Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Composites with Inorganic Nanoparticles for Biomedical Applications

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. GO Preparation
2.2. RGO Preparation
2.3. Preparation of Graphene Composites with Ag, Au, Ag2O, and TiO2 Nanoparticles
2.3.1. Composites with Nano-Ag
Composites with Nano-Ag[KA]
Composites with Nano-Ag[BS]
Composites with Nano-Ag[PP60]
2.3.2. Composites with Nano-Au
2.3.3. Composites with Nano-Ag2O
2.3.4. Composites with Nano-TiO2
3. Results and Discussion
3.1. GO and RGO Characterization
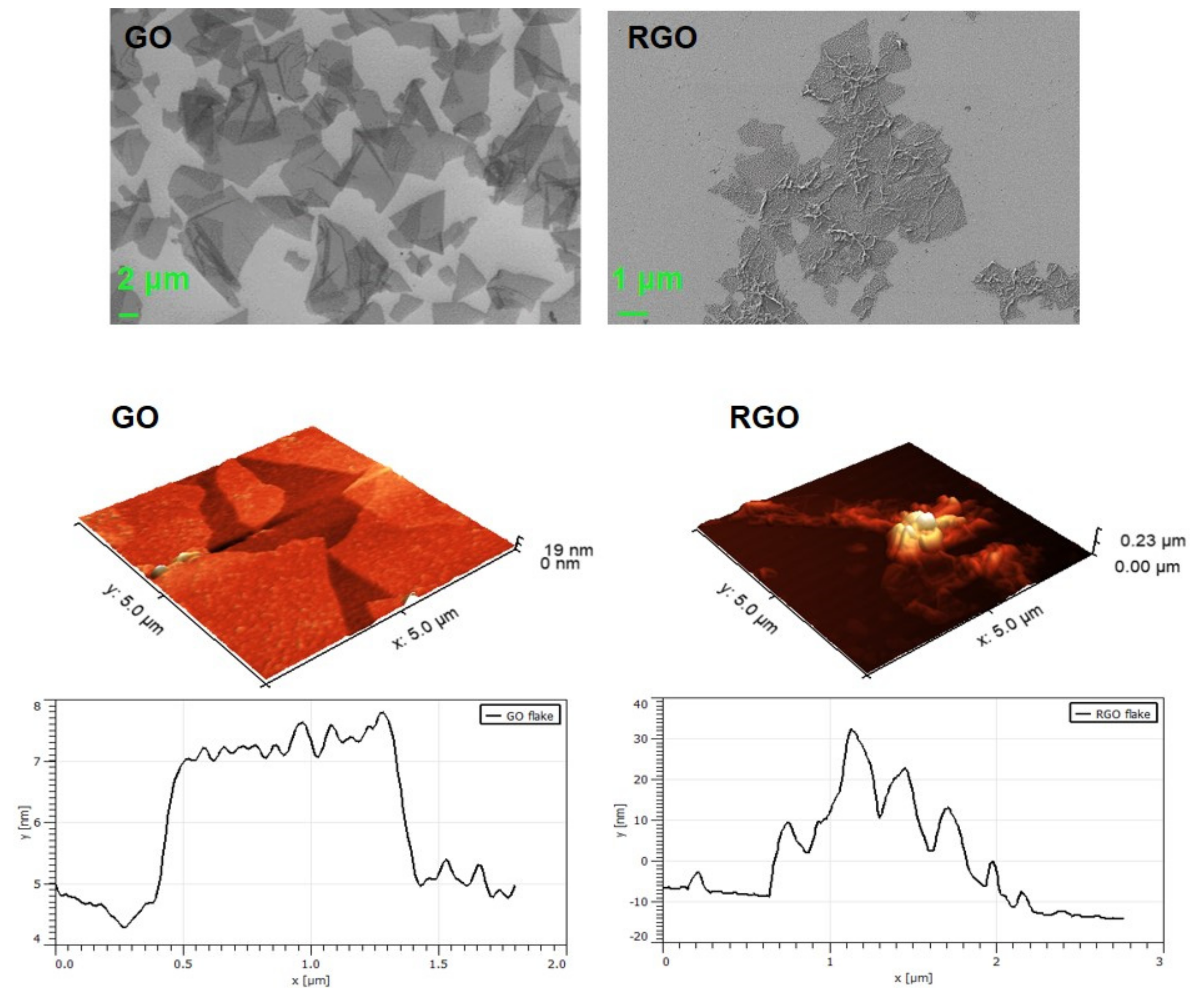
3.1.1. SEM and AFM Visualization
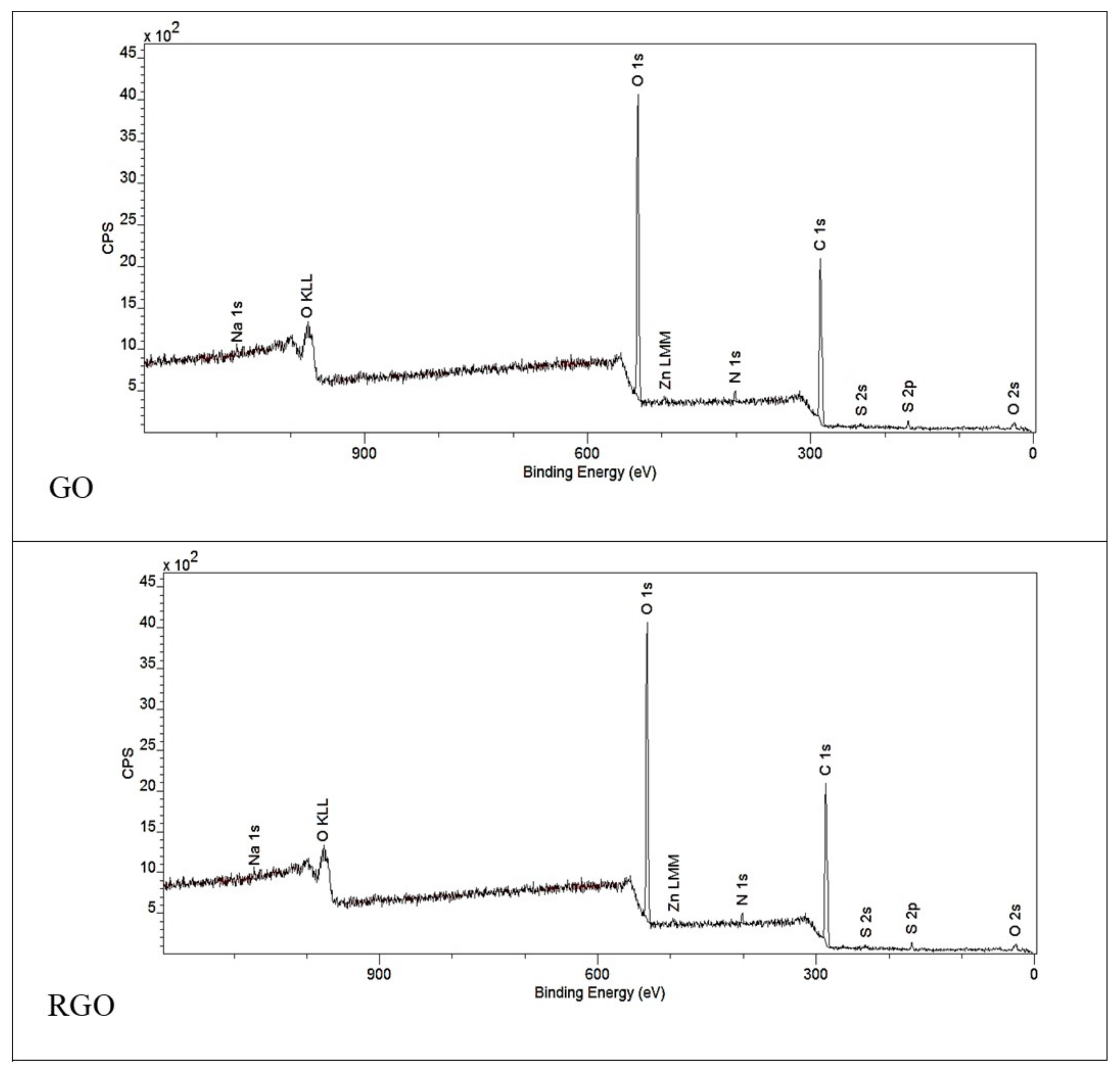
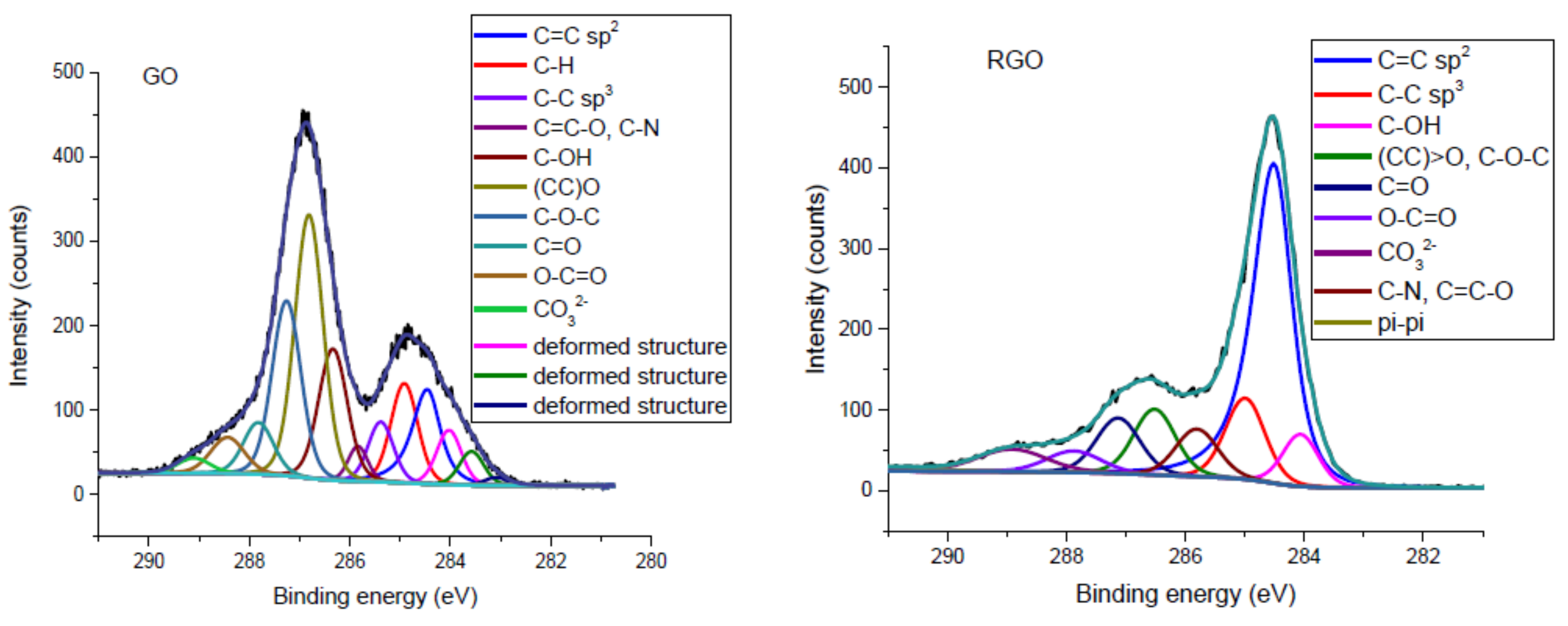
3.1.2. XPS Spectroscopy
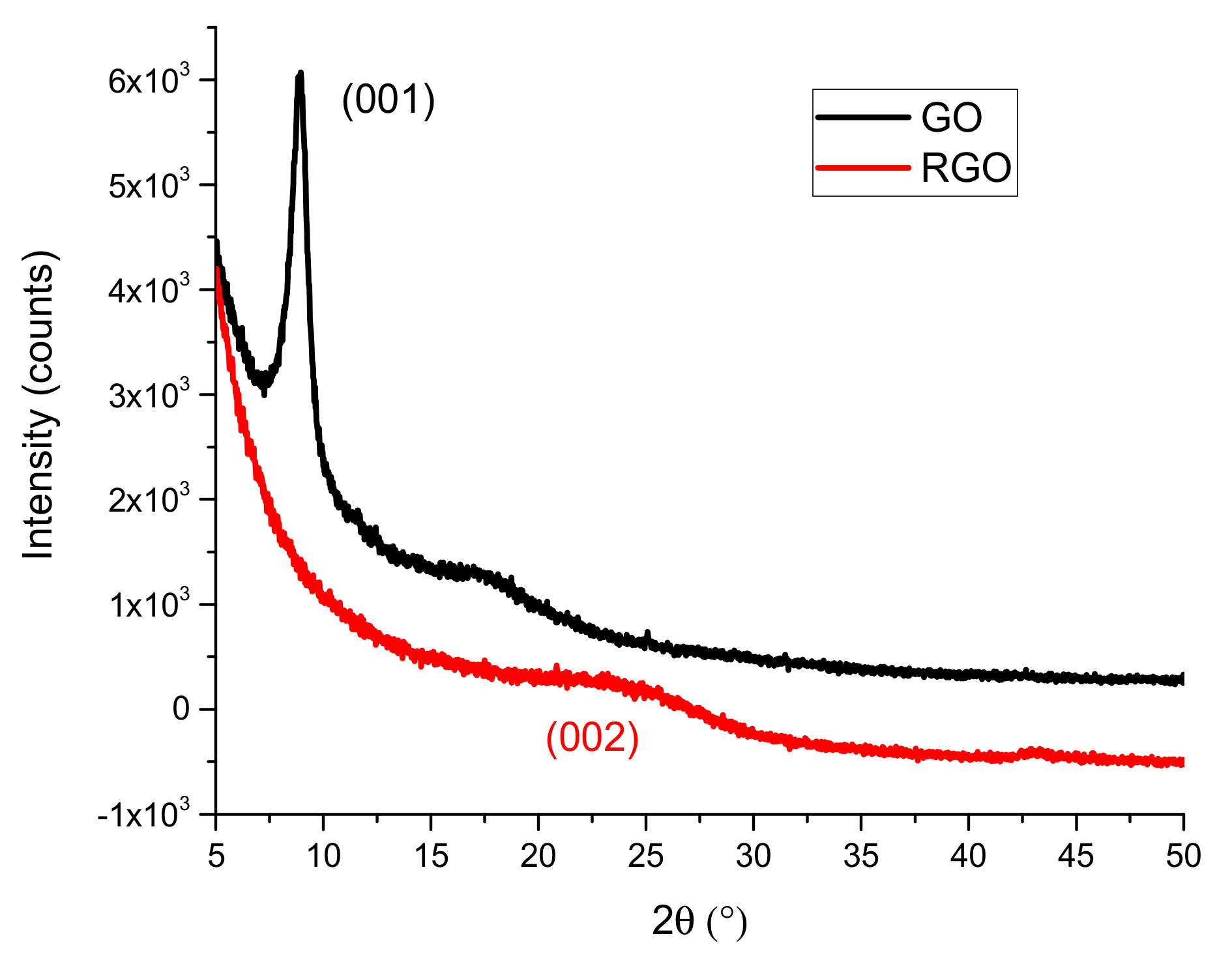
3.1.3. X-ray Diffraction (XRD)
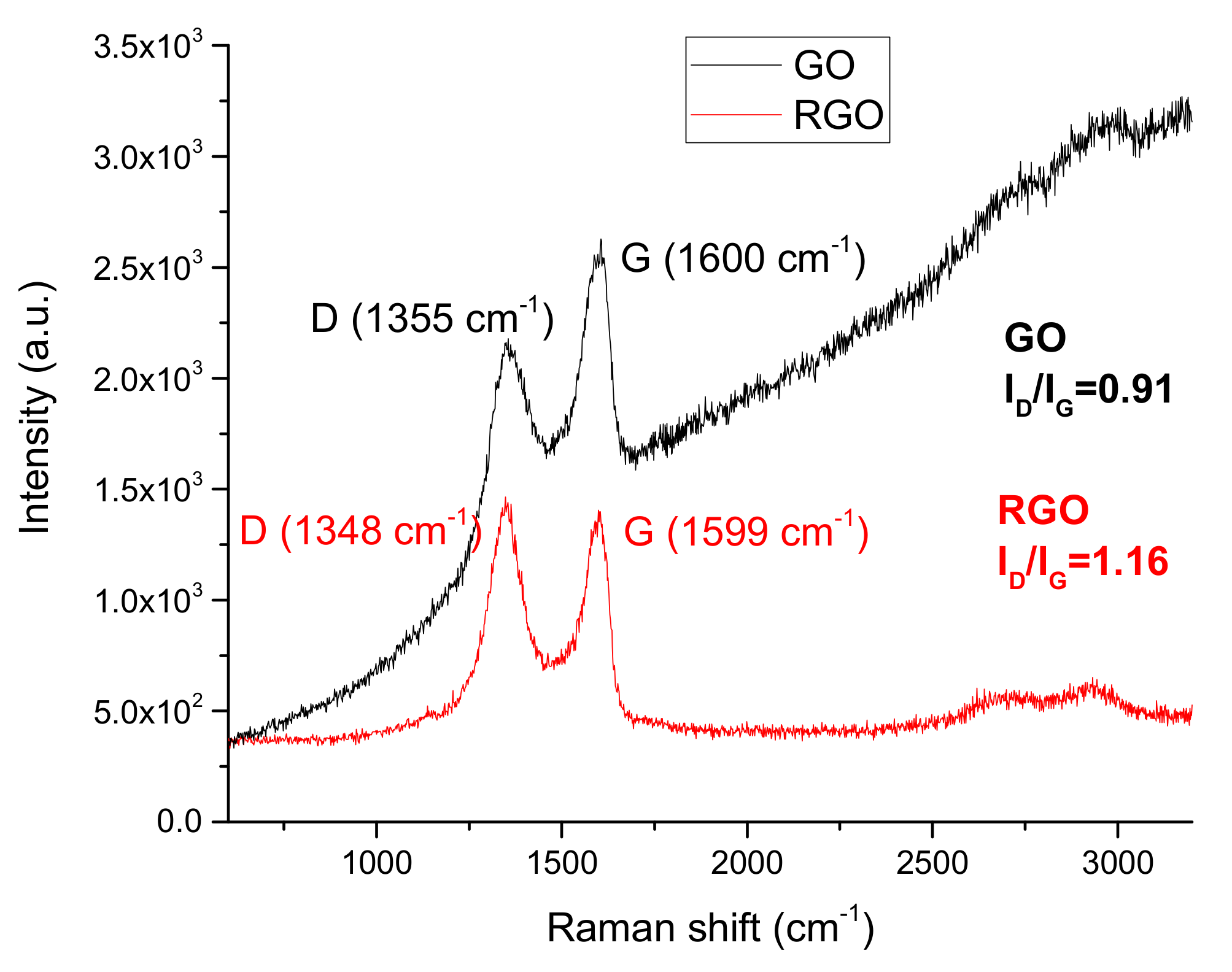
3.1.4. Raman Spectroscopy
3.2. Composite Characterization
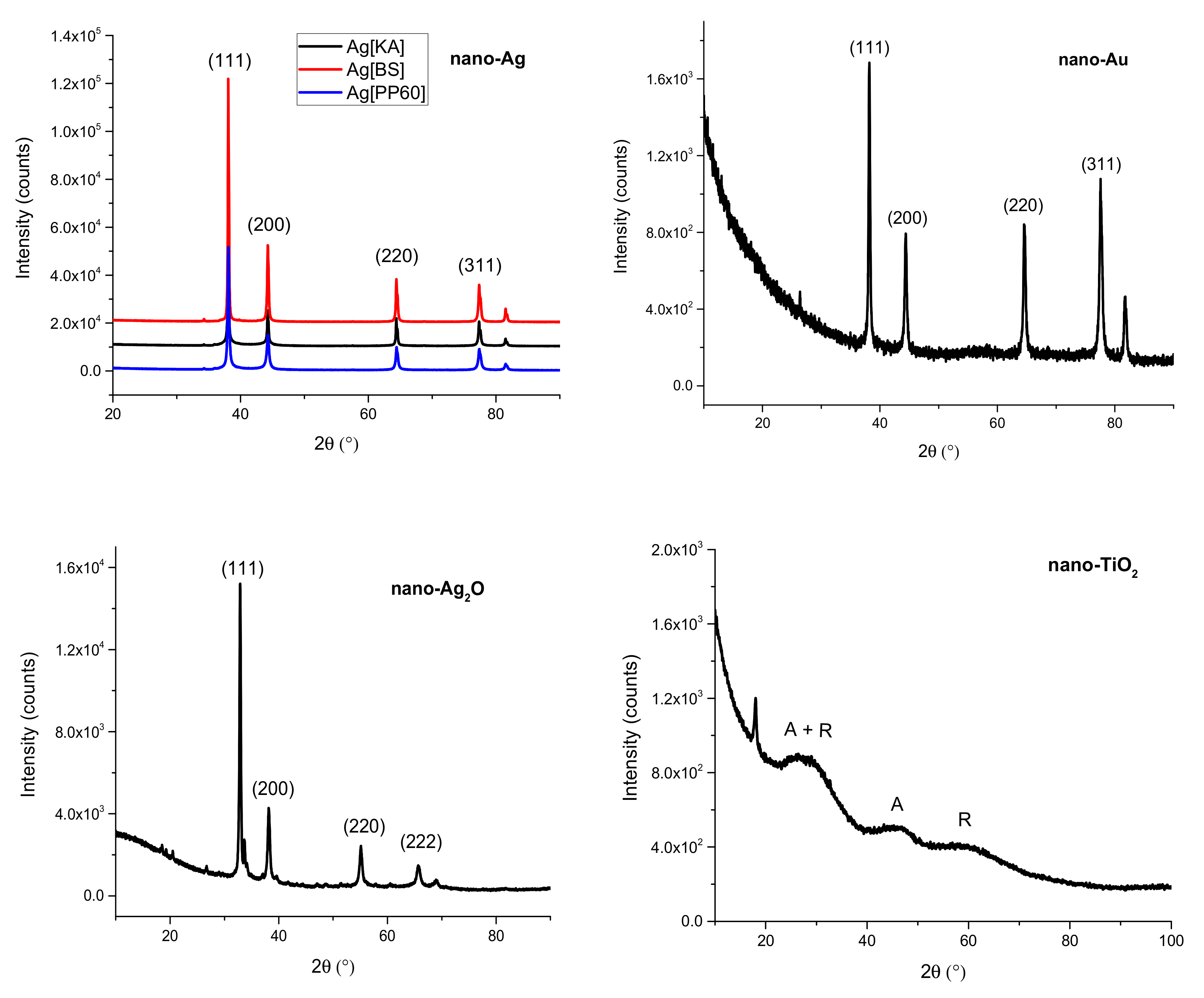
3.2.1. X-Ray Diffraction (XRD)
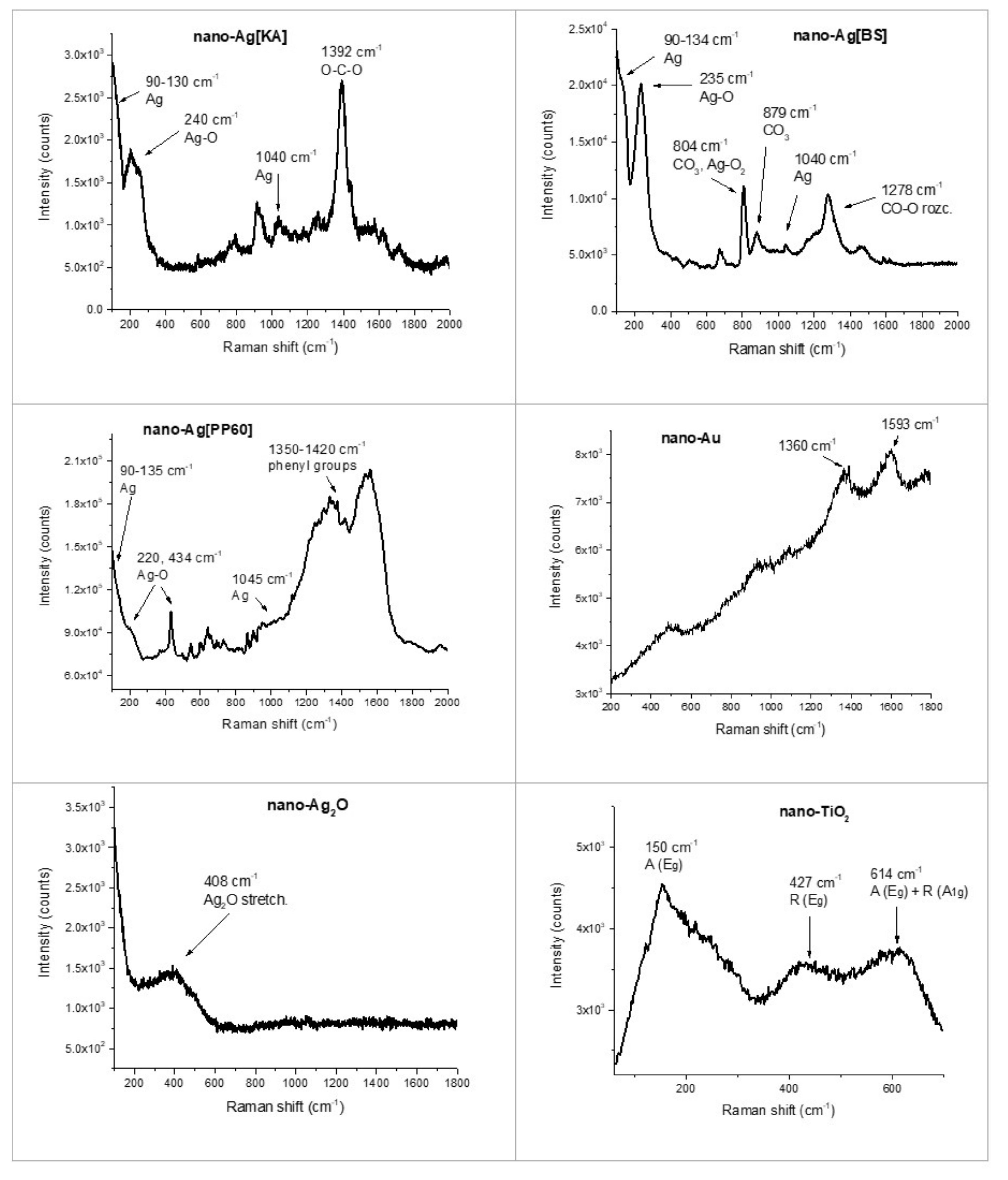
3.2.2. Raman Spectroscopy
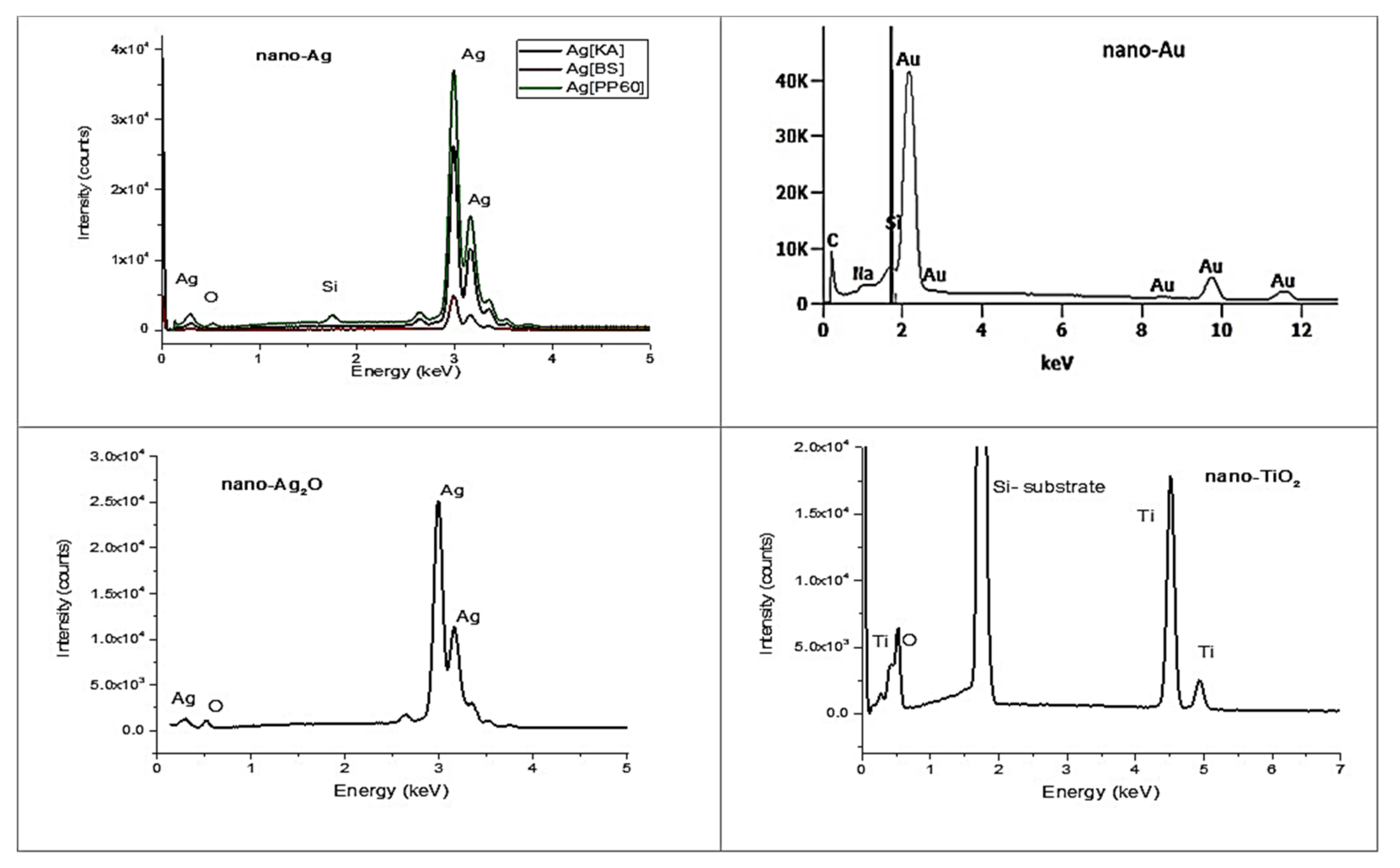
3.2.3. EDS Spectroscopy
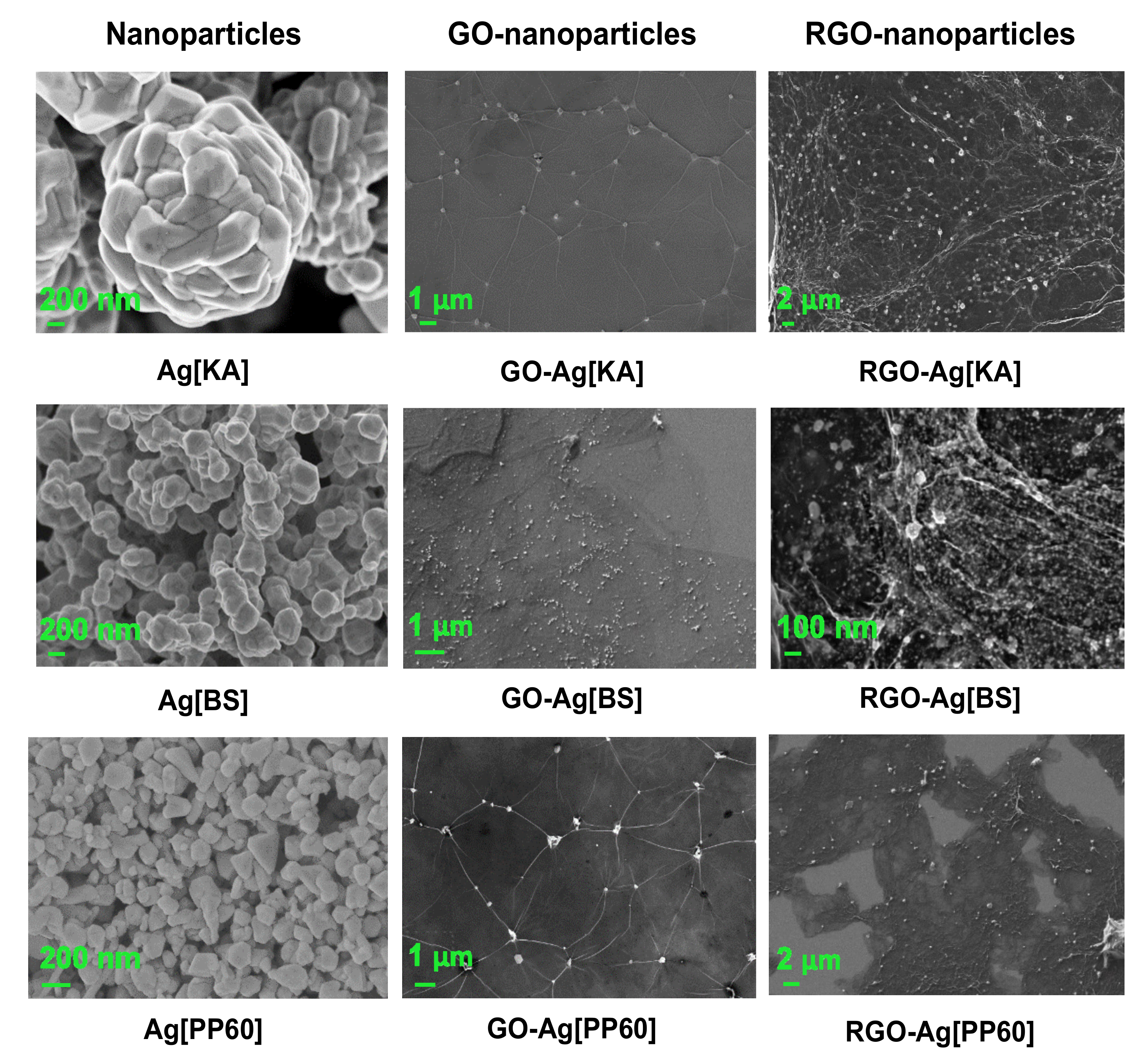
3.2.4. SEM Microscopy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, Y.-S.; Lu, Y.-J.; Chen, J.-P. Magnetic graphene oxide as a carrier for targeted delivery of chemotherapy drugs in cancer therapy. J. Magn. Magn. Mater. 2017, 427, 34–40. [Google Scholar] [CrossRef]
- Zhang, B.; Yan, Y.; Shen, Q.; Ma, D.; Huang, L.; Cai, X.; Tan, S. A colon targeted drug delivery system based on alginate modificated graphene oxide for colorectal liver metastasis. Mater. Sci. Eng. C 2017, 79, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Barahuie, F.; Saifullah, B.; Dorniani, D.; Fakurazi, S.; Karthivashan, G.; Hussein, M.Z.; Elfghi, F.M. Graphene oxide as a nanocarrier for controlled release and targeted delivery of an anticancer active agent, chlorogenic acid. Mater. Sci. Eng. C 2017, 74, 177–185. [Google Scholar] [CrossRef]
