, 731–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.J.; Russo, A.; Craswell, P.; Ward, M.; Steinhardt, I. Radioimmunoassay for serum and urinary lysozyme. Clin. Chem. 1981, 27, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; John, M.R.; Schmidt-Gayk, H.; Arndt, B.; Scheida, M.; Theuer, D. Solid-phase competitive luminescence immunoassay for lysozyme in faeces. Clin. Chim. Acta 1995, 239, 167–177. [Google Scholar] [CrossRef]
- Montagne, P.; Cuillière, M.L.; Molé, C.; Béné, M.C.; Faure, G. Microparticle-enhanced nephelometric immunoassay of lysozyme in milk and other human body fluids. Clin. Chem. 1998, 44, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Labella, C.; Lelario, F.; Bufo, S.A.; Musto, M.; Freschi, P.; Cosentino, C. Optimization and Validation of a Chromatographic Method for Quantification of Lysozyme in Jenny Milk. J. Food Nutr. Res. 2016, 55, 263–269. [Google Scholar]
- Huang, J.; Kailemia, M.J.; Goonatilleke, E.; Parker, E.A.; Hong, Q.; Sabia, R.; Smilowitz, J.T.; German, J.B.; Lebrilla, C.B. Quantitation of human milk proteins and their glycoforms using multiple reaction monitoring (MRM). Anal. Bioanal. Chem. 2016, 409, 589–606. [Google Scholar] [CrossRef]
- Fu, X.; Fu, X.; Wang, Q.; Sheng, L.; Huang, X.; Ma, M.; Cai, Z. Fluorescence switch biosensor based on quantum dots and gold nanoparticles for discriminative detection of lysozyme. Int. J. Biol. Macromol. 2017, 103, 1155–1161. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhang, L.; Gong, J. High-Throughput Signal-On Photoelectrochemical Immunoassay of Lysozyme Based on Hole-Trapping Triggered by Disintegrating Bioconjugates of Dopamine-Grafted Silica Nanospheres. ACS Sens. 2018, 3, 1480–1488. [Google Scholar] [CrossRef]
- Marlow, E.; Murphy, A.; Pollard, R. Optimisation of Gold Nanorod Array for Sensing Applications. In Proceedings of the 2018 IEEE British and Irish Conference on Optics and Photonics (BICOP), London, UK, 12–14 September 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 1–4. [Google Scholar] [CrossRef]
- Melinte, G.; Selvolini, G.; Cristea, C.; Marrazza, G. Aptasensors for lysozyme detection: Recent advances. Talanta 2021, 226, 122169. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, T.; Mazzotta, E.; Malitesta, C. Molecularly Imprinted Polyscopoletin for the Electrochemical Detection of the Chronic Disease Marker Lysozyme. Biosensors 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Meng, Z.; Xue, M.; Zhang, H.; Shea, K.J.; Kang, L. Detection of lysozyme in body fluid based on two-dimensional colloidal crystal sensor. Microchem. J. 2020, 157, 105073. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, X.; He, X.; Chen, L.; Zhang, Y. A self-assembled polydopamine film on the surface of magnetic nanoparticles for specific capture of protein. Nanoscale 2012, 4, 3141–3147. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dai, Q.; Wang, Y.; Hu, X.; Xu, P.; Ni, R.; Meng, J. Creating magnetic ionic liquid-molecularly imprinted polymers for selective extraction of lysozyme. RSC Adv. 2018, 8, 21850–21856. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Ma, S.; Ma, J.; Yang, Q.; Yi, Q.; Wu, Y.; Gu, Z. Superparamagnetic nanocomposites based on surface imprinting for biomacromolecular recognition. Mater. Sci. Eng. C 2017, 70, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yuan, D.; Fu, G. Enhanced surface imprinting of lysozyme over a new kind of magnetic chitosan submicrospheres. J. Colloid Interface Sci. 2015, 440, 53–59. