Heterofullerene MC59 (M = B, Si, Al) as Potential Carriers for Hydroxyurea Drug Delivery

Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
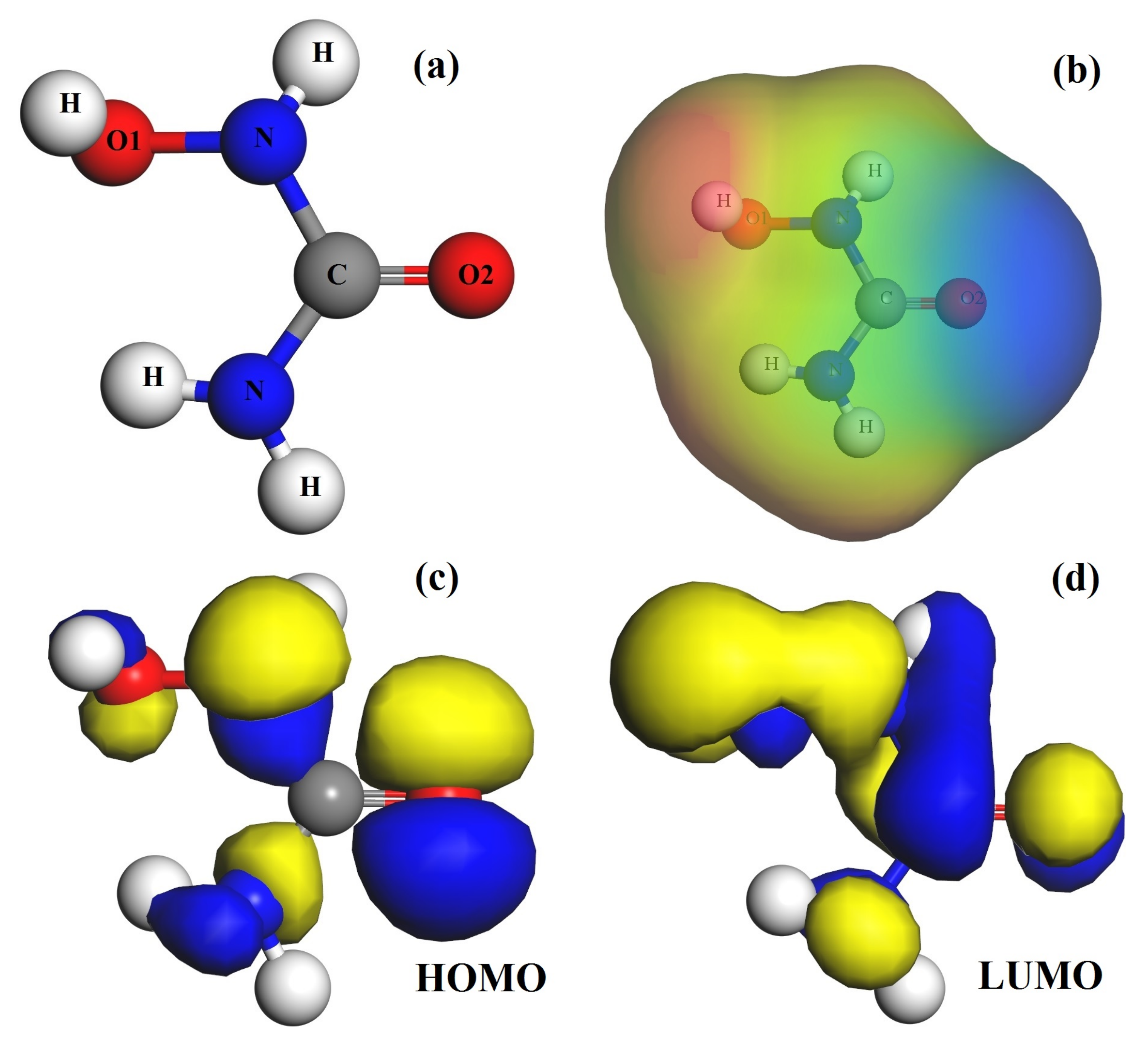
3.1. The Hydroxyurea Molecule Characterizations
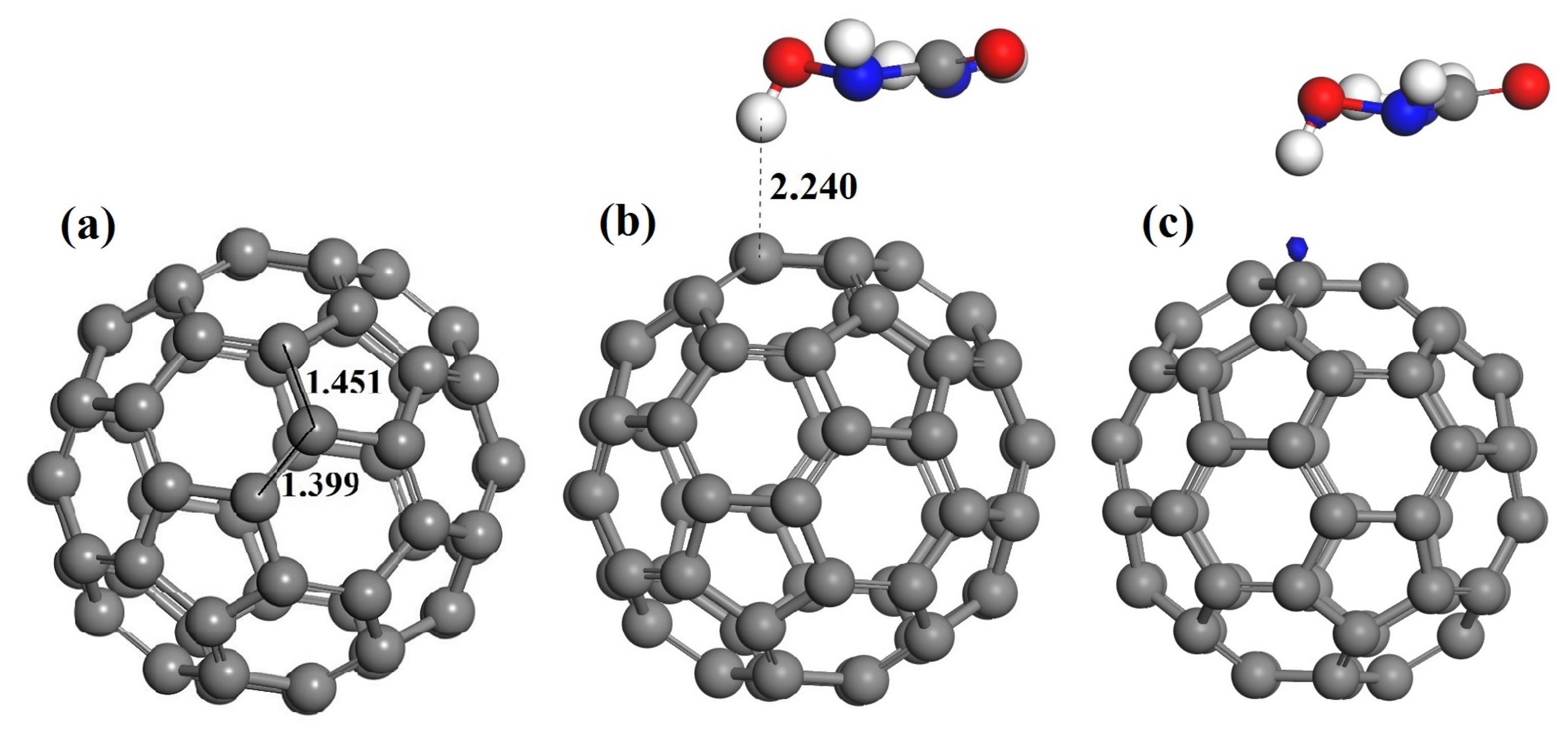
3.2. The Adsorption of HU on the Pristine C60
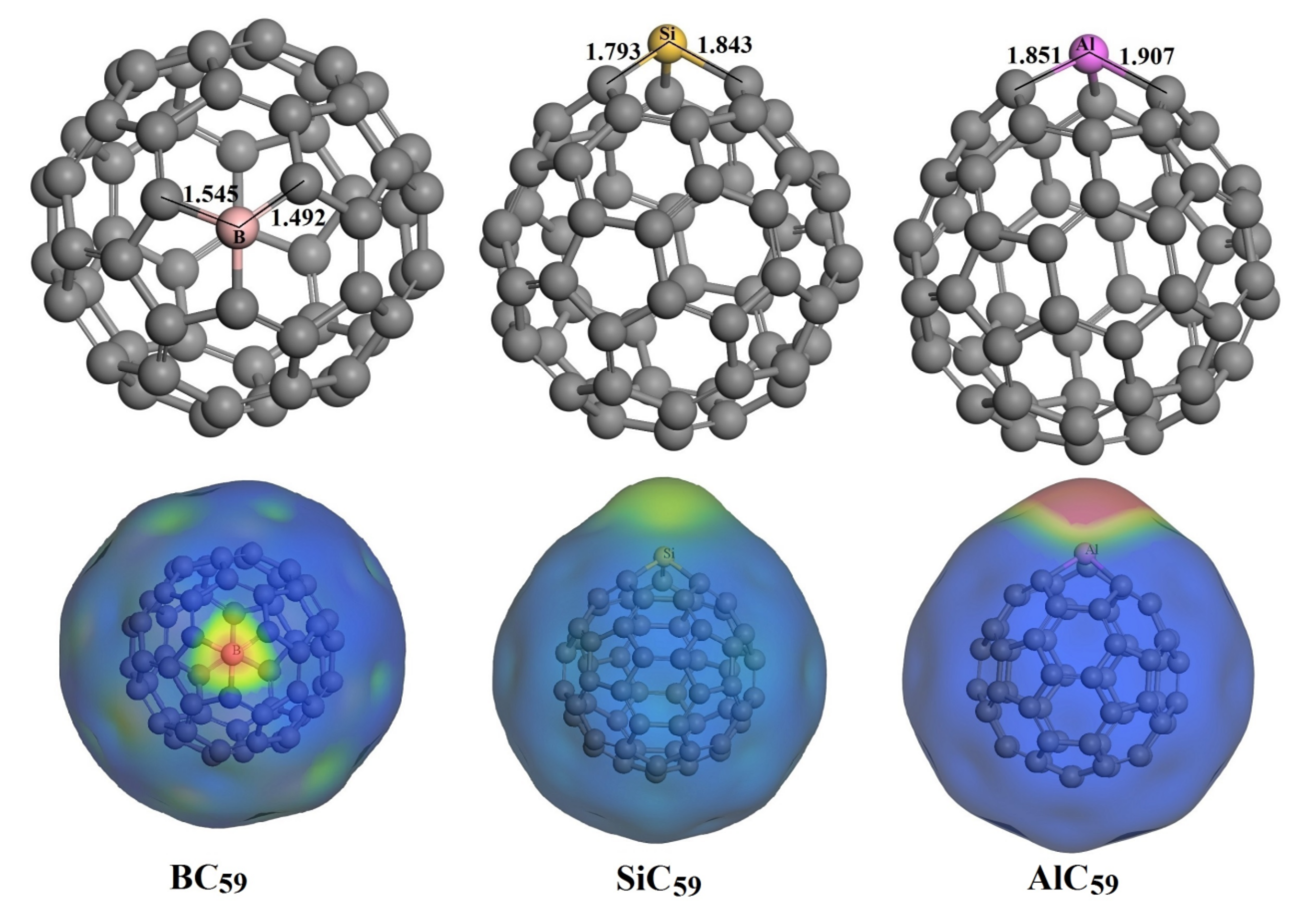
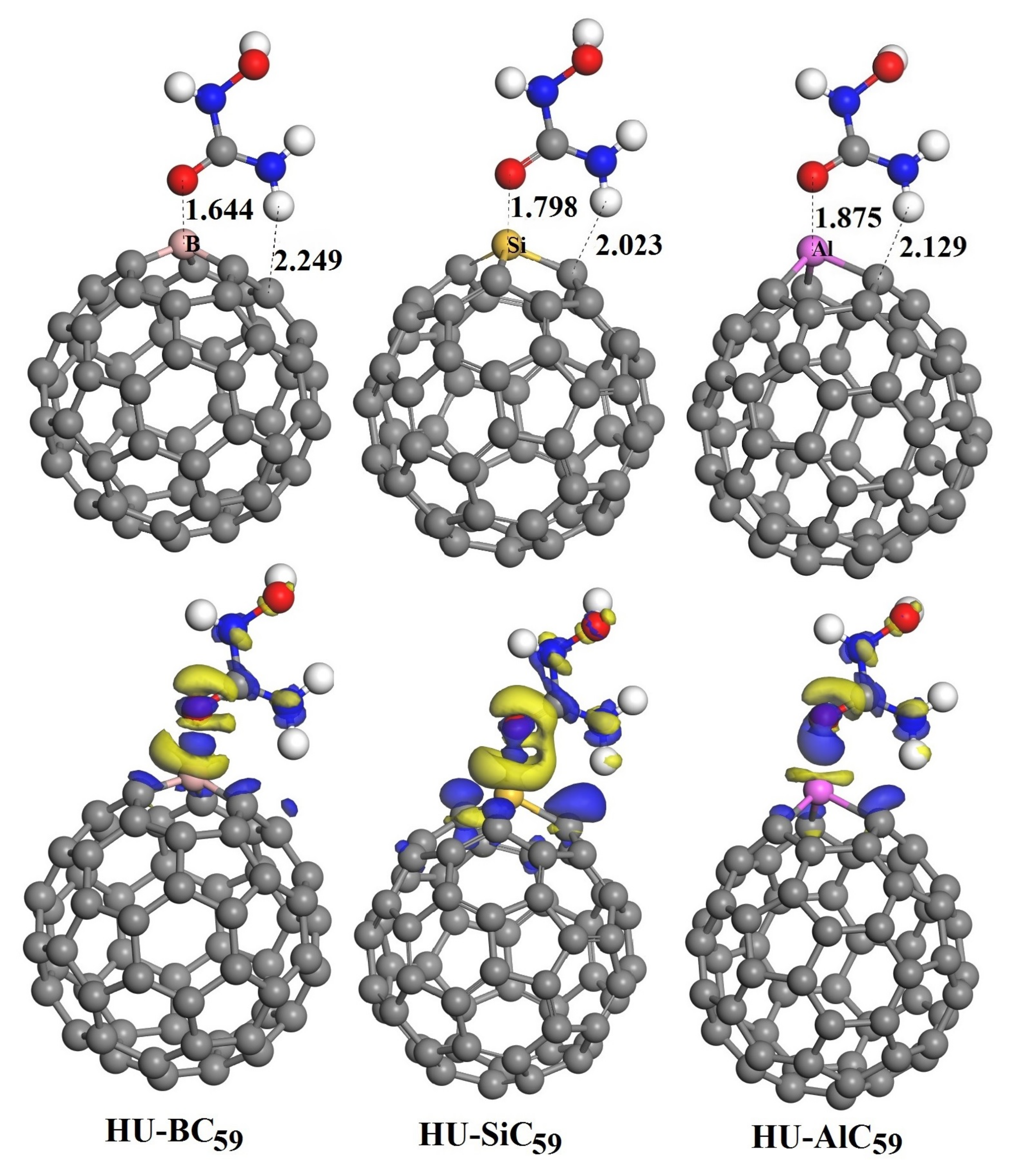
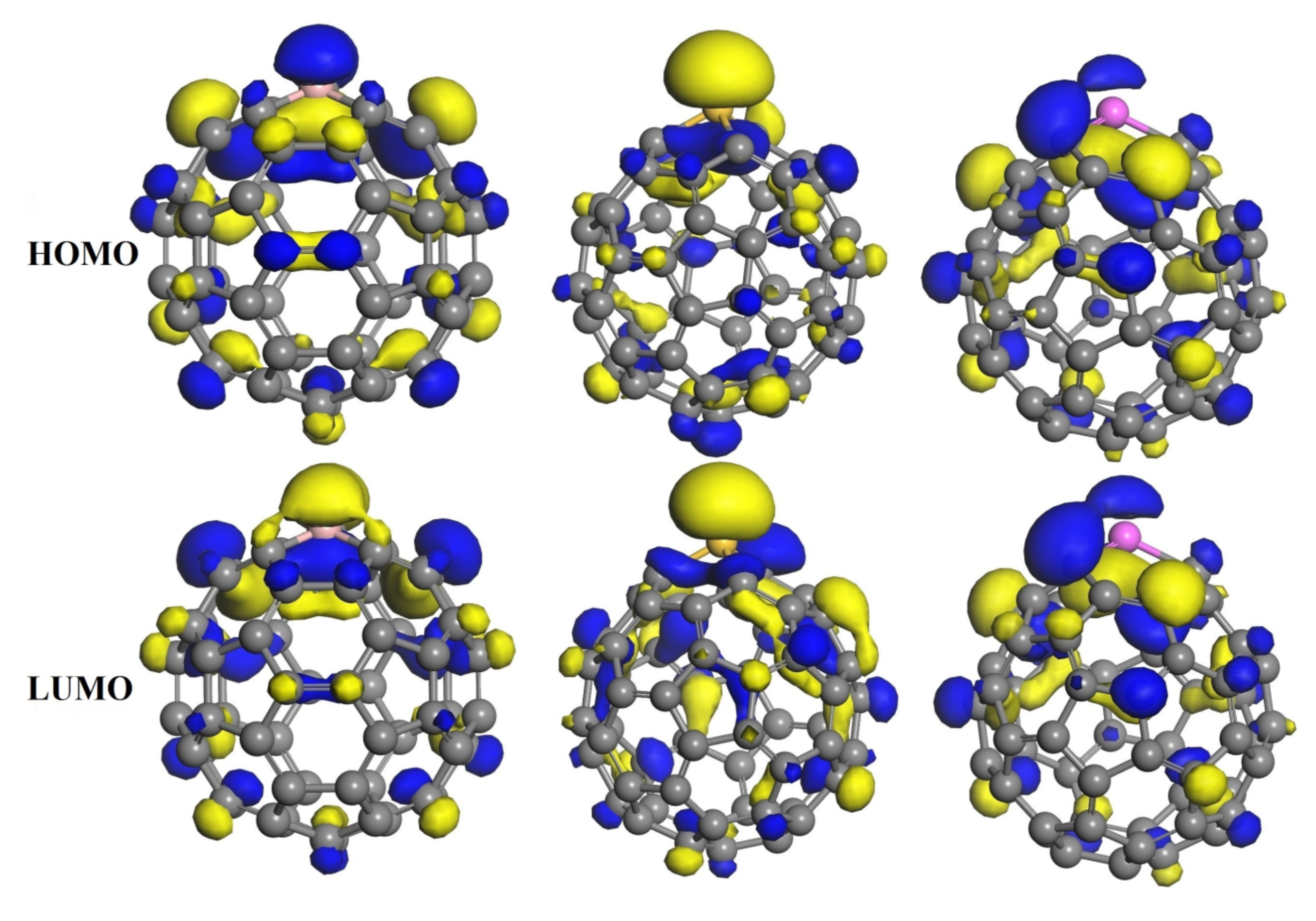
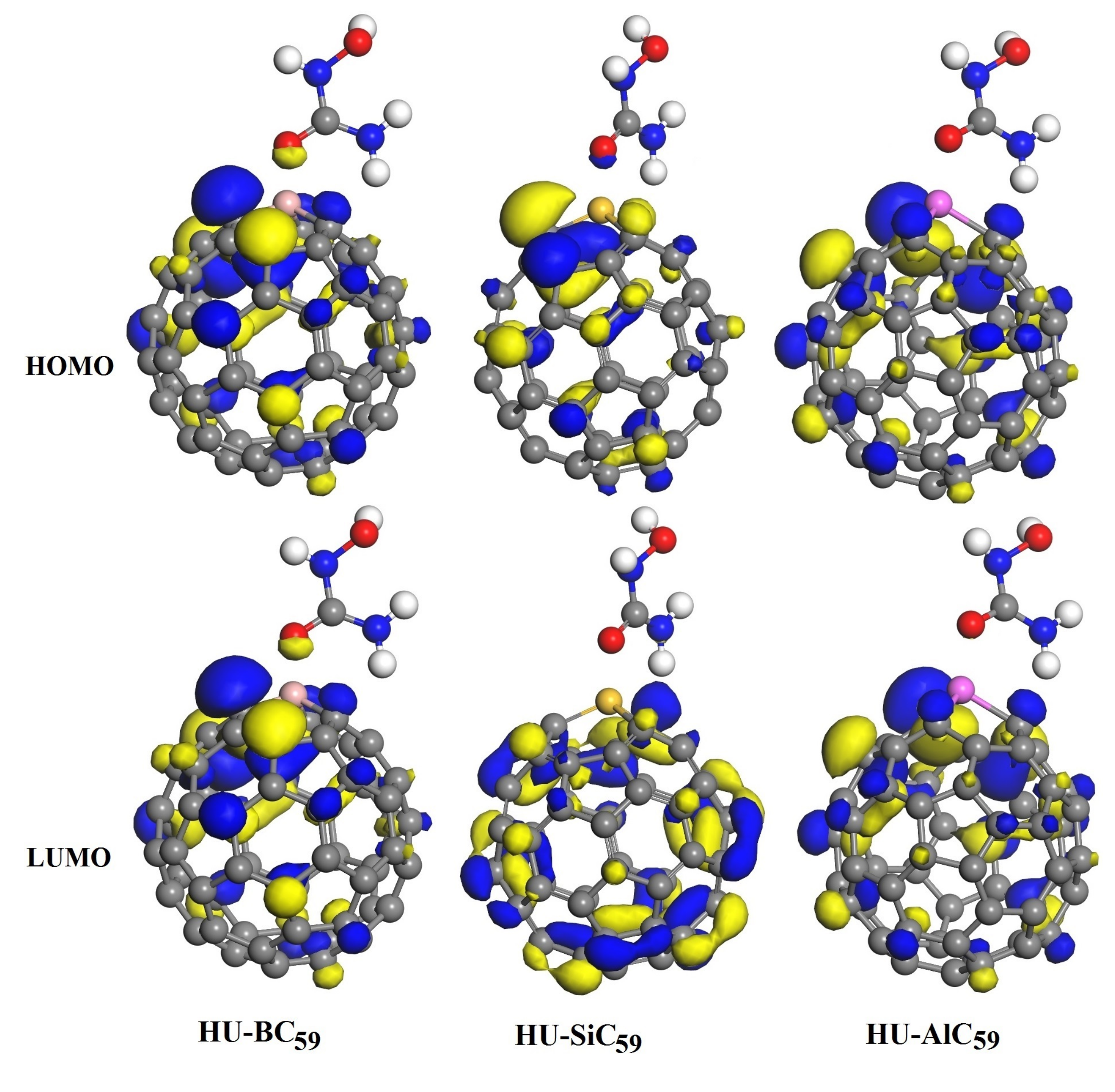
3.3. The Adsorption of HU on the Doped C60
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, K.; Davis, C.; Sherlock, S.; Cao, Q.Z.; Chen, X.Y.; Dai, H.J. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008, 68, 6652–6660. [Google Scholar] [CrossRef]
- Shi, J.J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Yong, Y.L.; Lv, S.; Li, X.H.; Li, T.W.; Cui, H.L. W@Si12 cluster as a potential sensor for CO and NO detection. Europhys. Lett. 2015, 111, 1. [Google Scholar] [CrossRef]
- Yong, Y.L.; Li, C.; Li, X.H.; Li, T.W.; Cui, H.L.; Lv, S.J. Ag7Au6 cluster as a Potential Gas Sensor for CO, HCN, and NO Detection. J. Phys. Chem. C 2015, 119, 7534–7540. [Google Scholar] [CrossRef]
- Yong, Y.L.; Lv, S.; Zhang, R.Z.; Zhou, Q.X.; Su, X.Y.; Li, T.W.; Cui, H.L. C54Si6 heterofullerene as a potential gas sensor for CO, NO, and HCN detection. RSC Adv. 2016, 6, 89080–89088. [Google Scholar] [CrossRef]