- Tran, T.H.; Nguyen, H.T.; Pham, T.T.; Choi, J.Y.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of a Graphene Oxide Nanocarrier for Dual-Drug Chemo-phototherapy to Overcome Drug Resistance in Cancer. ACS Appl. Mater. Interfaces 2015, 7, 28647–28655. [Google Scholar] [CrossRef]
- Kurantowicz, N.; Strojny, B.; Sawosz, E.; Jaworski, S.; Kutwin, M.; Grodzik, M.; Wierzbicki, M.; Lipińska, L.; Mitura, K.; Chwalibog, A. Biodistribution of a High Dose of Diamond, Graphite, and Graphene Oxide Nanoparticles After Multiple Intraperitoneal Injections in Rats. Nanoscale Res. Lett. 2015, 10. [Google Scholar] [CrossRef]
- Pinto, A.M.; Gonçalves, I.C.; Magalhães, F.D. Graphene-based materials biocompatibility: A review. Colloids Surf. B Biointerfaces 2013, 111, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Jagiełło, J.; Sekuła-Stryjewska, M.; Noga, S.; Adamczyk, E.; Dźwigońska, M.; Kurcz, M.; Kurp, K.; Winkowska-Struzik, M.; Karnas, E.; Boruczkowski, D.; et al. Impact of graphene-based surfaces on the basic biological properties of human umbilical cord mesenchymal stem cells: Implications for ex vivo cell expansion aimed at tissue repair. Int. J. Mol. Sci. 2019, 20, 4561. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Lojka, M.; Lochman, B. Oxidized Graphite Oxides. Materials (Basel) 2019, 12, 2367. [Google Scholar] [CrossRef]
- Jankovský, O.; Nováček, M.; Luxa, J.; Sedmidubský, D.; Fila, V.; Pumera, M.; Sofer, Z. A New Member of the Graphene Family: Graphene Acid. Chem. A Eur. J. 2016, 22, 17416–17424. [Google Scholar] [CrossRef] [PubMed]
- Jankovský, O.; Nováček, M.; Luxa, J.; Sedmidubský, D.; Boháčová, M.; Pumera, M.; Sofer, Z. Concentration of Nitric Acid Strongly Influences Chemical Composition of Graphite Oxide. Chem. A Eur. J. 2017, 23, 6432–6440. [Google Scholar] [CrossRef] [PubMed]
- Jankovský, O.; Jiříčková, A.; Luxa, J.; Sedmidubský, D.; Pumera, M.; Sofer, Z. Fast Synthesis of Highly Oxidized Graphene Oxide. ChemistrySelect 2017, 2, 9000–9006. [Google Scholar] [CrossRef]
- López-Miranda, J.L.; Cervantes-Chávez, J.A.; Hernández-Martínez, A.R.; Pérez, R.; Esparza, R.; Estévez-González, M. Study on the photocatalytic and antibacterial properties of silver nanoparticles synthesized by a green approach. Mater. Res. Express 2019, 6, 065066. [Google Scholar] [CrossRef]
