Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels †

 ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Synthesis of Cellulose/Polypyrrole Composite
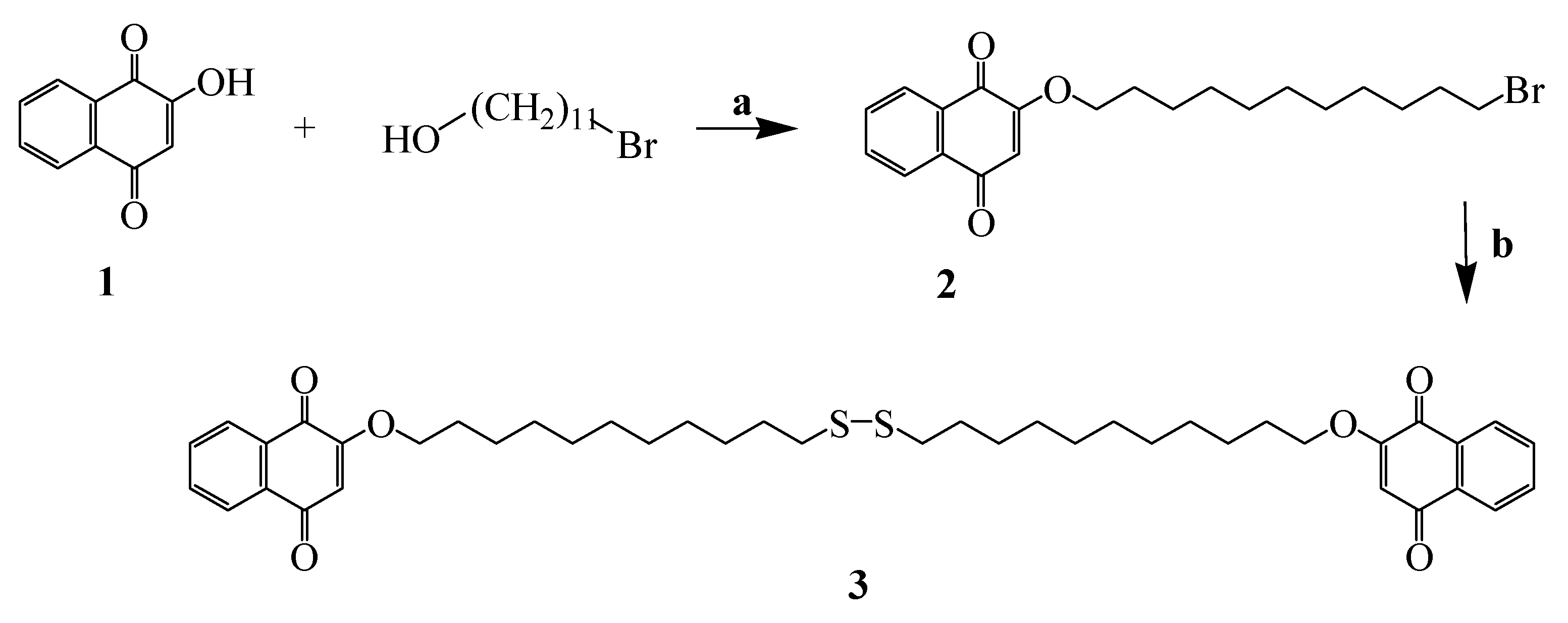
2.3. Synthesis of