Fabrication of Carbon-Like, π-Conjugated Organic Layer on a Nano-Porous Silica Surface

 , ,
, ,  , ,
, , 
Abstract
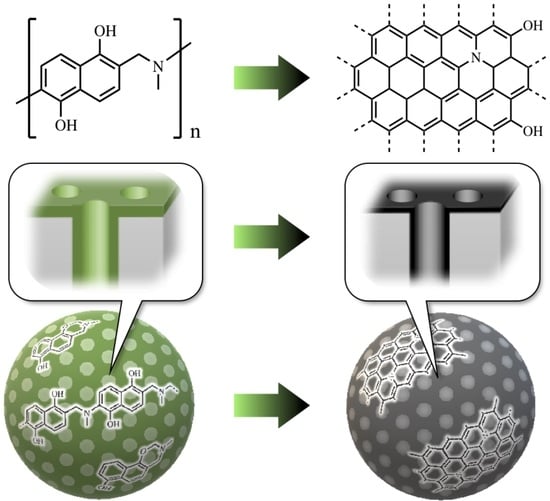
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Preparation of π-Conjugated Polymer-Silica Composites
2.3. Blackening of Polymer-Silica Composites
2.4. Instrumentations
2.5. Evaluation of Absorptivity
3. Results
3.1. Characterization of the Polymerization-Induced Assembly Method
3.2. Formation of a Carbon-Like Black Layer on Porous Carrier
3.3. Blackening and Carbonization of the Polymer Component
3.4. Pore Characterization of Black Polymer-Laminated Silica
3.5. Detection of Specific Interface Property by Selective Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kibbey, C.E.; Meyerhoff, M.E. Preparation and characterization of covalently bound tetraphenylporphyrin-silica gel stationary phases for reversed-phase and anion-exchange chromatography. Anal. Chem. 1993, 65, 2189–2196. [Google Scholar] [CrossRef]
- Xiao, J.; Kibbey, C.E.; Coutant, D.E.; Martin, G.B.; Meyerhoff, M.E. Immobilized porphyrins as versatile stationary phases in liquid chromatography. J. Liq. Chromatogr. Relat. Tech. 1996, 19, 2901–2932. [Google Scholar] [CrossRef]
- Záruba, K.; Tománková, Z.; Sýkora, D.; Charvátová, J.; Kavenová, I.; Bouř, P.; Matějka, P.; Fähnricha, J.; Volka, K.; Král, V. Interaction of porphyrin and sapphyrin macrocycles with nucleobases and nucleosides. Anal. Chim. Acta 2001, 437, 39–53. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tanaka, M.; Nakae, M.; Koichi, F.; Shono, T. Chemically bonded cyclodextrin stationary phases for liquid chromatographic separation of aromatic compounds. Anal. Chem. 1983, 55, 1852–1857. [Google Scholar] [CrossRef]
- Thuaud, N.; Sébille, B.; Deratani, A.; Pöpping, B.; Pellet, C. Enantiomer separations with chromatographic supports based on β-cyclodextrin polymers immobilized on porous silica. Role of the polymer structure in separating ability. Chromatographia 1993, 36, 373–380. [Google Scholar] [CrossRef]
- Lemr, K.; Jirovský, D.; Ševèík, J. Effect of some parameters on enantiomer separation of ephedrine, methamphetamine and selegiline using HPLC with β-cyclodextrin stationary phase. J. Liq. Chromatogr. Relat. Technol. 1996, 19, 3173–3191. [Google Scholar] [CrossRef]
- Hankins, M.G.; Hayashita, H.; Kasprzyk, S.P.; Bartsch, R.A. Immobilization of crown ether carboxylic acids on silica gel and their use in column concentration of alkali metal cations from dilute aqueous solutions. Anal. Chem. 1996, 68, 2811–2817. [Google Scholar] [CrossRef]
- Ng, S.; Ong, T.; Fu, P.; Ching, C. Enantiomer separation of flavour and fragrance compounds by liquid chromatography using novel urea-covalent bonded methylated β-cyclodextrins on silica. J. Chromatogr. A 2002, 968, 31–40. [Google Scholar] [CrossRef]
- Sinha, A.; Jana, N.R. Separation of microcystin-LR by cyclodextrin-functionalized magnetic composite of colloidal graphene and porous silica. ACS Appl. Mater. Interfaces 2015, 7, 9911–9919. [Google Scholar] [CrossRef]
- Li, Y.; Sheng, Z.; Zhu, C.; Yin, W.; Chu, C. Silica based click-dibenzo-18-crown-6-ether high performance liquid chromatography stationary phase and its application in separation of fullerenes. Talanta 2018, 178, 195–201. [Google Scholar] [CrossRef]