- Jain, P.K.; El-Sayed, I.H.; El-Sayed, M.A. Au nanoparticles target cancer. Nano Today 2007, 2, 18–29. [Google Scholar] [CrossRef]
- Peng, J.; Liang, X.; Calderon, L. Progress in research on gold nanoparticles in cancer management. Medicine (U.S.) 2019, 98. [Google Scholar] [CrossRef]
- Li, J.; Li, J.J.; Zhang, J.; Wang, X.; Kawazoe, N.; Chen, G. Gold nanoparticle size and shape influence on osteogenesis of mesenchymal stem cells. Nanoscale 2016, 8, 7992–8002. [Google Scholar] [CrossRef]
- Yin, Z.F.; Wu, L.; Yang, H.G.; Su, Y.H. Recent progress in biomedical applications of titanium dioxide. Phys. Chem. Chem. Phys. 2013, 15, 4844–4858. [Google Scholar] [CrossRef]
- Kulkarni, M.; Mazare, A.; Gongadze, E.; Perutkova, Š.; Kralj-Iglič, V.; Milošev, I.; Schmuki, P.; Iglič, A.; Mozetič, M. Titanium nanostructures for biomedical applications. Nanotechnology 2015, 26, 062002. [Google Scholar] [CrossRef]
- Belcaro, G.; Cesarone, M.R.; Errichi, B.M.; Ricci, A.; Antelman, P.; Dugall, M.; Pellegrini, L.; Ledda, A.; Viscardi, G. Silver oxide ointment wound dressing in venous ulcerations: Home, self-management. Panminerva Med. 2011, 53 (Suppl. 3), 29–33. [Google Scholar]
- Sun, L.; Du, T.; Hu, C.; Chen, J.; Lu, J.; Lu, Z.; Han, H. Antibacterial Activity of Graphene Oxide/g-C3N4 Composite through Photocatalytic Disinfection under Visible Light. ACS Sustain. Chem. Eng. 2017, 5, 8693–8701. [Google Scholar] [CrossRef]
- Wang, Y.W.; Cao, A.; Jiang, Y.; Zhang, X.; Liu, J.H.; Liu, Y.; Wang, H. Superior antibacterial activity of zinc oxide/graphene oxide composites originating from high zinc concentration localized around bacteria. ACS Appl. Mater. Interfaces 2014, 6, 2791–2798. [Google Scholar] [CrossRef]
- Perdikaki, A.; Galeou, A.; Pilatos, G.; Karatasios, I.; Kanellopoulos, N.K.; Prombona, A.; Karanikolos, G.N. Ag and Cu Monometallic and Ag/Cu Bimetallic Nanoparticle-Graphene Composites with Enhanced Antibacterial Performance. ACS Appl. Mater. Interfaces 2016, 8, 27498–27510. [Google Scholar] [CrossRef]