- Yong, Y.L.; Su, X.Y.; Zhou, Q.X.; Kuang, Y.N.; Li, X.H. The Zn12O12 cluster-assembled nanowires as a highly sensitive and selective gas sensor for NO and NO2. Sci. Rep. 2017, 7, 17505. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Nojoomi, A.; Mozafari, M.; Dubnika, A.; Inayathullah, M.; Rajadas, J. Nanomaterials engineering for drug delivery: A hybridization approach. J. Mater. Chem. B 2017, 5, 3995–4018. [Google Scholar] [CrossRef]
- Kazemzadeh, H.; Mozafari, M. Fullerene-based delivery systems. Drug Discov. Today 2019, 24, 898–905. [Google Scholar] [CrossRef]
- Kargozar, S.; Mozafari, M. Nanotechnology and nanomedicine: Start small, think big. Mater. Today 2018, 5, 15492–15500. [Google Scholar] [CrossRef]
- Goodarzi, S.; Ros, T.D.; Conde, J.; Sefat, F.; Mozafari, M. Fullerene: Biomedical engineers get to revisit an old friend. Mater. Today 2017, 20, 460–480. [Google Scholar]
- Wang, H.; Agarwal, P.; Zhao, S.; Yu, J.; Lu, X.; He, X. A biomimetic hybrid nanoplatform for encapsulation and precisely controlled delivery of theranostic agents. Nat. Commun. 2015, 6, 10081. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.J.; Zhang, H.L.; Wang, L.; Li, L.L.; Wang, H.H.; Wang, Z.Z.; Li, Z.; Chen, C.Q.; Hou, L.; Zhang, C.F.; et al. PEI-derivatized fullerene drug delivery using folate as a homing device targeting to tumor. Biomaterials 2013, 34, 251–261. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Cho, C.R.; Manzhos, S. Lithium attachment to C60 and nitrogen-and boron-doped C60: A mechanistic study. Materials 2019, 12, 2136. [Google Scholar] [CrossRef]
