
Insights into the Structure and Dynamics of Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures: A Molecular Dynamics Approach




Abstract
1. Introduction
2. Materials and Methods
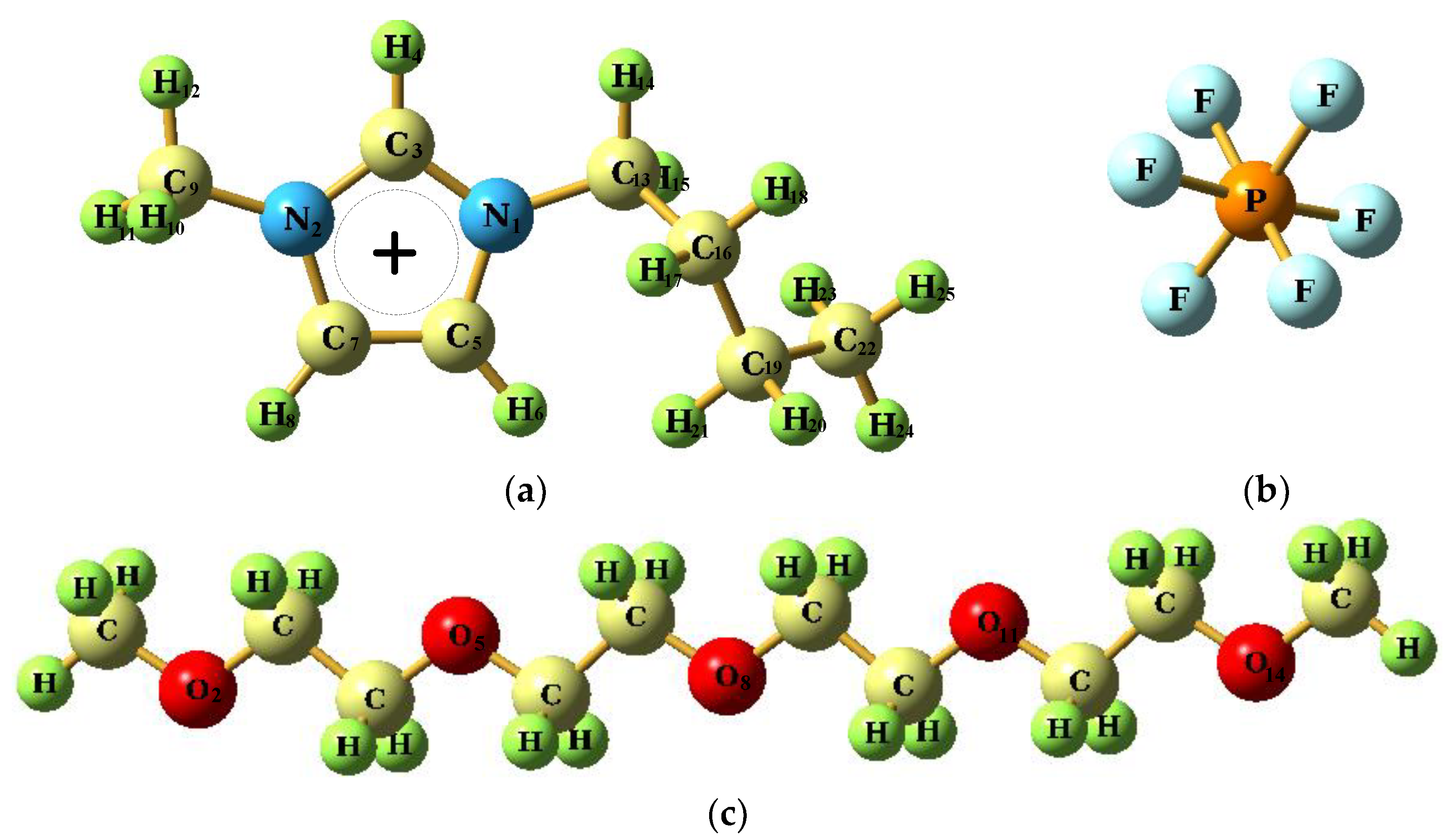
2.1. Force Field Parameters
2.2. Simulation Details
3. Results
3.1. Density for Evaluation of Forcefields
3.2. Structural Analysis
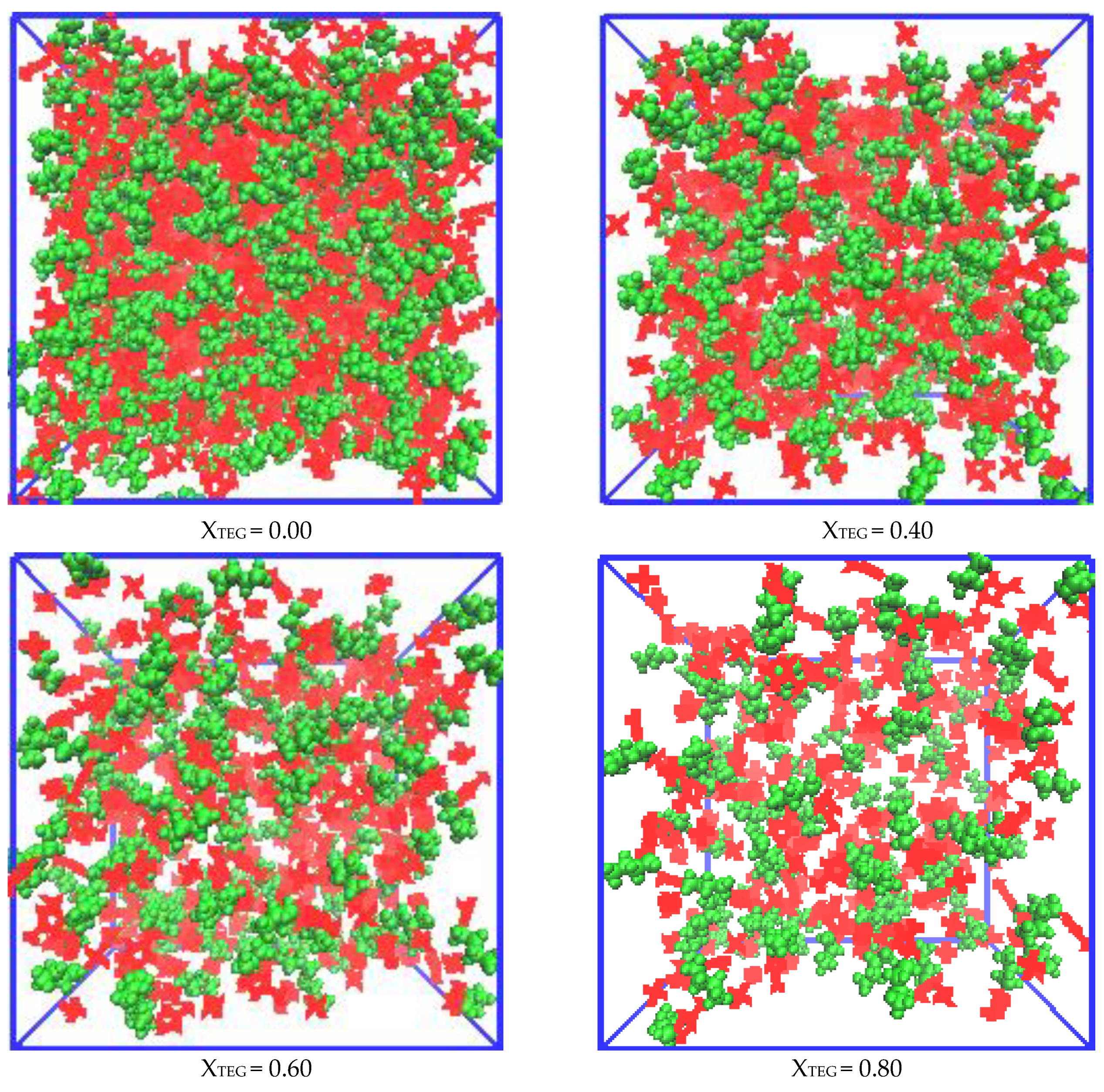
3.2.1. Morphologies of Microscopic Structure in IL/TEGDME Mixtures

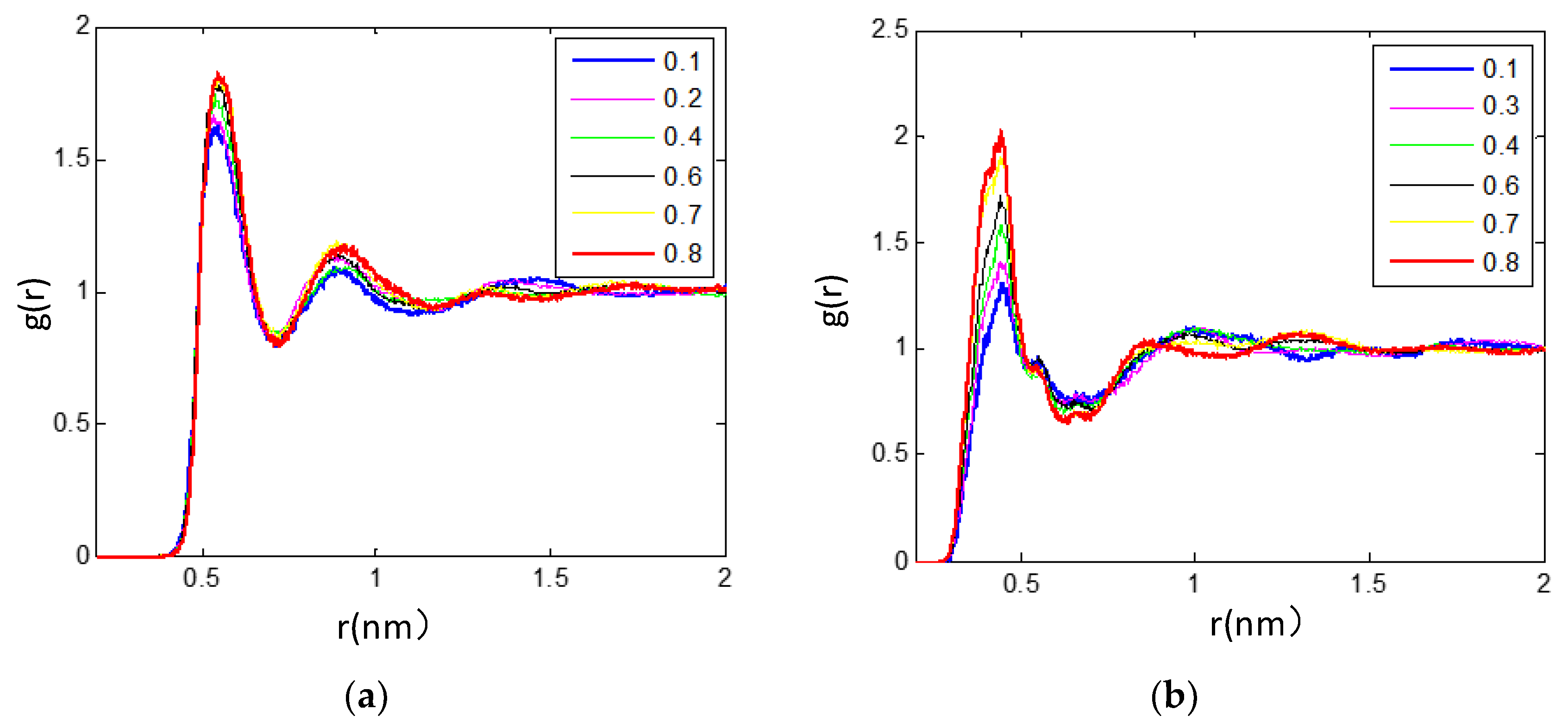
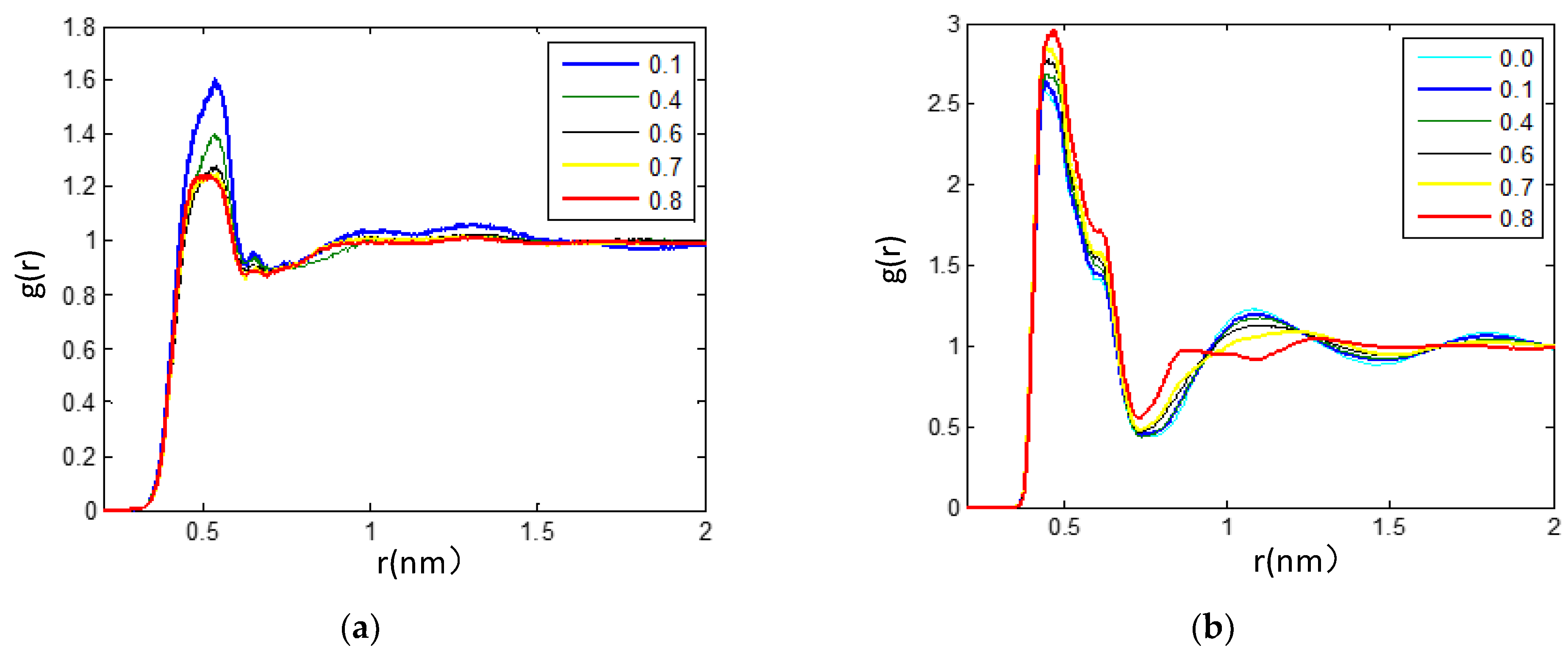
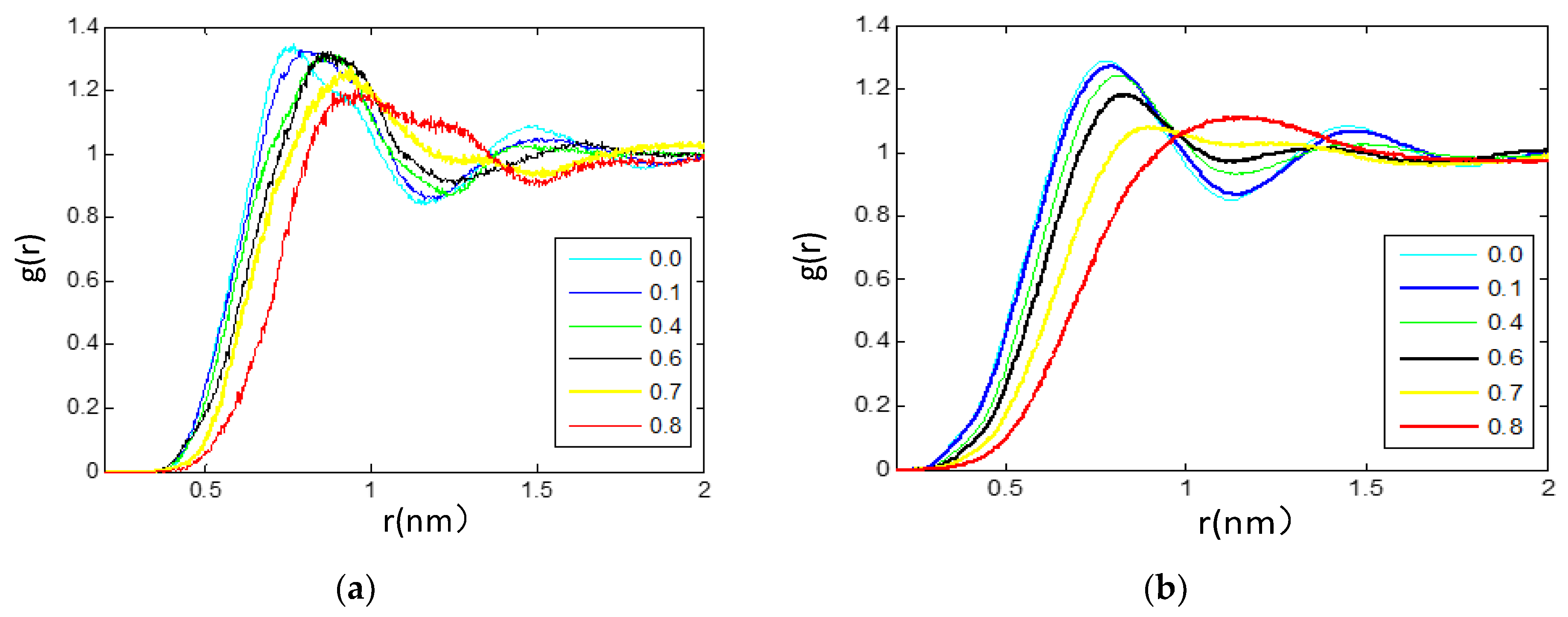
3.2.2. Radial Distribution Functions Analysis of Microscopic Ionic Structure






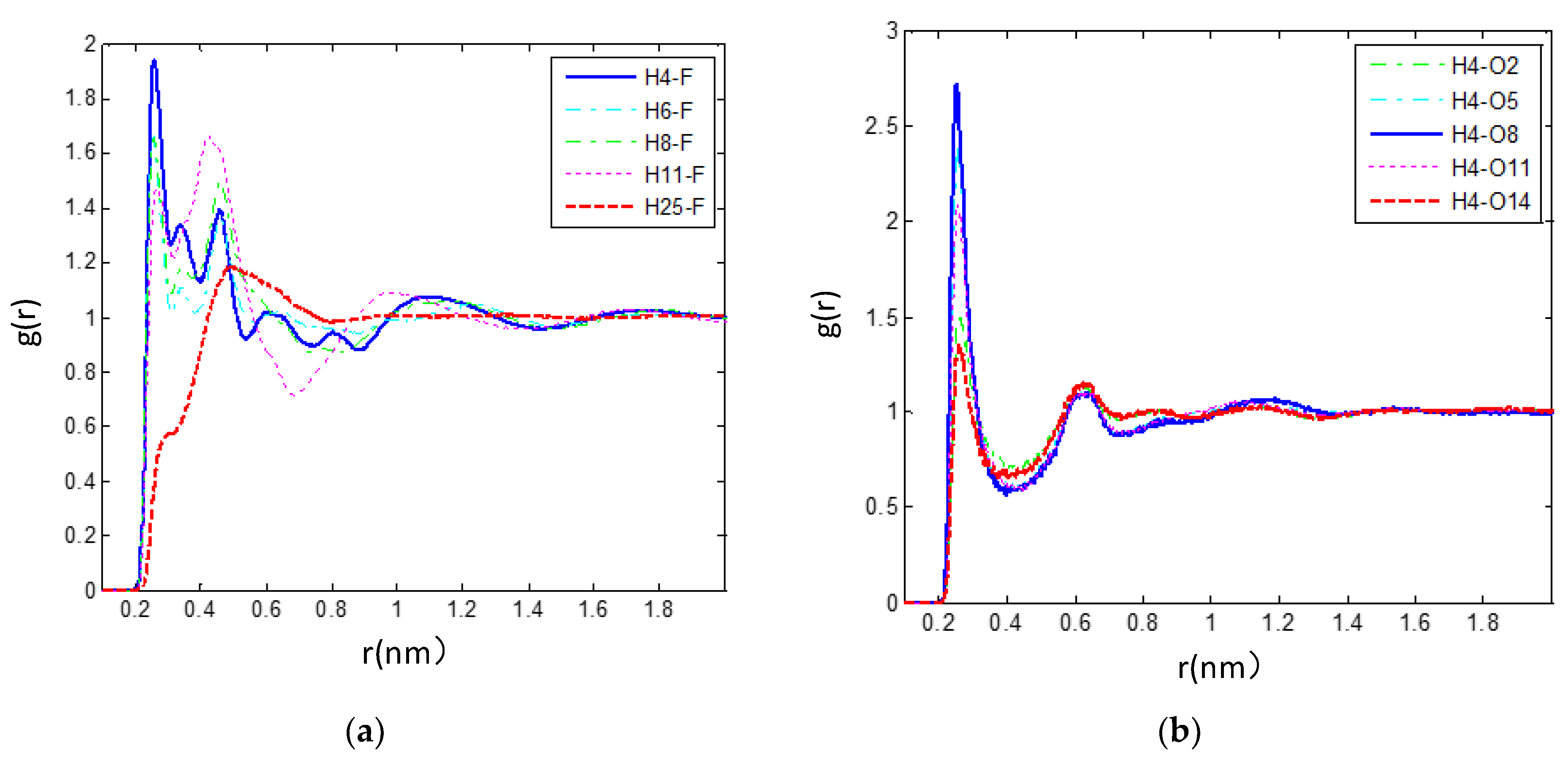
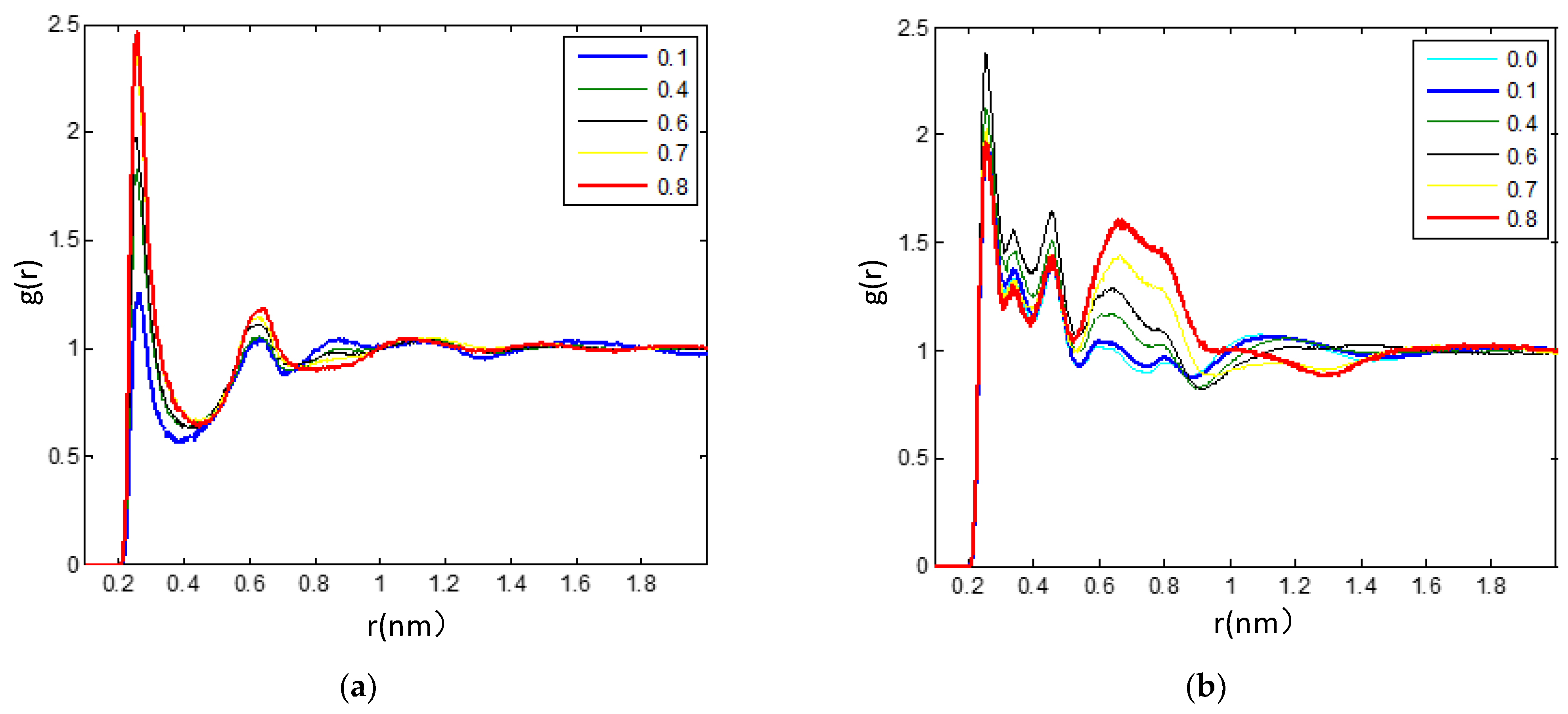
3.2.3. The Hydrogen Bonds in the IL/TEGDME Mixture
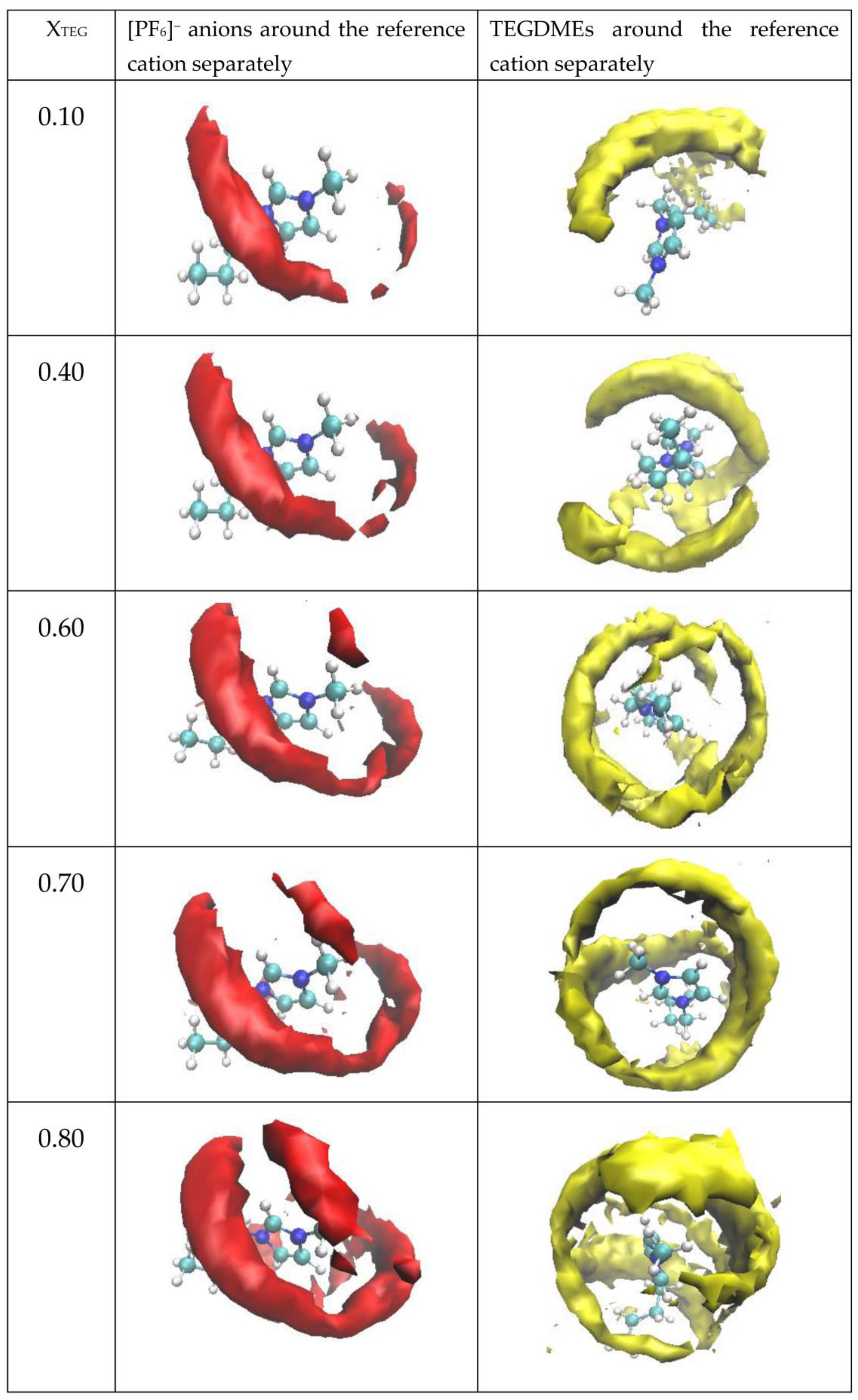
3.2.4. Spatial Distribution Functions
3.3. Dynamical and Transport Properties of [bmim][PF6]/TEGDME Mixtures
3.3.1. Rotational Dynamics of C153 in [bmim][PF6] and TEGDME Mixtures

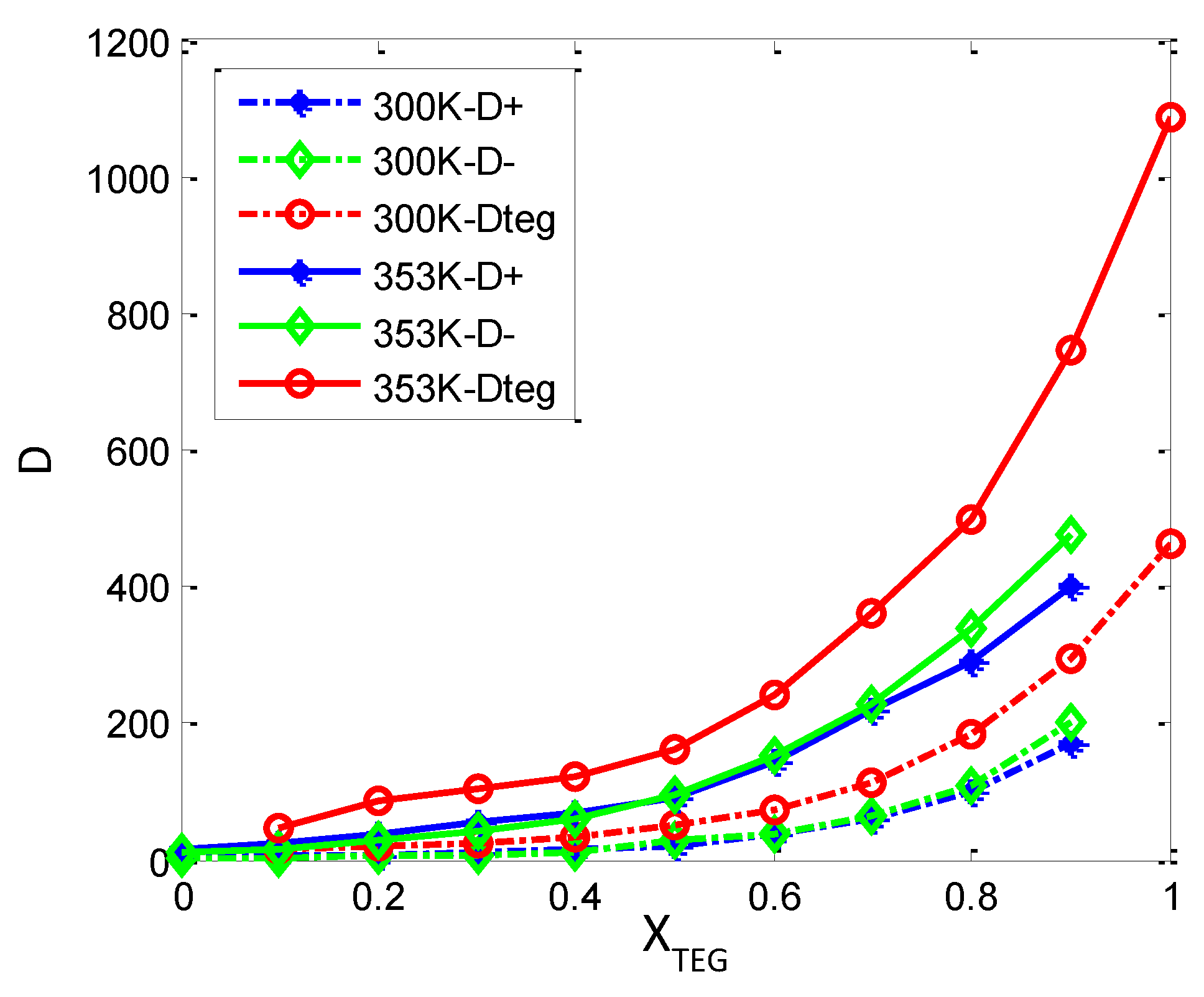
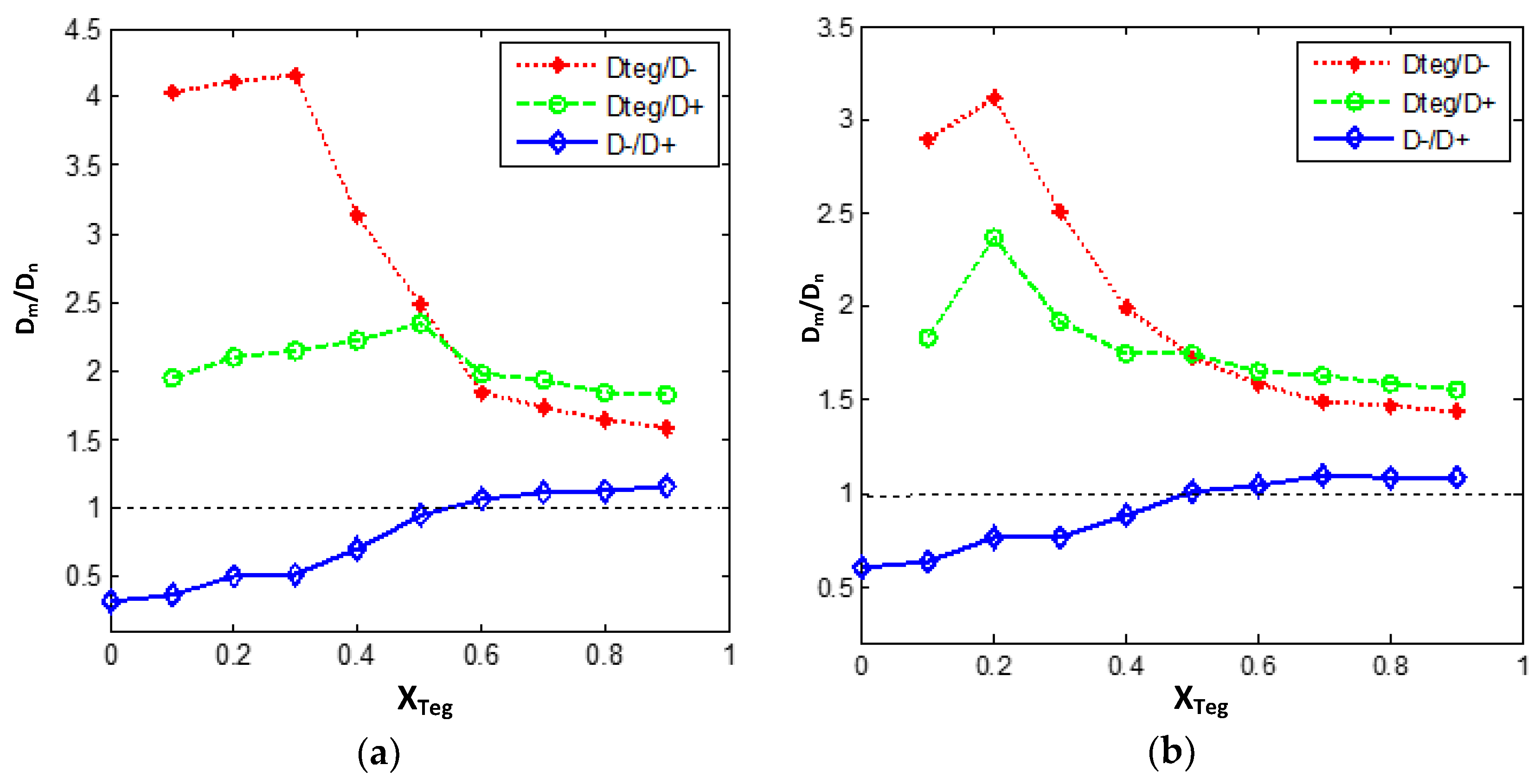
3.3.2. Diffusion Dynamics of Ions in [bmim][PF6] and TEGDME Mixtures


4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogers, R.D.; Seddon, K.R. Ionic liquids-solvents of the future. Science 2003, 302, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Fadeev, A.G.; Qi, B.; Smela, E.; Mattes, B.R.; Ding, J.; Spinks, G.M.; Mazurkiewicz, J.; Zhou, D.; Wallace, G.G.; et al. Use of ionic liquids for pi-conjugated polymer electrochemical devices. Science 2002, 297, 983–987. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-Liquid Materials for the Electrochemical Challenges of the Future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Liu, Y.A. Selection of Prediction Methods for Thermophysical Properties for Process Modeling and Product Design of Biodiesel Manufacturing. Ind. Eng. Chem. Res. 2011, 50, 6809–6836. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- Niedermeyer, H.; Hallett, J.P.; Villar-Garcia, I.J.; Hunt, P.A.; Welton, T. Mixtures of Ionic Liquids. Chem. Soc. Rev. 2012, 41, 7780–7802. [Google Scholar] [CrossRef] [PubMed]