- Hirayama, C.; Ihara, H.; Mukai, T. Lipid membrane analogs. Specific retention behavior in comb-shaped telomer-immobilized porous silica gels. Macromolecules 1992, 25, 6375–6376. [Google Scholar] [CrossRef]
- Ihara, H.; Tanaka, H.; Nagaoka, S.; Sakaki, K.; Hirayama, C. Lipid membrane analogue-immobilized silica gels for separation with molecular recognition. J. Liq. Chromatogr. Relat. Technol. 1996, 19, 2967–2984. [Google Scholar] [CrossRef]
- Nagase, K.; Kobayashi, J.; Kikuchi, A.; Akiyama, Y.; Kanazawa, H.; Okano, T. High stability of thermoresponsive polymer-brush-grafted silica beads as chromatography matrices. ACS Appl. Mater. Interfaces 2012, 4, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Mallik, A.K.; Sawada, T.; Takafuji, M.; Ihara, H. Novel approach for the separation of shape-constrained isomers with alternating copolymer-grafted silica in reversed-phase liquid chromatography. Anal. Chem. 2010, 82, 3320–3328. [Google Scholar] [CrossRef] [PubMed]
- Shundo, A.; Sakurai, T.; Takafuji, M.; Nagaoka, S.; Ihara, H. Molecular-length and chiral discriminations by β-structural poly(L-alanine) on silica. J. Chromatogr. A 2005, 1073, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Bocian, S.; Skoczylas, M.; Buszewski, B. Amino acids, peptides, and proteins as chemically bonded stationary phases. A review. J. Sep. Sci. 2015, 39, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P.; Janas, P. Recent advances in stationary phases and understanding of retention in hydrophilic interaction chromatography. A review. Anal. Chim. Acta 2017, 967, 12–32. [Google Scholar] [CrossRef]
- Buszewski, B.; Skoczylas, M. Multi-Parametric Characterization of amino acid- and peptide-silica stationary phases. Chromatographia 2019, 82, 153–166. [Google Scholar] [CrossRef]
- Rahman, M.M.; Takafuji, M.; Ansarian, H.R.; Ihara, H. Molecular shape selectivity through multiple carbonyl−π interactions with noncrystalline solid phase for RP-HPLC. Anal. Chem. 2005, 77, 6671–6681. [Google Scholar] [CrossRef]
- Mallik, A.K.; Qiu, H.; Oishi, T.; Kuwahara, Y.; Takafuji, M.; Ihara, H. Molecular shape recognition through self-assembled molecular ordering: Evaluation with determining architecture and dynamics. Anal. Chem. 2012, 84, 6577–6585. [Google Scholar] [CrossRef]
- Mallik, A.K.; Qiu, H.; Oishi, T.; Kuwahara, Y.; Takafuji, M.; Ihara, H. Design of C18 organic phases with multiple embedded polar groups for ultraversatile applications with ultrahigh selectivity. Anal. Chem. 2015, 87, 6614–6621. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, S.; Kato, K.; Miyazaki, T.; Musashi, Y.; Ohkubo, K.; Ihara, H. Hirayama, C. Structures and binding energies of benzene-methane and benzene-benzene complexes. An ab initio SCF/MP2 studies. J. Chem. Soc. Faraday Trans. 1993, 89, 659–664. [Google Scholar] [CrossRef]
- Goto, Y.; Nakashima, K.; Mitsuishi, K.; Takafuji, M.; Sakaki, S.; Ihara, H. Selectivity enhancement for diastereomer separation in RPLC Using crystalline-organic phase-bonded silica instead of simply-hydrophobized silica. Chromatographia 2002, 56, 19–23. [Google Scholar] [CrossRef]
- Ihara, H.; Fukui, M.; Mimaki, T.; Shundo, A.; Dong, W.; Derakhshan, M.; Sakurai, T.; Takafuji, M.; Nagaoka, S. Poly(4-vinylpyridine) as a novel organic end-capping reagent for silica and its specific selectivity for PAHs and dinitropyrenes in a reversed phase. Anal. Chim. Acta 2005, 548, 51–57. [Google Scholar] [CrossRef]