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Wang, H.; Wu, C.; Li, M.; Li, L. Fabrication and evaluation of molecularly imprinted magnetic nanoparticles for selective recognition and magnetic separation of lysozyme in human urine. Analyst 2018, 143, 5849–5856. [Google Scholar] [CrossRef]
- He, H.; Fu, G.; Wang, Y.; Chai, Z.; Jiang, Y.; Chen, Z. Imprinting of protein over silica nanoparticles via surface graft copolymerization using low monomer concentration. Biosens. Bioelectron. 2010, 26, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Jing, T.; Du, H.; Dai, Q.; Xia, H.; Niu, J.; Hao, Q.; Mei, S.; Zhou, Y. Magnetic molecularly imprinted nanoparticles for recognition of lysozyme. Biosens. Bioelectron. 2010, 26, 301–306. [Google Scholar] [CrossRef]
- Li, N.; Qi, L.; Shen, Y.; Qiao, J.; Chen, Y. Novel Oligo(ethylene glycol)-Based Molecularly Imprinted Magnetic Nanoparticles for Thermally Modulated Capture and Release of Lysozyme. ACS Appl. Mater. Interfaces 2014, 6, 17289–17295. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Guo, P.; Luo, Z.; Ge, Y.; Zhou, Y.; Chang, R.; Du, W.; Chang, C.; Fu, Q. Preparation and characterization of surface molecularly imprinted films coated on multiwall carbon nanotubes for recognition and separation of lysozyme with high binding capacity and selectivity. RSC Adv. 2017, 7, 18765–18774. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Xia, Z.; Zheng, J.; Zheng, D.; Zhang, L.; Yang, H.; Chen, G. Synthesis of uniformly sized molecularly imprinted polymer-coated silica nanoparticles for selective recognition and enrichment of lysozyme. J. Mater. Chem. 2012, 22, 17914–17922. [Google Scholar] [CrossRef]
- Jing, T.; Xia, H.; Guan, Q.; Lu, W.; Dai, Q.; Niu, J.; Lim, J.-M.; Hao, Q.; Lee, Y.-I.; Zhou, Y.; et al. Rapid and selective determination of urinary lysozyme based on magnetic molecularly imprinted polymers extraction followed by chemiluminescence detection. Anal. Chim. Acta 2011, 692, 73–79. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Erdőssy, J.; Horváth, V. The Role of the Initiator System in the Synthesis of Acidic Multifunctional Nanoparticles Designed for Molecular Imprinting of Proteins. Period. Polytech. Chem. Eng. 2020, 65, 28–41. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Bioanalytical Method Validation; European Medicines Agency: Amsterdam, The Netherlands, 2011.
- Deng, H.; Li, X.; Peng, Q.; Wang, X.; Chen, J.; Li, Y. Monodisperse Magnetic Single-Crystal Ferrite Microspheres. Angew. Chem. Int. Ed. 2005, 44, 2782–2785. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Nagy, F.; Tasnádi, G.; Balogh-Weiser, D.; Bell, E.; Hall, M.; Faber, K.; Poppe, L. Smart Nanoparticles for Selective Immobilization of Acid Phosphatases. ChemCatChem 2018, 10, 3490–3499. [Google Scholar] [CrossRef] [Green Version]
- Erdőssy, J.; Kassa, E.; Farkas, A.; Horváth, V. Enzymatic digestion as a tool for removing proteinaceous templates from molecularly imprinted polymers. Anal. Methods 2017, 9, 4496–4503. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Ogawa, K.; Suzuki, H.; Wang, B.; Yoshida, R.; Kokufuta, E. Preparation of Thermosensitive Submicrometer Gel Particles with Anionic and Cationic Charges. Langmuir 1999, 15, 4289–4294. [Google Scholar] [CrossRef]