- Doroodian, A.; Dengler, J.E.; Genest, A.; Rösch, N.; Rieger, B. Methylguanidinium Borohydride: An Ionic-Liquid-Based Hydrogen-Storage Material. Angew. Chem. Int. Ed. 2010, 49, 1871–1873. [Google Scholar] [CrossRef]
- Aparicio, S.; Atilhan, M.; Karadas, F. Thermophysical Properties of Pure Ionic Liquids: Review of Present Situation. Ind. Eng. Chem. Res. 2010, 49, 9580–9595. [Google Scholar] [CrossRef]
- Forse, A.C.; Griffin, J.M.; Merlet, C.; Bayley, P.M.; Wang, H.; Simon, P.; Grey, C.P. NMR study of ion dynamics and charge storage in ionic liquid supercapacitors. J. Am. Chem. Soc. 2015, 137, 7231–7242. [Google Scholar] [CrossRef]
- Gouverneur, M.; Kopp, J.; Wüllen, L.; Van Schönho, M. Direct determination of ionic transference numbers in ionic liquids by electrophoretic NMR. Phys. Chem. Chem. Phys. 2015, 17, 30680–30686. [Google Scholar] [CrossRef]
- Borodin, O.; Gorecki, W.; Smith, G.D.; Armand, M. Molecular Dynamics Simulation and Pulsed-Field Gradient NMR Studies of Bis(fluorosulfonyl)imide (FSI) and Bis[(trifluoromethyl)sulfonyl]imide (TFSI)-Based Ionic Liquids. J. Phys. Chem. B 2010, 114, 6786–6798. [Google Scholar] [CrossRef]
- Hardacre, C.; Holbrey, J.D.; Mullan, C.L.; Youngs, T.G.A.; Bowron, D.T. Small Angle Neutron Scattering from 1-Alkyl-3- methylimidazolium Hexafluorophosphate Ionic Liquids ([Cnmim]-[PF6], n = 4, 6, and 8). J. Chem. Phys. 2010, 133, 074510. [Google Scholar] [CrossRef]
- Annapureddy, H.V.; Kashyap, H.K.; De Biase, P.M.; Margulis, C.J. What is the Origin of the Prepeak in the X-ray Scattering of Imidazolium-Based Room-Temperature Ionic Liquids? J. Phys. Chem. B 2010, 114, 16838–16846. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, H.; Maeda, Y. LCST and UCST behavior of poly(Nisopropylacrylamide) in DMSO/water mixed solvents studied by IR and micro-raman spectroscopy. J. Phys. Chem. B 2007, 111, 12964–12968. [Google Scholar] [CrossRef] [PubMed]
- Li, B.X.; Qiu, M.; Long, S.R.; Wang, X.F.; Guo, Q.J.; Xia, A.D. Unraveling heterogeneous microviscosities of the 1-alkyl-3- methylimidazolium hexafluorophosphate ionic liquids with different chain lengths. Phys. Chem. Chem. Phys. 2013, 15, 16074–16081. [Google Scholar] [CrossRef] [PubMed]