- Qiu, H.; Jiang, S.; Takafuji, M.; Ihara, H. Polyanionic and polyzwitterionic azobenzene ionic liquid-functionalized silica materials and their chromatographic applications. Chem. Commun. 2013, 49, 2454–2456. [Google Scholar] [CrossRef]
- Hiruta, Y.; Kanazashi, R.; Ayano, E.; Okano, T.; Kanazawa, H. Temperature-responsive molecular recognition chromatography using phenylalanine and tryptophan derived polymer modified silica beads. Analyst 2016, 141, 910–917. [Google Scholar] [CrossRef]
- Xu, Z.; Uddin, K.M.A.; Kamra, T.; Schnadt, J.; Ye, L. Fluorescent boronic acid polymer grafted on silica particles for affinity separation of saccharides. ACS Appl. Mater. Interfaces 2014, 6, 1406–1414. [Google Scholar] [CrossRef]
- Kimita, K.; Hirose, T.; Hosoya, K.; Araki, T.; Tanaka, N. High-capacity stationary phases containing heavy atoms for HPLC separation of fullerenes. Anal. Chem. 1995, 67, 2556–2561. [Google Scholar] [CrossRef]
- Kartsova, L.A.; Makarov, A.A. New fullerene-based stationary phases for gas chromatography. J. Anal. Chem. 2004, 59, 724–729. [Google Scholar] [CrossRef]
- Kubo, T.; Murakami, Y.; Tsuzuki, M.; Kobayashi, T.; Naito, H.; Sano, T.; Yan, T.; Otsuka, K. Unique separation behavior of a C60 fullerene-bonded silica monolith prepared by an effective thermal coupling agent. Chem. Eur. J. 2015, 21, 18095–18098. [Google Scholar] [CrossRef]
- Noguchi, H.; Takafuji, M.; Maurizot, V.; Huc, I.; Ihara, H. Chiral separation by terminal chirality triggered P-helical quinoline oligoamide foldamer. J. Chromatogr. A 2016, 1437, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Speltini, A.; Merli, D.; Profumo, A. Analytical application of carbon nanotubes, fullerenes and nanodiamonds in nanomaterials-based chromatographic stationary phases: A review. Anal. Chim. Acta 2013, 783, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, C.; Qi, M.; Qu, L. A graphene-based porous carbon material as a stationary phase for gas chromatographic separations. RSC Adv. 2017, 7, 32126–32132. [Google Scholar] [CrossRef]
- Yang, L.; Guo-Xiao, T.; Yang, J.; Xu, Z.; Qin, M. Graphene-based materials used in as stationary phase for chromatography: A Mini Review. J. Chromatogr. Sep. Tech. 2018, 9, 1000399. [Google Scholar]
- Song, L.; Zhang, H.; Cai, T.; Chen, J.; Li, Z.; Guan, M.; Qiu, H. Porous graphene decorated silica as a new stationary phase for separation of sulfanilamide compounds in hydrophilic interaction chromatography. Chin. Chem. Lett. 2019, 30, 863–866. [Google Scholar] [CrossRef]
- Lee, S.; Moon, B.J.; Lee, H.J.; Bae, S.; Kim, T.; Jung, Y.C.; Park, J.H.; Lee, S.H. Enhancement of adsorption performance for organic molecules by combined effect of intermolecular interaction and morphology in porous rGO-incorporated hydrogels. ACS Appl. Mater. Interfaces 2018, 10, 17335–17344. [Google Scholar] [CrossRef]
- Ciccioli, P.; Tappa, R.; Corcia, A.; Liberti, A. Graphitized carbon black columns for high-performance liquid chromatography. J. Chromatogra. A 1981, 206, 35–42. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Han, Q.; Ding, M. Preparation and retention mechanism study of graphene and graphene oxide bonded silica microspheres as stationary phases for high performance liquid chromatography. J. Chromatogr. A 2013, 1307, 135–143. [Google Scholar] [CrossRef]