- Hassani, F.; Tavakol, H. A DFT, AIM and NBO study of adsorption and chemical sensing ofiodine by S-doped fullerenes. Sens. Actuators B 2014, 196, 624–630. [Google Scholar]
- Esrafili, M.D.; Janebi, H.B. N-doped and BN codoped C60 heterofullerenes for environmental monitoring of NO and NO2: A DFT study. Mol. Phys. 2020, 118, 5. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadirad, N. A first-principles study on the adsorption behaviour of methanol and ethanol over C59B heterofullerene. Mol. Phys. 2017, 115, 1633–1641. [Google Scholar]
- Parlak, C.; Alver, O. A density functional theory investigation on amantadine drug interaction with pristine and B, Al, Si, Ga, Ge doped C60 fullerenes. Chem. Phys. Lett. 2017, 678, 85–90. [Google Scholar] [CrossRef]
- Hazrati, M.K.; Hadipour, L.N. Adsorption behavior of 5-fluorouracil on pristine, B-, Si-, and Al-doped C60 fullerenes: A first-principles study. Phys. Lett. A 2016, 380, 937–941. [Google Scholar]
- Bashiri, S.; Vessally, E.; Bekhradnia, A.; Hosseinian, A.; Edjlali, L. Utility of extrinsic [60] fullerenes as work function type sensors for amphetamine drug detection: DFT studies. Vacuum 2017, 136, 156–162. [Google Scholar] [CrossRef]
- Moradi, M.; Nouraliei, M.; Moradi, R. Theoretical study on the phenylpropanolamine drug interaction with the pristine, Si and Al doped [60] fullerenes. Physica E 2017, 87, 186–191. [Google Scholar] [CrossRef]
- Gokpek, Y.; Bilge, M.; Bilge, D.; Alver, O.; Parlak, C. Adsorption mechanism, structural and electronic properties: 4-Phenylpyridine & undoped or doped (B or Si) C60. J. Mol. Liq. 2017, 238, 225–228. [Google Scholar]