- Li, B.X.; Wang, Y.Y.; Wang, X.F.; Vdovic, S.; Guo, Q.J.; Xia, A.D. Spectroscopic Evidence for Unusual Microviscosity in Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures. J. Phys. Chem. B 2012, 116, 13272–13281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stoppa, A.; Hunger, J.; Buchner, R. Conductivities of Binary Mixtures of Ionic Liquids with Polar Solvents. J. Chem. Eng. Data 2009, 54, 472–479. [Google Scholar] [CrossRef]
- Hogan, C.J., Jr.; de la Mora, J.F. Tandem Ion Mobility-Mass Spectrometry (IMS-MS) Study of Ion Evaporation from Ionic Liquid-Acetonitrile Nanodrops. Phys. Chem. Chem. Phys. 2009, 11, 8079–8090. [Google Scholar] [CrossRef]
- Richey, F.W.; Tran, C.; Kalra, V.; Elabd, Y.A. Ionic liquid dynamics in nanoporous carbon nanofibers in supercapacitors measured with in operando infrared spectroelectrochemistry. J. Phys. Chem. C 2014, 118, 21846–21855. [Google Scholar] [CrossRef]
- Chakrabarty, D.; Seth, D.; Chakraborty, A.; Sarkar, N. Dynamics of solvation and rotational relaxation of coumarin 153 in ionic liquid confined nanometer-sized microemulsions. J. Phys. Chem. B 2005, 109, 5753–5758. [Google Scholar] [CrossRef]
- Seth, D.; Sarkar, S.; Sarkar, N. Dynamics of solvent and rotational relaxation of coumarin-153 in room-temperature ionic liquid 1-butyl-3-methyl imidazolium tetrafluoroborate confined in poly(oxyethylene glycol) ethers containing micelles. J. Phys. Chem. B 2007, 111, 4781–4787. [Google Scholar] [CrossRef]
- Fumino, K.; Wulf, A.; Ludwig, R. Strong, Localized and Directional Hydrogen Bonds Fluidize Ionic Liquids. Angew. Chem. Int. Ed. 2008, 47, 8731–8734. [Google Scholar] [CrossRef]
- Peppel, T.; Roth, C.; Paschek, D.; Kocherling, M.; Ludwig, R. The Influence of Hydrogen-Bond Defects on the Properties of Ionic Liquids. Angew. Chem. Int. Ed. 2011, 50, 6661–6665. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, N.; Luo, J.; Zhou, Y.; Yu, Z. Hydrogen-Bonding Interactions Between [BMIM][BF4] and Acetonitrile. Phys. Chem. Chem. Phys. 2013, 15, 18055–18064. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, R.; Rao, V.G.; Sarkar, S.; Ghatak, C.; Setua, P.; Sarkar, N.J. To Probe the Interaction of Methanol and Acetonitrile with the Ionic Liquid N,N,N-Trimethyl-N-propyl Ammonium Bis(trifluoromethanesulfonyl) Imide at Different Temperatures by Solvation Dynamics Study. Phys. Chem. B 2009, 113, 8626–8634. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Pandey, S. Interactions within a Ionic Liquid plus Poly(ethylene glycol) Mixture Revealed by Temperature-Dependent Synergistic Dynamic Viscosity and Probe-Reported Microviscosity. J. Phys. Chem. B 2011, 115, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Trivedi, S.; Pandey, S. Polymer Molecular Weight-Dependent Unusual Fluorescence Probe Behavior within 1-Butyl-3-methylimidazolium Hexafluorophosphate plus Poly(ethylene glycol). J. Phys. Chem. B 2009, 113, 7606–7614. [Google Scholar] [CrossRef]
- Sarkar, A.; Trivedi, S.; Baker, G.A.; Pandey, S. Multiprobe Spectroscopic Evidence for “Hyperpolarity” within 1-Butyl-3- methylimidazolium Hexafluorophosphate Mixtures with Tetraethylene glycol. J. Phys. Chem. B 2008, 112, 14927–14936. [Google Scholar] [CrossRef]
- Stoppa, A.; Hunger, J.; Hefter, G.; Buchner, R. Structure and Dynamics of 1-N-Alkyl-3-NMethylimidazolium Tetrafluoroborate + Acetonitrile Mixtures. J. Phys. Chem. B 2012, 116, 7509–7521. [Google Scholar] [CrossRef]
- Kalugin, O.N.; Voroshylova, I.V.; Riabchunova, A.V.; Lukinova, E.V.; Chaban, V.V. Conductometric Study of Binary Systems Based on Ionic Liquids and Acetonitrile in a Wide Concentration Range. Electrochim. Acta 2013, 105, 188–199. [Google Scholar] [CrossRef]
- Coadou, E.; Timperman, L.; Jacquemin, J.; Galiano, H.; Hardacre, C.; Anouti, M. Comparative Study on Performances of Trimethyl-Sulfonium and Trimethyl-Ammonium Based Ionic Liquids in Molecular Solvents as Electrolyte for Electrochemical Double Layer Capacitors. J. Phys. Chem. C 2013, 117, 10315–10325. [Google Scholar] [CrossRef]
- Chevrot, G.; Fileti, E.E.; Chaban, V.V. Enhanced stability of the model mini-protein in amino acid ionic liquids and their aqueous solutions. J. Comput. Chem. 2015, 36, 2044–2051. [Google Scholar] [CrossRef]