- Bapiro, T.E.; Richards, F.M.; Jodrell, D.I. Understanding the complexity of porous graphitic carbon (PGC) chromatography: Modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability. Anal. Chem. 2016, 88, 6190–6194. [Google Scholar] [CrossRef]
- Nozato, S.; Mallik, A.; Satoh, E.; Fukuda, R.; Ganapathy, H.; Kuwahara, Y.; Takafuji, M.; Ihara, H. A facile and green method to prepare conductive carbon-coated polymer microspheres using supercritical carbon dioxide. Chem. Lett. 2016, 45, 92–94. [Google Scholar] [CrossRef]
- Khan, M.N.; Orimoto, Y.; Ihara, H. Amphiphilic spherical nanoparticles with nitrogen-enriched carbon-like surface by using ß-lactoglobulin as template. Chem. Commun. 2018, 54, 13204–13207. [Google Scholar] [CrossRef] [PubMed]
- Hano, N.; Takafuji, M.; Noguchi, H.; Ihara, H. Surface charge-controlled, monodisperse black-colored nanoparticles exhibiting selective reflectance of NIR wavelengths. ACS Appl. Nano Mater. 2019, 2, 3597–3605. [Google Scholar] [CrossRef]
- Yamada, N.; Noguchi, H.; Orimoto, Y.; Kuwahara, Y.; Takafuji, M.; Pathan, S.; Oda, R.; Rahmuri, A.; Ramzanov, M.; Ihara, H. Emission color control in polymer film by memorized fluorescence solvatochromism due to a new class of totally-organic fluorescent nanogel particles. Chem. Eur. J. 2019, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ihara, H.; Sagawa, T.; Goto, Y.; Nagaoka, S. Crystalline polymer on silica. Geometrical selectivity for azobenzenes through highly-oriented structure. Polymer 1999, 40, 2555–2560. [Google Scholar] [CrossRef]
- Murakami, A.; Noguchi, H.; Kuwahara, Y.; Takafuji, M.; Nozato, S.; Sun, R.; Nakasuga, A.; Ihara, H. Non-conductive, size-controlled monodisperse black particles prepared by a one-pot polymerization and low-temperature calcination. Chem. Lett. 2017, 46, 680–682. [Google Scholar] [CrossRef]
- Shen, S.B.; Ishida, H. Synthesis and characterization of polyfunctional naphthoxazines and related polymers. J. Appl. Polym. Sci. 1996, 61, 1595–1605. [Google Scholar] [CrossRef]
- Jawhari, T.; Roid, A.; Casado, J. Raman spectroscopic characterization of some commercially available carbon black materials. Carbon 1995, 33, 1561–1565. [Google Scholar] [CrossRef]
- Deng, L.; Young, R.J.; Kinlich, I.A.; Abdelkader, A.M.; Holmes, S.M.; Rio, D.A.D.H.; Eichhorn, S.J. Supercapacitance from cellulose and carbon nanotube nanocomposite fibers. ACS Appl. Mater. Interfaces 2013, 5, 9983–9990. [Google Scholar] [CrossRef]
- Petris, A.; Vasiliu, I.C.; Gheorghe, P.; Iordache, A.M.; Ionel, L.; Rusen, L.; Iordache, S.; Elisa, M.; Trusca, R.; Ulieru, D.; et al. Graphene oxide-based silico-phosphate composite films for optical limiting of ultrashort near-infrared laser pulses. Nanomaterials 2020, 10, 1638. [Google Scholar] [CrossRef]
- Dimovski, S.; Nikitin, A.; Ye, H.; Gogotsi, Y. Synthesis of graphite by chlorination of iron carbide at moderate temperatures. J. Mater. Chem. 2004, 14, 238–243. [Google Scholar] [CrossRef]