- Muhr, H.J.; Nesper, R.; Schnyder, B.; Koetz, R. The boron heterofullerenes C59B and C69B: Generation, extraction, mass spectrometric and XPS characterization. Chem. Phys. Lett. 1996, 249, 399–405. [Google Scholar] [CrossRef]
- Hummelen, J.C.; Knight, B.; Pavlovich, J.; Gonzalez, R.; Wudl, F. Isolation of the heterofullerene C59N as its dimer (C59N)2. Science 1995, 269, 1554–1556. [Google Scholar] [CrossRef] [PubMed]
- Ray, C.; Pellarin, M.; Lerme, J.L.; Vialle, J.L.; Broyer, M. Synthesis and Structure of Silicon-doped Heterofullerenes. Phys. Rev. Lett. 1998, 80, 5365. [Google Scholar] [CrossRef]
- Billas, I.M.L.; Tast, F.; Branz, W.; Malinowski, N.; Heinebrodt, M.; Martin, T.P.; Boero, M.; Massobrio, C.; Parrinello, M. Experimental and computational studies of Si-doped fullerenes. Eur. Phys. J. D 1999, 9, 337–340. [Google Scholar] [CrossRef]
- Stry, J.J.; Garvey, J.F. Generation of C59O− via collision induced dissociation of oxy-fullerene anions. Chem. Phys. Let. 1995, 243, 199–204. [Google Scholar] [CrossRef]
- Moschel, C.; Jansen, M. Generation of Stable Phosphorus Heterofullerenes in a Radiofrequency Furnace. Z. Anorg. Allg. Chem. 1999, 625, 175–177. [Google Scholar]
- Ohtsuki, T.; Ohno, K.; Shiga, K.; Kawazoe, Y.; Maruyama, Y.; Masumoto, K. Systematic study of foreign-atom-doped fullerenes by using a nuclear recoil method and their MD simulation. J. Chem. Phys. 2000, 112, 2834. [Google Scholar] [CrossRef]