the Naphthoquinone Thiol Derivative
2.4. Gold Nanoparticles Modified with the Naphthoquinone Derivative
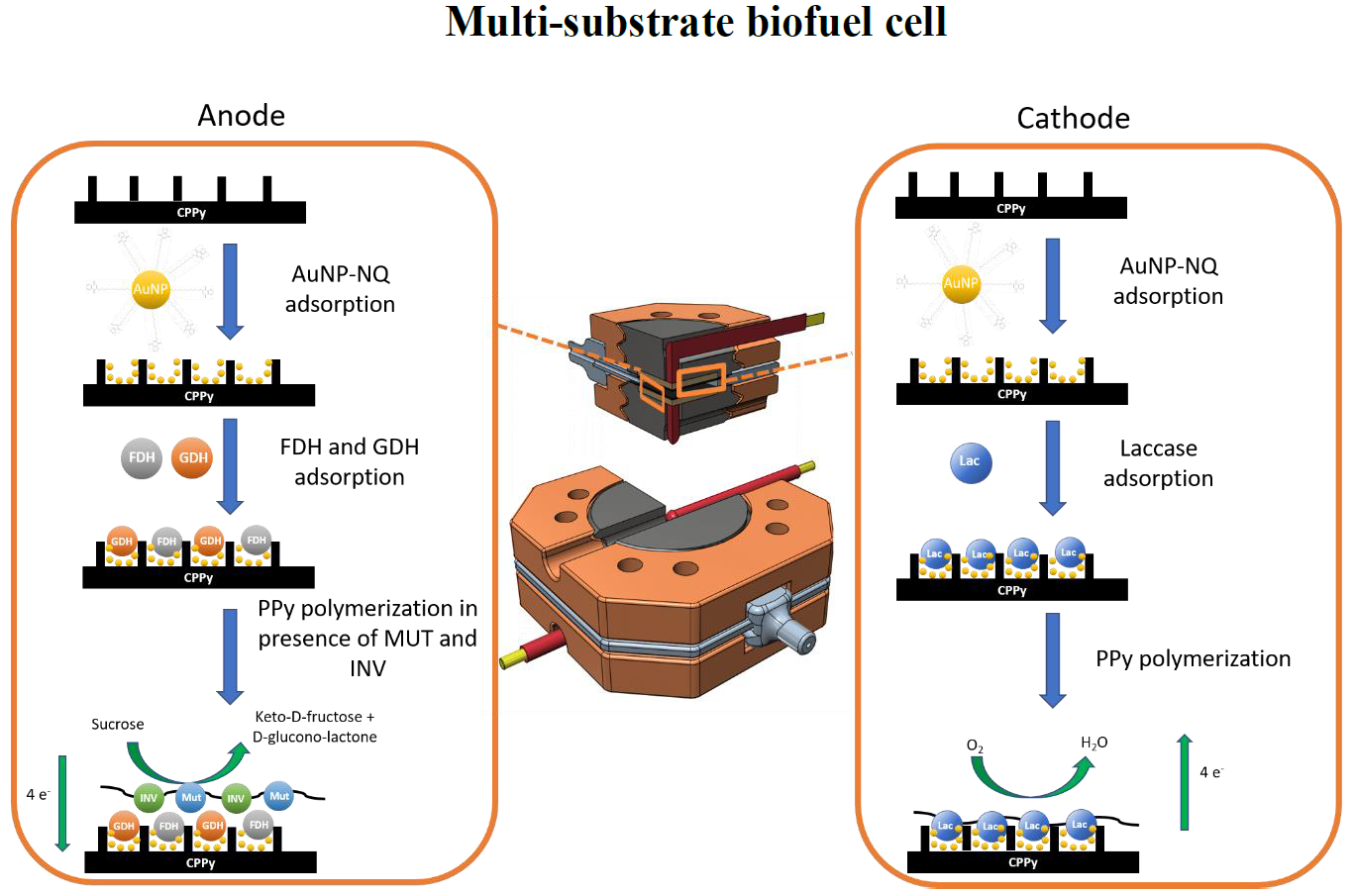
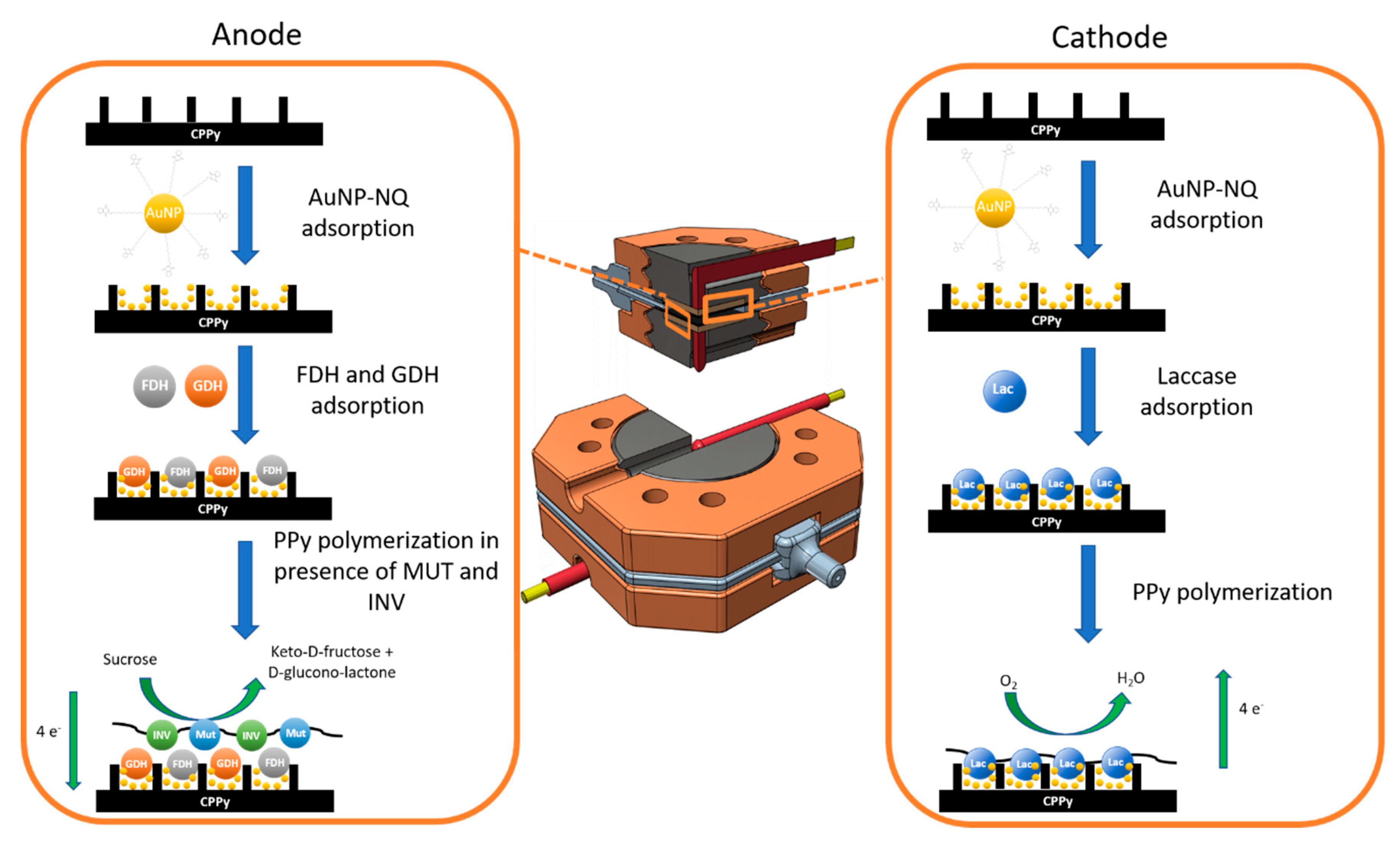
2.5. Electrode Preparation