- Sulpizi, M.; Gaigeot, M.; Sprik, M. The Silica—water interface: How the silanols determine the surface acidity and modulate the water properties. J. Chem. Theory Comput. 2012, 8, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, T.; Ihara, H.; Sakaki, S.; Shosenji, H.; Hirayama, C. Chromatographic separation of geometrical isomers using highly oriented polymer-immobilized silica gels. J. Chromatogr. A 1994, 672, 237–241. [Google Scholar] [CrossRef]
- Noguchi, H.; Charoenraks, T.; Takafuji, M.; Ihara, H. Effects of substitution groups of glutamide-derived molecular gels on molecular shape recognition. J. Chromatogr. A 2015, 1392, 56–62. [Google Scholar] [CrossRef]
- Ihara, H.; Okazaki, S.; Ohmori, K.; Uemura, S.; Hirayama, C.; Nagaoka, S. Polymer-silica hybrids: Evaluation of grafted poly(acrylonitrile) as organic phase for high-performance liquid chromatography. Anal. Sci. 1998, 14, 349–354. [Google Scholar] [CrossRef][Green Version]
- Gautam, U.G.; Sawada, T.; Gautam, M.P.; Takafuji, M.; Ihara, H. Poly(2-N-carbazolylethyl acrylate)-modified silica as a new polymeric stationary phase for reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 7422–7426. [Google Scholar] [CrossRef] [PubMed]
- Mallik, A.K.; Shingo, K.; Gautam, U.G.; Sawada, T.; Takafuji, M.; Ihara, H. Complete chromatographic separation of steroids including 17α- and 17β-estradioles using carbazole-based polymeric organic phase in both revered and normal phase HPLC. Anal. Bioanal. Chem. 2010, 397, 623–629. [Google Scholar] [CrossRef]
- Qiu, H.; Liu, X.; Jiang, S.; Takafuji, M.; Ihara, H. Investigation of π-π and ion-dipole interactions on 1-allyl-3-butylimidazolium ionic liquid-modified silica stationary phase in reversed-phase liquid chromatography. J. Chromatogr. A 2010, 1217, 5190–5196. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Takafuji, M.; Sawada, T.; Liu, X.; Jiang, S.; Ihara, H. New strategy for drastic enhancement of selectivity via chemical modification of counter anions in ionic liquid polymer phase. Chem. Commun. 2010, 46, 8740–8742. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbr. | Method | Solvent (Vol %) | Conc. (mM) | Reaction Temp. (°C) | Reaction Time | Average Particle Diameter (μm) | CV (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| EtOH | H2O | THF | IPA | |||||||
| Pm-1 | Microwave | - | 30 | 70 | - | 10 | 150 | 3 min | 0.18 | 33 |
| Pm-2 | Microwave | 100 | - | - | - | 30 | 150 | 3 min | 0.69 | 18 |
| Ph-1 | Heating | 100 | - | - | - | 1 | 75 | 24 h | 0.31 | 19 |
| Ph-2 | Heating | 100 | - | - | - | 10 | 75 | 12 h | 0.92 | 10 |
| Ph-3 | Heating | 100 | - | - | - | 30 | 75 | 6 h | 1.33 | 7 |
| Ph-4 | Heating | 50 | - | - | 50 | 150 | 75 | 6 h | 2.06 | 6 |
| Ph-5 | Heating | 100 | - | - | - | 150 | 75 | 6 h | 2.41 | 25 |
| Abbrev. a | Process I: Polymerization b | Process II: Heat Treatment c | ||||||
|---|---|---|---|---|---|---|---|---|
| Method | Initial wt. ratio (%) | Temp. (°C) | Time | Method | Medium | Temp. (°C) | Time | |
| Sil-0 d | – | – | – | – | – | – | – | – |
| Sil-P14/M100-0 | Microwave | 14 | 100 | 3 min | – | – | – | – |
| Sil-P14/M100-M200 | Microwave | 14 | 100 | 3 min | Microwave | in EG | 200 | 10 min |
| Sil-P14/M100-M300 | Microwave | 14 | 100 | 3 min | Microwave | in EG | 300 | 10 min |
| Sil-P17/H75-0 | Heating | 17 | 75 | 4 h | – | – | – | – |
| Sil-P17/H75-H200 | Heating | 17 | 75 | 4 h | Heating | under N2 | 200 | 2 h |