- Branz, W.; Billas, I.M.L.; Malinowski, N.; Tast, F.; Heinebrodt, M.; Martin, T.P. Cage substitution in metal–fullerene clusters. J. Chem. Phys. 1998, 109, 3425. [Google Scholar] [CrossRef]
- Poblet, J.M.; Munoz, J.; Winkler, K.; Cancilla, M.; Hayashi, A.; Lebrilla, C.B.; Balch, A.L. Geometric and electronic structure of metal-cage fullerenes, C59M (M = Pt, Ir) obtained by laser ablation of electrochemically deposited films. Chem. Commun. 1999, 64, 493–494. [Google Scholar] [CrossRef]
- Platt, O.S. Hydroxyurea for the treatment of sickle cell anemia. N. Engl. J. Med. 2008, 358, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Angona, A.; Bellosillo, B.; Alvarez-Larrán, A.; Martinez-Aviles, L.; Camacho, L.; Pairet, S.; Fernandez-Rodriguez, M.C.; Ancochea, A.; Besses, C. Genetic predisposition to molecular response in patients with myeloproliferative neoplasms treated with hydroxycarbamide. Leuk. Res. 2013, 37, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Daoud, M.S.; Gibson, L.E.; Pittelkow, M.R. Hydroxyurea dermopathy: A unique lichenoid eruption complicating long-term therapy with hydroxyurea. J. Am. Acad. Dermatol. 1997, 36, 178–182. [Google Scholar] [CrossRef]
- Best, P.J.; Daoud, M.S.; Pittelkow, M.R.; Petitt, R.M. Hydroxyurea-Induced Leg Ulceration in 14 Patients. Ann. Intern. Med. 1998, 128, 29–32. [Google Scholar] [CrossRef]
- Chen, X.Y.; Sun, Z.P.; Zhang, H.R.; Onsori, S. Effect of metal atoms on the electronic properties of metal oxide nanoclusters for use in drug delivery applications: A density functional theory study. Mol. Phys. 2020, 118, 13. [Google Scholar] [CrossRef]