2.6. Experimental Techniques
3. Results
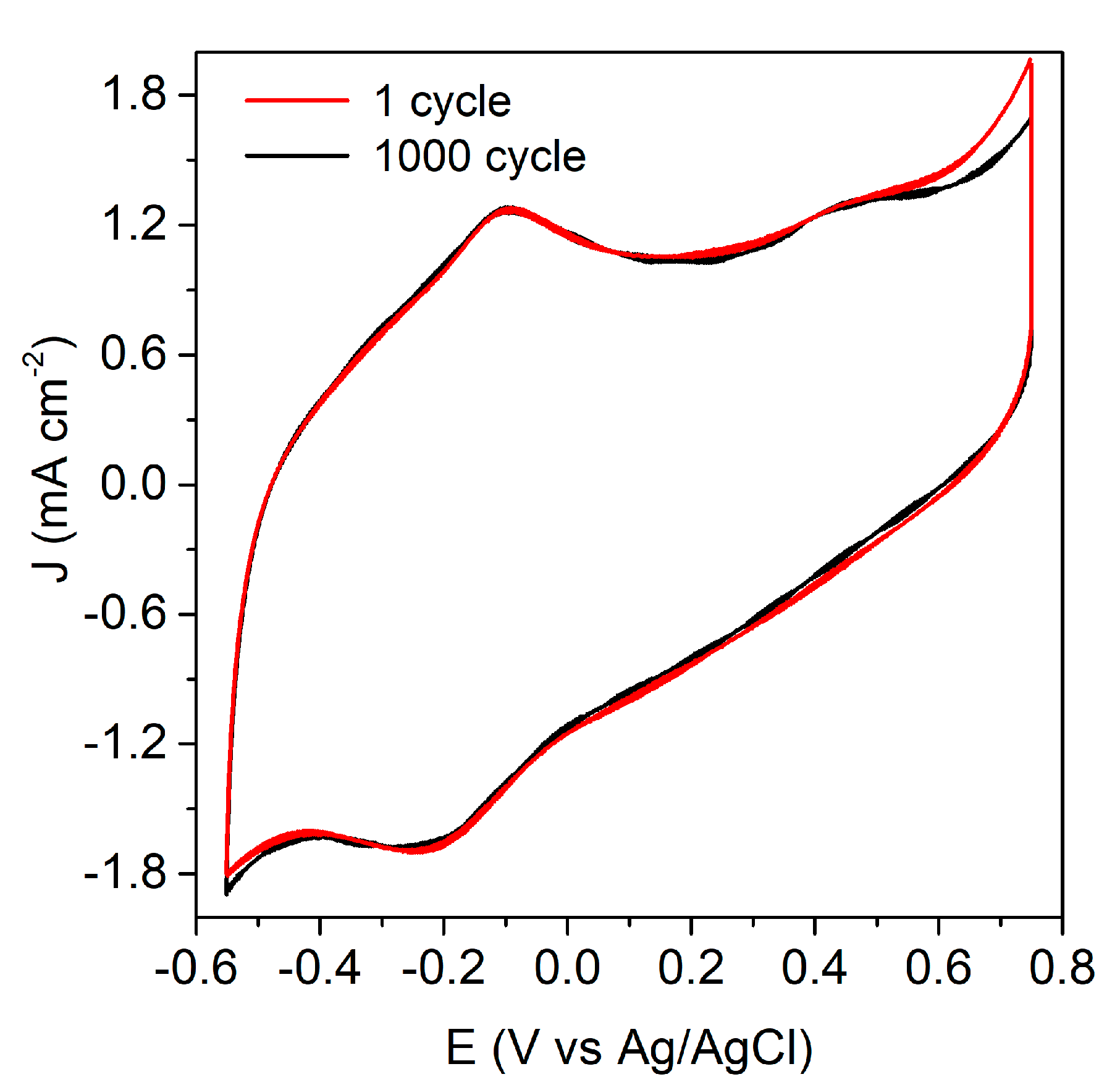
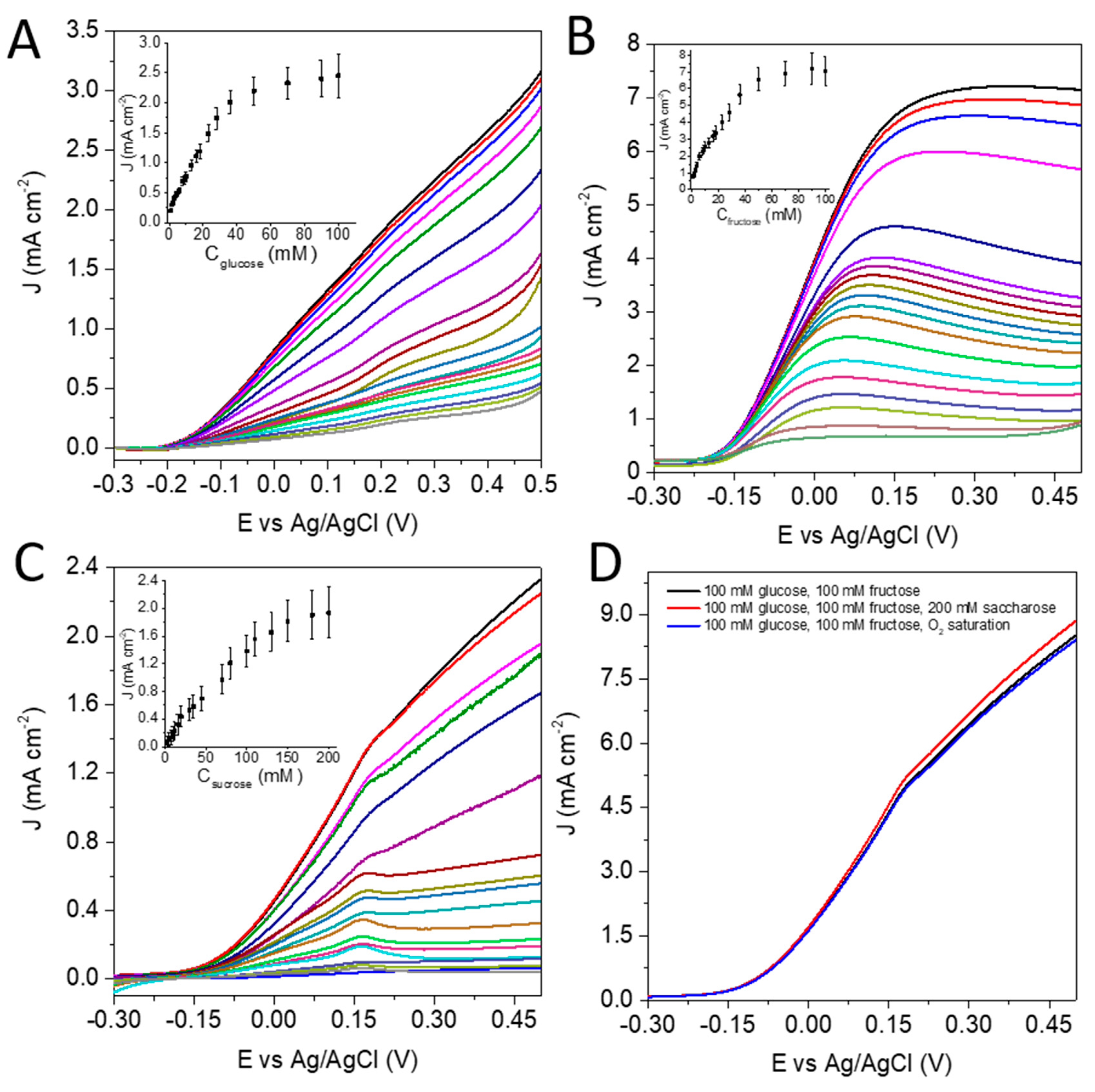