- Ahmad, M.Z.; Bhatti, I.A.; Qureshi, K.; Ahmad, N.; Nisar, J.; Zuber, M.; Ashar, A.; Khan, M.I.; Iqbal, M. Graphene oxide supported Fe2(MoO4)3 nano rods assembled round-ball fabrication via hydrothermal route and photocatalytic degradation of nonsteroidal anti-inflammatory drug. J. Mol. Liq. 2020, 301. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, Q.; Han, Q.; Wan, W.; Ding, M. Metal-organic frameworks@graphene hybrid aerogels for solid-phase extraction of non-steroidal anti-inflammatory drugs and selective enrichment of proteins. Analyst 2016, 141, 4219–4226. [Google Scholar] [CrossRef]
- Amiri, A.; Mirzaei, M.; Derakhshanrad, S. A nanohybrid composed of polyoxotungstate and graphene oxide for dispersive micro solid-phase extraction of non-steroidal anti-inflammatory drugs prior to their quantitation by HPLC. Microchim. Acta 2019, 186. [Google Scholar] [CrossRef] [PubMed]
- Shie, M.Y.; Chiang, W.H.; Chen, I.W.P.; Liu, W.Y.; Chen, Y.W. Synergistic acceleration in the osteogenic and angiogenic differentiation of human mesenchymal stem cells by calcium silicate–graphene composites. Mater. Sci. Eng. C 2017, 73, 726–735. [Google Scholar] [CrossRef]
- Tatavarty, R.; Ding, H.; Lu, G.; Taylor, R.J.; Bi, X. Synergistic acceleration in the osteogenesis of human mesenchymal stem cells by graphene oxide–calcium phosphate nanocomposites. Chem. Commun. 2014, 50, 8484–8487. [Google Scholar] [CrossRef]
- Akhavan, O.; Ghaderi, E. Flash photo stimulation of human neural stem cells on graphene/TiO2 heterojunction for differentiation into neurons. Nanoscale 2013, 5, 10316–10326. [Google Scholar] [CrossRef]
- Bagri, A.; Mattevi, C.; Acik, M.; Chabal, Y.J.; Chhowalla, M.; Shenoy, V.B. Structural evolution during the reduction of chemically derived graphene oxide. Nat. Chem. 2010, 2, 581–587. [Google Scholar] [CrossRef]
- Gao, J.; Liu, F.; Liu, Y.; Ma, N.; Wang, Z.; Zhang, X. Environment-friendly method to produce graphene that employs vitamin C and amino acid. Chem. Mater. 2010, 22, 2213–2218. [Google Scholar] [CrossRef]