- Mortazavifar, A.; Raissi, H.; Shahabi, M. Comparative prediction of binding affinity of Hydroxyurea anti-cancer to boron nitride and carbon nanotubes as smart targeted drug delivery vehicles. J. Biomol. Struct. Dyn. 2019, 37, 4852–4862. [Google Scholar] [CrossRef]
- Delley, B. An allelectron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–518. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Buke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar]
- Hedberg, K.; Hedberg, L.; Bethune, D.S.; Brown, C.A.; Dorn, H.C.; Johnson, R.D.; Devries, M. Bond lengths in free molecules of buckminsterfullerene, C60, from gas phase electron diffraction. Science 1992, 254, 410–412. [Google Scholar]
- Samanta, P.N.; Das, K.K. Noncovalent interaction assisted fullerene for the transportation of some brain anticancer drugs: A theoretical study. J. Mol. Graph. Model. 2017, 72, 187–200. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chen. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gergen, B.; Nienhaus, H.; Weinberg, W.H.; McFarland, E.W. Chemically induced electronic excitations at metal surfaces. Science 2001, 294, 2521–2523. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; pp. 205–209. [Google Scholar]
- Ahmadi, A.; Hadipour, N.L.; Kamfiroozi, M.; Bagheri, Z. Theoretical study of aluminum nitride nanotubes for chemical sensing of formaldehyde. Sens. Actuators B 2012, 161, 1025–1029. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Hadipour, N.L.; Bagheri, Z. Effects of Al doping and double-antisite defect on the adsorption of HCN on a BC2N Nanotube: Density functional theory studies. J. Phys. Chem. C 2013, 117, 2427–2432. [Google Scholar] [CrossRef]