- Chevrot, G.; Fileti, E.E.; Chaban, V.V. Protein remains stable at unusually high temperatures when solvated in aqueous mixtures of amino acid based ionic liquids. J. Mol. Modeling 2016, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maginn, E.J. Direct Correlation Between Ionic Liquid Transport Properties and Ion Pair Lifetimes: A Molecular Dynamics Study. J. Phys. Chem. Lett. 2015, 6, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, Y.; Zeng, S.; Zhang, X.; Zhang, S. Density Prediction of Mixtures of Ionic Liquids and Molecular Solvents Using Two New Generalized Models. Ind. Eng. Chem. Res. 2014, 53, 15270–15277. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Z.; Han, B.; Hu, S.; Xie, Y.; Yang, G. Effect of Water and Organic Solvents on the Ionic Dissociation of Ionic Liquids. J. Phys. Chem. B 2007, 111, 6452–6456. [Google Scholar] [CrossRef]
- Menjoge, A.; Dixon, J.; Brennecke, J.F.; Maginn, E.J.; Vasenkov, S. Influence of Water on Diffusion in Imidazolium-Based Ionic Liquids: A Pulsed Field Gradient NMR study. J. Phys. Chem. B 2009, 113, 6353–6359. [Google Scholar] [CrossRef]
- Rollet, A.L.; Porion, P.; Vaultier, M.; Billard, I.; Deschamps, M.; Bessada, C.; Jouvensal, L. Anomalous Diffusion of Water in [BMIM][TFSI] Room-Temperature Ionic Liquid. J. Phys. Chem. B 2007, 111, 11888–11891. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.A.; Le, K.A.; Rudaz, C.; Radhi, A.; Lovell, C.S.; Damion, R.A.; Budtova, T.; Ries, M.E. Macroscopic and Microscopic Study of 1-Ethyl-3-methyl-imidazolium Acetate−Water Mixtures. J. Phys. Chem. B 2012, 116, 12810–12818. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.M.; Dreyer, D.R.; Bielawski, C.W.; Ruoff, R.S. Combined Measurement of Translational and Rotational Diffusion in Quaternary Acyclic Ammonium and Cyclic Pyrrolidinium Ionic Liquids. J. Phys. Chem. B 2013, 117, 1967–1977. [Google Scholar] [CrossRef]
- Liu, H.; Maginn, E. A Molecular Dynamics Investigation of the Structural and Dynamic Properties of the Ionic Liquid 1-n-Butyl-3- Methylimidazolium Bis(Trifluoromethanesulfonyl)Imide. J. Chem. Phys. 2011, 135, 124507. [Google Scholar] [CrossRef]
- Lopes, J.N.C.; Deschamps, J.; Padua, A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Lopes, J.N.C.; Padua, A.H.; Shimizu, K. Molecular Force Field for Ionic Liquids IV: Trialkylimidazolium and Alkoxycarbonyl-Imidazolium Cations; Alkylsulfonate and Alkylsulfate Anions. J. Phys. Chem. B 2008, 112, 5039–5046. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.N.C.; Padua, A.H. CL&P: A Generic and Systematic Force Field for Ionic Liquids Modeling. Theor. Chem. Acc. 2012, 131, 1129–1140. [Google Scholar]