- Sobczak-Kupiec, A.; Malina, D.; Wzorek, Z.; Zimowska, M. Influence of silver nitrate concentration on the properties of silver nanoparticles. Micro Nano Lett. 2011, 6, 656–660. [Google Scholar] [CrossRef]
- Shih, C.J.; Lin, S.; Sharma, R.; Strano, M.S.; Blankschtein, D. Understanding the pH-dependent behavior of graphene oxide aqueous solutions: A comparative experimental and molecular dynamics simulation study. Langmuir 2012, 28, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.; Duch, M.C.; Mansukhani, N.D.; Hersam, M.C.; Bouchard, D. Colloidal properties and stability of graphene oxide nanomaterials in the aquatic environment. Environ. Sci. Technol. 2013, 47, 6288–6296. [Google Scholar] [CrossRef]
- Shearer, C.J.; Slattery, A.D.; Stapleton, A.J.; Shapter, J.G.; Gibson, C.T. Accurate thickness measurement of graphene. Nanotechnology 2016, 27, 125704. [Google Scholar] [CrossRef]
- Rabchinskii, M.K.; Dideikin, A.T.; Kirilenko, D.A.; Baidakova, M.V.; Shnitov, V.V.; Roth, F.; Konyakhin, S.V.; Besedina, N.A.; Pavlov, S.I.; Kuricyn, R.A.; et al. Facile reduction of graphene oxide suspensions and films using glass wafers. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Koinuma, M.; Tateishi, H.; Hatakeyama, K.; Miyamoto, S.; Ogata, C.; Funatsu, A.; Taniguchi, T.; Matsumoto, Y. Analysis of Reduced Graphene Oxides by X-ray Photoelectron Spectroscopy and Electrochemical Capacitance. Chem. Lett. 2013, 42, 924–926. [Google Scholar] [CrossRef]
- Larciprete, R.; Gardonio, S.; Petaccia, L.; Lizzit, S. Atomic oxygen functionalization of double walled C nanotubes. Carbon N. Y. 2009, 47, 2579–2589. [Google Scholar] [CrossRef]
- Morais, A.; Alves, J.P.C.; Lima, F.A.S.; Lira-Cantu, M.; Nogueira, A.F. Enhanced photovoltaic performance of inverted hybrid bulk-heterojunction solar cells using TiO2/reduced graphene oxide films as electron transport layers. J. Photonics Energy 2015, 5, 057408. [Google Scholar] [CrossRef]