- Hjiri, M.; Mir, L.E.; Leonardi, S.G.; Pistone, A.; Mavilia, L.; Neri, G. Al-doped ZnO for highly sensitive CO gas sensors. Sens. Actuators B 2014, 196, 413–420. [Google Scholar] [CrossRef]
- Hadipour, N.L.; Peyghan, A.A.; Soleymanabadi, H. Theoretical Study on the Al-Doped ZnO Nanoclusters for CO Chemical Sensors. J. Phys. Chem. C 2015, 119, 6398–6404. [Google Scholar] [CrossRef]
- Trpkovic, A.; Todorovic-Markovic, B.; Trajkovic, V. Toxicity of pristine versus functionalized fullerenes: Mechanisms of cell damage and the role of oxidative stress. Arch. Toxicol. 2012, 86, 1809–1827. [Google Scholar] [CrossRef] [PubMed]
- Partha, R.; Conyers, J.L. Biomedical applications of functionalized fullerene-based nanomaterials. Int. J. Nanomed. 2009, 4, 261–275. [Google Scholar]
- Prylutska, S.V.; Grebinyk, A.G.; Lynchak, O.V.; Byelinska, I.V.; Cherepanov, V.V.; Tauscher, E.; Matyshevska, O.P.; Prylutskyy, Y.I.; Rybalchenko, V.K.; Ritter, U.; et al. In vitro and in vivo toxicity of pristine C60 fullerene aqueous colloid solution. Fullerenes Nanotubes Carbon Nanostruct. 2019, 27, 715–728. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.Y.; Qian, P.X.; Lu, X.F.; Sun, B.Y.; Zhang, X.; Wang, L.M.; Gao, X.F.; Li, H.; Chen, Z.Y.; et al. Gd-metallofullerenol nanomaterial as non-toxic breast cancer stem cell-specific inhibitor. Nat. Commun. 2016, 12, 510. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Eads (eV) | EHOMO (eV) | ELUMO (eV) | Eg (eV) | ∆Eg (%) | qCT (e) |