- Wetter, L.R.; Deutsch, H.F. Immunological studies on egg white proteins. IV. Immunochemical and physical studies of lysozyme. J. Biol. Chem. 1951, 192, 237–242. [Google Scholar] [CrossRef]
- Tokuhiro, T. Temperature Dependence of Density of Polymer Gels 1. A Pycnometry Method Applied to Poly(N-isopropylacrylamide)−Water System. J. Phys. Chem. B 1999, 103, 7097–7101. [Google Scholar] [CrossRef]
- Greer, R.M.; Scruggs, B.A.; May, R.A.; Chandler, B.D. Patterning High Surface Area Silica with Lysozyme: Adsorption Kinetics, Fluorescence Quenching, and Protein Readsorption Studies To Evaluate the Templated Surface. Langmuir 2009, 25, 7161–7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debord, J.D.; Lyon, L.A. Synthesis and Characterization of pH-Responsive Copolymer Microgels with Tunable Volume Phase Transition Temperatures. Langmuir 2003, 19, 7662–7664. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Step | Release Step | ||||||
|---|---|---|---|---|---|---|---|
| PB Concentration [mM] | pH | Phase Ratio [mg/mL] | PB Concentration [mM] | pH | Additive | Phase Ratio [mg/mL] | Recovery in the Release Step [%] |
| 10 | 8.6 | 1.6 | 10 | 8.4 | - | 10.67 | 31.2 |
| 50 | 7 | - | 10.67 | 70.0 | |||
| 10 | 8.4 | 0.2 M NaCl | 10.67 | 81.2 | |||
| 50 | 7 | 0.2 M NaCl | 10.67 | 90.6 | |||
| 50 | 7 | - | 1.6 | 104.2 | |||
| Urine Concentration [µg/mL] | Nominal Sample Concentration [µg/mL] | Measured Concentration [µg/mL] | Precision [%] | Accuracy [%] |
|---|---|---|---|---|
| 25 | 2.5 | 2.94 | 3.44 | 117.6 |
| 100 | 10 | 9.35 | 8.20 | 93.5 |
| 250 | 25 | 25.8 | 4.25 | 103.0 |
| 1000 | 100 | 89.9 | 2.24 | 89.9 |
| Urine Concentration [µg/mL] | Nominal Sample Concentration [µg/mL] | Measured Concentration [µg/mL] | Precision [%] | Accuracy [%] |
|---|---|---|---|---|
| 25 | 2.5 | 2.774 | 1.08 | 111.0 |
| 100 | 10 | 9.442 | 3.21 | 94.4 |
| 250 | 25 | 24.81 | 5.04 | 99.2 |
| 1000 | 100 | 88.80 | 4.40 | 88.8 |
| 2000 | 200 | 128.8 | 2.25 | 64.4 |
| Urine Concentration [µg/mL] | Standard Solution Concentration [µg/mL] | Recovery [%] | RSD [%] |
|---|---|---|---|
| 100 | 10 | 91.7 ± 3.8 | 4.19 |
| 250 | 25 | 103.1 ± 3.9 | 3.80 |
| 1000 | 100 | 87.5 ± 9.4 | 10.81 |
| 2000 | 200 | 62.3 ± 6.3 | 10.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.A.; Erdőssy, J.; Horvath, V. Temperature-Responsive Magnetic Nanoparticles for Bioanalysis of Lysozyme in Urine Samples. Nanomaterials 2021, 11, 3015. https://doi.org/10.3390/nano11113015
Ahmed MA, Erdőssy J, Horvath V. Temperature-Responsive Magnetic Nanoparticles for Bioanalysis of Lysozyme in Urine Samples. Nanomaterials. 2021; 11(11):3015. https://doi.org/10.3390/nano11113015
Chicago/Turabian StyleAhmed, Marwa A., Júlia Erdőssy, and Viola Horvath. 2021. "Temperature-Responsive Magnetic Nanoparticles for Bioanalysis of Lysozyme in Urine Samples" Nanomaterials 11, no. 11: 3015. https://doi.org/10.3390/nano11113015
APA StyleAhmed, M. A., Erdőssy, J., & Horvath, V. (2021). Temperature-Responsive Magnetic Nanoparticles for Bioanalysis of Lysozyme in Urine Samples. Nanomaterials, 11(11), 3015. https://doi.org/10.3390/nano11113015