- Maddi, C.; Bourquard, F.; Barnier, V.; Avila, J.; Asensio, M.C.; Tite, T.; Donnet, C.; Garrelie, F. Nano-Architecture of nitrogen-doped graphene films synthesized from a solid CN source. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Permatasari, F.A.; Aimon, A.H.; Iskandar, F.; Ogi, T.; Okuyama, K. Role of C-N Configurations in the Photoluminescence of Graphene Quantum Dots Synthesized by a Hydrothermal Route. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, X.; Wang, J.; Dong, F.; Chu, P.K.; Zhang, T.; Zhang, Y. Anionic Group Self-Doping as a Promising Strategy: Band-Gap Engineering and Multi-Functional Applications of High-Performance CO32--Doped Bi2O2CO3. ACS Catal. 2015, 5, 4094–4103. [Google Scholar] [CrossRef]
- Barinov, A.; Malcioǧlu, O.B.; Fabris, S.; Sun, T.; Gregoratti, L.; Dalmiglio, M.; Kiskinova, M. Initial stages of oxidation on graphitic surfaces: Photoemission study and density functional theory calculations. J. Phys. Chem. C 2009, 113, 9009–9013. [Google Scholar] [CrossRef]
- Saleem, H.; Haneef, M.; Abbasi, H.Y. Synthesis route of reduced graphene oxide via thermal reduction of chemically exfoliated graphene oxide. Mater. Chem. Phys. 2018, 204, 1–7. [Google Scholar] [CrossRef]
- Soomro, S.A.; Gul, I.H.; Naseer, H.; Marwat, S.; Mujahid, M. Improved Performance of CuFe2O4/rGO Nanohybrid as an Anode Material for Lithium-ion Batteries Prepared Via Facile One-step Method. Curr. Nanosci. 2018, 15, 420–429. [Google Scholar] [CrossRef]
- Gupta, V.; Sharma, N.; Singh, U.; Arif, M.; Singh, A. Higher oxidation level in graphene oxide. Optik (Stuttg) 2017, 143, 115–124. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hirsch, A. Visualization of defect densities in reduced graphene oxide. Carbon N. Y. 2012, 50, 3666–3673. [Google Scholar] [CrossRef]
- Vanaja, M.; Annadurai, G. Coleus aromaticus leaf extract mediated synthesis of silver nanoparticles and its bactericidal activity. Appl. Nanosci. 2013, 3, 217–223. [Google Scholar] [CrossRef]
- Yang, X.; Wu, X.; Li, J.; Liu, Y. TiO2-Au composite nanofibers for photocatalytic hydrogen evolution. RSC Adv. 2019, 9, 29097–29104. [Google Scholar] [CrossRef]