- Jorgensen, W.L.; Briggs, J.M.; Contreras, M.L. Relative partition coefficients for organic solutes from fluid simulations. J. Phys. Chem. 1990, 94, 1683–1686. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225. [Google Scholar] [CrossRef]
- Voroshylova, I.V.; Chaban, V.V. Atomistic Force Field for Pyridinium-Based Ionic Liquids: Reliable Transport Properties. J. Phys. Chem. B 2014, 118, 10716–10724. [Google Scholar] [CrossRef] [PubMed]
- Chaban, V.V.; Voroshylova, I.V. Systematic Refinement of Canongia Lopes–Pádua Force Field for Pyrrolidinium-Based Ionic Liquids. J. Phys. Chem. B 2015, 119, 6242–6249. [Google Scholar] [CrossRef] [PubMed]
- Chaban, V.V. Polarizability versus mobility: Atomistic force field for ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 16055–16062. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D.; Swenson, C.J. Optimized intermolecular potential functions for liquid hydrocarbons. J. Am. Chem. Soc. 1984, 106, 6638–6646. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Kirchner, B. TRAVIS—A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Morrow, T.I.; Maginn, E.J. Molecular Dynamics Study of the Ionic Liquid 1-n-Butyl-3-methylimidazolium Hexafluorophosphate. J. Phys. Chem. B 2002, 106, 12807–12813. [Google Scholar] [CrossRef]
- Liu, Z.P.; Huang, S.P.; Wang, W.C. A Refined Force Field for Molecular Simulation of Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2004, 108, 12978–12989. [Google Scholar] [CrossRef]
- Liu, Z.P.; Wu, X.P.; Wang, W.C. A novel united-atom force field for imidazolium-based ionic liquids. Phys. Chem. Chem. Phys. 2006, 8, 1096–1104. [Google Scholar] [CrossRef]
- Borodin, O. Polarizable Force Field Development and Molecular Dynamics Simulations of Ionic Liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Bell, A.; Smit, B. Improved United-Atom Force Field for 1-Alkyl-3-methylimidazolium Chloride. J. Phys. Chem. B 2010, 114, 4572–4582. [Google Scholar] [CrossRef]