|---|---|---|---|---|---|---|
| C60 | - | −5.745 | −4.104 | 1.641 | - | - |
| HU-C60 | −0.271 | −5.738 | −4.183 | 1.555 | −5.241 | −0.047 |
| BC59 | - | −5.636 | −5.363 | 0.273 | - | - |
| HU-BC59 | −1.122 | −4.803 | −4.438 | 0.365 | 33.700 | 0.373 |
| SiC59 | - | −5.806 | −4.619 | 1.187 | - | - |
| HU-SiC59 | −1.567 | −4.720 | −3.709 | 1.011 | −14.827 | 0.429 |
| AlC59 | - | −5.445 | −5.124 | 0.321 | - | - |
| HU-AlC59 | −2.174 | −4.884 | −4.520 | 0.364 | 13.396 | 0.346 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Yan, G.; Zhu, X.; Du, Y.; Chen, D.; Zhang, J. Heterofullerene MC59 (M = B, Si, Al) as Potential Carriers for Hydroxyurea Drug Delivery. Nanomaterials 2021, 11, 115. https://doi.org/10.3390/nano11010115
Wang P, Yan G, Zhu X, Du Y, Chen D, Zhang J. Heterofullerene MC59 (M = B, Si, Al) as Potential Carriers for Hydroxyurea Drug Delivery. Nanomaterials. 2021; 11(1):115. https://doi.org/10.3390/nano11010115
Chicago/Turabian StyleWang, Peng, Ge Yan, Xiaodong Zhu, Yingying Du, Da Chen, and Jinjuan Zhang. 2021. "Heterofullerene MC59 (M = B, Si, Al) as Potential Carriers for Hydroxyurea Drug Delivery" Nanomaterials 11, no. 1: 115. https://doi.org/10.3390/nano11010115
APA StyleWang, P., Yan, G., Zhu, X., Du, Y., Chen, D., & Zhang, J. (2021). Heterofullerene MC59 (M = B, Si, Al) as Potential Carriers for Hydroxyurea Drug Delivery. Nanomaterials, 11(1), 115. https://doi.org/10.3390/nano11010115