- Reddy, P.N.; Reddy, M.H.P.; Pierson, J.F.; Uthanna, S. Characterization of Silver Oxide Films Formed by Reactive RF Sputtering at Different Substrate Temperatures. ISRN Opt. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Sutrisno, H.; Ariswan, A.; Purwaningsih, D. Qualitative and quantitative phase-analysis of undoped titanium dioxide and chromium doped titanium dioxide from powder x-ray diffraction data. Indones. J. Chem. 2018, 18, 486–495. [Google Scholar] [CrossRef]
- Kora, A.J.; Arunachalam, J. Green fabrication of silver nanoparticles by gum tragacanth (Astragalus gummifer): A dual functional reductant and stabilizer. J. Nanomater. 2012, 2012. [Google Scholar] [CrossRef]
- Martina, I.; Wiesinger, R.; Schreiner, M. Micro-Raman investigations of early stage silver corrosion products occurring in sulfur containing atmospheres. J. Raman Spectrosc. 2013, 44, 770–775. [Google Scholar] [CrossRef]
- Aguilar-Hernández, I.; Afseth, N.K.; López-Luke, T.; Contreras-Torres, F.F.; Wold, J.P.; Ornelas-Soto, N. Surface enhanced Raman spectroscopy of phenolic antioxidants: A systematic evaluation of ferulic acid, p-coumaric acid, caffeic acid and sinapic acid. Vib. Spectrosc. 2017, 89, 113–122. [Google Scholar] [CrossRef]
- Liu, G.; Liu, Y.; Tang, L.; Liu, X.; Fu, G.; Liu, Z. Semiconductor-enhanced Raman scattering sensors via quasi-three-dimensional Au/Si/Au structures. Nanophotonics 2019, 8, 1095–1107. [Google Scholar] [CrossRef]
- Waterhouse, G.I.N.; Bowmaker, G.A.; Metson, J.B. The thermal decomposition of silver (I, III) oxide: A combined XRD, FT-IR and Raman spectroscopic study. Phys. Chem. Chem. Phys. 2001, 3, 3838–3845. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, L.; Shao, M.; Huang, J.; Ding, M.; Deng, X.; Wei, X.; Xu, X. Anodic oxidation synthesis of one-dimensional TiO2 nanostructures for photocatalytic and field emission properties. J. Nanomater. 2014, 2014. [Google Scholar] [CrossRef]
- Wypych, A.; Bobowska, I.; Tracz, M.; Opasinska, A.; Kadlubowski, S.; Krzywania-Kaliszewska, A.; Grobelny, J.; Wojciechowski, P. Dielectric properties and characterisation of titanium dioxide obtained by different chemistry methods. J. Nanomater. 2014, 2014. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Area | % Atoms | |