| Sil-P17/H75-H400 | Heating | 17 | 75 | 4 h | Heating | under N2 | 400 | 2 h |
| Sil-P17/H75-H200 | Heating | 17 | 75 | 4 h | Heating | under N2 | 560 | 2 h |
| Sil-P17/H75-H900 | Heating | 17 | 75 | 4 h | Heating | under N2 | 900 | 2 h |
| Sil-P50/H75-0 | Heating | 50 | 75 | 4 h | – | – | – | – |
| Polymer (wt %) | Diameter (μm) | PS (nm) | SA (m2g−1) | PV (mL g−1) | |
|---|---|---|---|---|---|
| Sil-0 | – | 4.37 | 12.0 | 330 | 1.01 |
| Sil-P17/H75-0 | 15.2 | 4.65 | 9.7 | 229 | 0.69 |
| Sil-P50/H75-0 | 48.3 | 5.02 | 6.6 | 124 | 0.21 |
| Sil-P17/H75-H200 | 15.0 | 4.48 | 9.7 | 235 | 0.72 |
| Sil-P17/H75-H560 | 9.83 | 4.42 | 12.5 | 279 | 0.75 |
| Separation Agents | Organic Phase a | Mobile Phase | α b | k′ for Stilbenes | Note | ||
|---|---|---|---|---|---|---|---|
| Solvent | Temp. | trans | cis | ||||
| Sil-P17/H75-H560 | p | CH3CN only | 20 °C | 14.0 | 13.7 | 0.98 | This work |
| Sil-P17/H75-H560 | p | EtOH:THF (8:2) | 20 °C | 9.80 | 1.96 | 0.20 | This work |
| Sil-P17/H75-H560 | p | EtOH only | 20 °C | incal. c | not eluted | 4.53 | This work |
| Sil-P17/H75-H560 | p | MeOH:H2O (9:1) | 20 °C | incal. c | not eluted | not eluted | This work |
| Sil-P17/H75-H200 | p | MeOH:H2O (9:1) | 20 °C | 3.67 | 1.77 | 0.48 | This work |
| Sil-P17/H75-0 | p | MeOH:H2O (9:1) | 20 °C | 3.60 | 1.60 | 0.62 | This work |
| ODS-m | m | MeOH:H2O (9:1) | 25 °C | 1.04 | 0.72 | 0.69 | Ref [52] |
| ODS-p | p | MeOH:H2O (9:1) | 25 °C | 1.19 | 3.05 | 2.57 | Ref [53] |
| MA-based | p | MeOH:H2O (7:3) | 25 °C | 1.97 | 5.51 | 2.80 | Ref [52] |
| ODA-based | p | MeOH:H2O (7:3) | 15 °C | 2.34 | 7.47 | 3.19 | Ref [52] |
| poly(L-Alanine) | p | MeOH:H2O (6:4) | 20 °C | 1.84 | 1.42 | 0.77 | Ref [15] |
| Glutamide-based | m | MeOH:H2O (7:3) | 20 °C | 3.04 | n.d. d | n.d. d | Ref [19] |
| CN-based | p | MeOH:H2O (7:3) | 25 °C | 2.25 | 0.74 | 0.33 | Ref [54] |
| Pyridine-based | p | MeOH:H2O (9:1) | 35 °C | 2.28 | 0.41 | 0.18 | Ref [55] |
| Carbazole-based | p | MeOH:H2O (9:1) | 35 °C | 1.79 | 1.59 | 0.89 | Ref [56] |
| Im+-based IL | p | MeOH:H2O (6:4) | 25 °C | 1.68 | 0.69 | 0.41 | Ref [57] |
| Im+-based IL/MO | p | MeOH only | 25 °C | 3.65 | n.d. d | n.d. d | Ref [58] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguchi, H.; Sultana, M.; Hano, N.; Kuwahara, Y.; Takafuji, M.; Nagaoka, S.; Qiu, H.; Ihara, H. Fabrication of Carbon-Like, π-Conjugated Organic Layer on a Nano-Porous Silica Surface. Nanomaterials 2020, 10, 1882. https://doi.org/10.3390/nano10091882
Noguchi H, Sultana M, Hano N, Kuwahara Y, Takafuji M, Nagaoka S, Qiu H, Ihara H. Fabrication of Carbon-Like, π-Conjugated Organic Layer on a Nano-Porous Silica Surface. Nanomaterials. 2020; 10(9):1882. https://doi.org/10.3390/nano10091882
Chicago/Turabian StyleNoguchi, Hiroki, Marzia Sultana, Nanami Hano, Yutaka Kuwahara, Makoto Takafuji, Shoji Nagaoka, Hongdeng Qiu, and Hirotaka Ihara. 2020. "Fabrication of Carbon-Like, π-Conjugated Organic Layer on a Nano-Porous Silica Surface" Nanomaterials 10, no. 9: 1882. https://doi.org/10.3390/nano10091882
APA StyleNoguchi, H., Sultana, M., Hano, N., Kuwahara, Y., Takafuji, M., Nagaoka, S., Qiu, H., & Ihara, H. (2020). Fabrication of Carbon-Like, π-Conjugated Organic Layer on a Nano-Porous Silica Surface. Nanomaterials, 10(9), 1882. https://doi.org/10.3390/nano10091882