- Endo, T.; Kato, T.; Nishikawa, K. Effects of Methylation at the 2 Position of the Cation Ring on Phase Behaviors and Conformational Structural of Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2010, 114, 9201–9208. [Google Scholar] [CrossRef]
- Fredlake, C.P.; Crosthwaite, J.M.; Hert, D.G.; Aki, S.N.V.K.; Brennecke, J.F. Thermodynamical properties of imidazolium-based ionic liquids. J. Chem. Eng. Data 2004, 49, 954–961. [Google Scholar] [CrossRef]
- Ahn, H.J.; Kim, K.W.; Ahn, J.H.; Cho, K.K.; Nam, T.H.; Kim, J.U.; Cho, G.B.; Ryu, H.S. Discharge behavior of lithium/sulfur cell with TEGDME based electrolyte at low temperature. J. Power Sources 2006, 163, 201–206. [Google Scholar]
- Taylor, R.; Kennard, O. Crystallographic evidence for the existence of C-H···O, C-H···N, and C-H···C1 hydrogen-bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. [Google Scholar] [CrossRef]
- Hinze, G.; Diezemann, G.; Basché, T. Rotational Correlation Functions of Single Molecules. Phys. Rev. Lett. 2004, 93, 203001. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Hsu, W.Y.; Lin, M.Y.; Tai, C.C.; Wang, S.P.; Sun, I.W. Isolated BMI + Cations are More than Isolated PF6 Anions in the Room Temperature 1-Butyl-3-Methylimidazolium Hexafluorophosphate (BMI-PF6) Ionic Liquid. J. Chin. Chem. Soc. 2010, 57, 1293–1298. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| xTEG | NTEG | Nion Pairs | Total Atoms | Box Sizes (X, Y, Z) /Å | Rcutoff/nm | Density g/cm3 |
|---|---|---|---|---|---|---|
| 0.00 | - | 400 | 12,823 | 52.67 × 52.67 × 50.01 | 1.0 | 1.351 |
| 0.10 | 40 | 360 | 12,143 | 53.33 × 53.33 × 49.06 | 1.0 | 1.316 |
| 0.40 | 160 | 240 | 10,103 | 51.59 × 51.59 × 51.10 | 1.0 | 1.213 |
| 0.60 | 240 | 160 | 8743 | 51.03 × 51.03 × 51.56 | 1.0 | 1.146 |
| 0.70 | 280 | 120 | 8063 | 51.31 × 51.31 × 51.35 | 1.0 | 1.113 |
| 0.80 | 320 | 80 | 7383 | 51.26 × 51.26 × 51.07 | 1.0 | 1.078 |
| 1.00 | 400 | - | 6023 | 51.94 × 51.94 × 50.48 | 1.0 | 1.012 |
| TEGDME Mole Fraction | Experiment | MD Simulation | ||||
|---|---|---|---|---|---|---|
| τ1 (ps) | τ2 (ps) | τ1 (ps) | τ2 (ps) | |||
| 300 K | 300 K | 300 K | 353 K | 300 K | 353 K | |
| 0.0 | 0.32 | 4.90 | 0.41 | 0.26 | 8.67 | 4.97 |
| 0.1 | 0.39 | 4.45 | 0.45 | 0.37 | 7.85 | 4.56 |
| 0.2 | 0.48 | 3.90 | 0.52 | 0.41 | 6.56 | 4.01 |
| 0.3 | 0.65 | 3.61 | 0.78 | 0.52 | 5.97 | 3.51 |