|---|---|---|
| GO | RGO | |
| C 1s | 65.3 | 79.5 |
| O 1s | 33.0 | 17.8 |
| N 1s | 1.5 | 1.1 |
| Other | 0.2 | 1.6 |
| Chemical Group | Position (eV) | % Atoms | Literature | ||
|---|---|---|---|---|---|
| GO | RGO | GO | RGO | ||
| C=C sp2 | 284.46 | 284.61 | 10.22 | 43.70 | [36,37,38,39] |
| C–H | 284.91 | 285.44 | 8.98 | 7.40 | [36,40] |
| C–C sp3 | 285.38 | 284.99 | 5.09 | 11.30 | [36,37,38,39] |
| C=C–O, C–N | 285.84 | 285.81 | 2.29 | 3.40 | [40,41] |
| C–OH | 286.34 | 286.44 | 12.69 | 7.80 | [36,37,39] |
| (CC)>O | 286.81 | 287.04 | 24.95 | 10.40 | [36,37,38,39] |
| C–O–C | 287.26 | 16.87 | |||
| CC=CO | 287.82 | 287.65 | 5.22 | 5.00 | |
| O–CC=CO | 288.43 | 289.01 | 4.11 | 4.90 | |
| CO32− | 289.10 | 285.51 | 1.66 | 1.40 | [42] |
| π–π shake-up | − | 290.94 | − | − | [38,39] |
| Deformed structure | 283.08 | 0.63 | − | [36,43] | |
| Deformed structure | 283.57 | 284.09 | 2.77 | − | |
| Deformed structure | 284.02 | 4.53 | 4.70 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagiełło, J.; Chlanda, A.; Baran, M.; Gwiazda, M.; Lipińska, L. Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Composites with Inorganic Nanoparticles for Biomedical Applications. Nanomaterials 2020, 10, 1846. https://doi.org/10.3390/nano10091846
Jagiełło J, Chlanda A, Baran M, Gwiazda M, Lipińska L. Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Composites with Inorganic Nanoparticles for Biomedical Applications. Nanomaterials. 2020; 10(9):1846. https://doi.org/10.3390/nano10091846
Chicago/Turabian StyleJagiełło, Joanna, Adrian Chlanda, Magdalena Baran, Marcin Gwiazda, and Ludwika Lipińska. 2020. "Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Composites with Inorganic Nanoparticles for Biomedical Applications" Nanomaterials 10, no. 9: 1846. https://doi.org/10.3390/nano10091846
APA StyleJagiełło, J., Chlanda, A., Baran, M., Gwiazda, M., & Lipińska, L. (2020). Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Composites with Inorganic Nanoparticles for Biomedical Applications. Nanomaterials, 10(9), 1846. https://doi.org/10.3390/nano10091846