| 0.4 | 0.73 | 2.88 | 0.81 | 0.61 | 4.89 | 2.71 |
| 0.5 | 0.77 | 2.64 | 0.85 | 0.56 | 4.31 | 2.59 |
| 0.6 | 0.76 | 2.13 | 0.83 | 0.52 | 3.56 | 2.07 |
| 0.7 | 0.74 | 1.63 | 0.79 | 0.50 | 2.21 | 1.52 |
| 0.8 | 0.70 | 1.19 | 0.77 | 0.47 | 1.67 | 1.21 |
| 0.9 | 0.63 | 0.63 | 0.73 | 0.43 | 0.81 | 0.57 |
| 1.0 | 0.46 | 0.46 | 0.71 | 0.39 | 0.71 | 0.39 |
| System (Xteg) | 300 K | 353 K | ||||||
|---|---|---|---|---|---|---|---|---|
| D+ | D− | Dteg | Dsys | D+ | D− | Dteg | Dsys | |
| 0 | 2.97 | 1.04 | -- | 1.94 | 8.81 | 5.30 | 7.02 | |
| 0.1 | 3.49 | 1.39 | 6.82 | 2.73 | 12.1 | 7.67 | 22.2 | 10.8 |
| 0.2 | 4.21 | 2.26 | 9.04 | 4.16 | 17.8 | 13.5 | 42.1 | 19.9 |
| 0.3 | 5.86 | 3.03 | 12.6 | 6.45 | 27.1 | 20.7 | 51.9 | 30.9 |
| 0.4 | 7.69 | 5.45 | 17.1 | 10.5 | 34.6 | 30.4 | 60.6 | 42.0 |
| 0.5 | 9.92 | 10.5 | 24.6 | 17.6 | 46.1 | 46.5 | 80.5 | 61.3 |
| 0.6 | 18.5 | 19.8 | 36.6 | 28.6 | 72.6 | 75.5 | 119.7 | 98.4 |
| 0.7 | 29.7 | 33.1 | 57.3 | 48.1 | 110.2 | 115.1 | 179.4 | 156.2 |
| 0.8 | 49.6 | 53.6 | 91.7 | 82.0 | 145.9 | 168.9 | 248.2 | 225.8 |
| 0.9 | 84.8 | 99.8 | 177.7 | 166.8 | 201.1 | 238.9 | 373.5 | 355.3 |
| 1.0 | - | - | 230.7 | 230.9 | 543.7 | 541.7 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Liu, Q.; Zhao, Y. Insights into the Structure and Dynamics of Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures: A Molecular Dynamics Approach. Nanomaterials 2021, 11, 2512. https://doi.org/10.3390/nano11102512
Guo Q, Liu Q, Zhao Y. Insights into the Structure and Dynamics of Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures: A Molecular Dynamics Approach. Nanomaterials. 2021; 11(10):2512. https://doi.org/10.3390/nano11102512
Chicago/Turabian StyleGuo, Qianjin, Qiang Liu, and Yixin Zhao. 2021. "Insights into the Structure and Dynamics of Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures: A Molecular Dynamics Approach" Nanomaterials 11, no. 10: 2512. https://doi.org/10.3390/nano11102512
APA StyleGuo, Q., Liu, Q., & Zhao, Y. (2021). Insights into the Structure and Dynamics of Imidazolium Ionic Liquid and Tetraethylene Glycol Dimethyl Ether Cosolvent Mixtures: A Molecular Dynamics Approach. Nanomaterials, 11(10), 2512. https://doi.org/10.3390/nano11102512