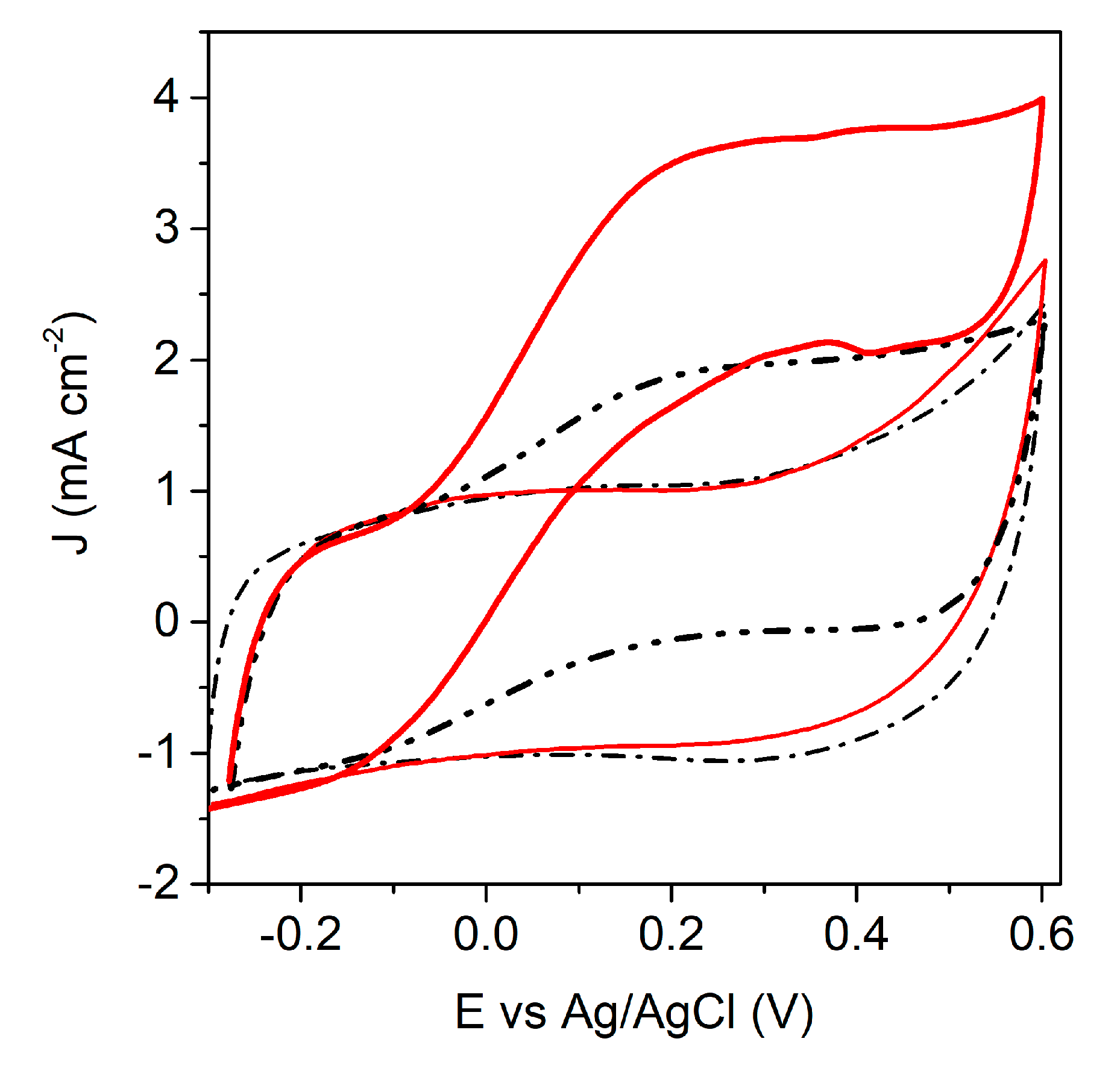
3.1. Characterization of the Anode and the Catalytic Process
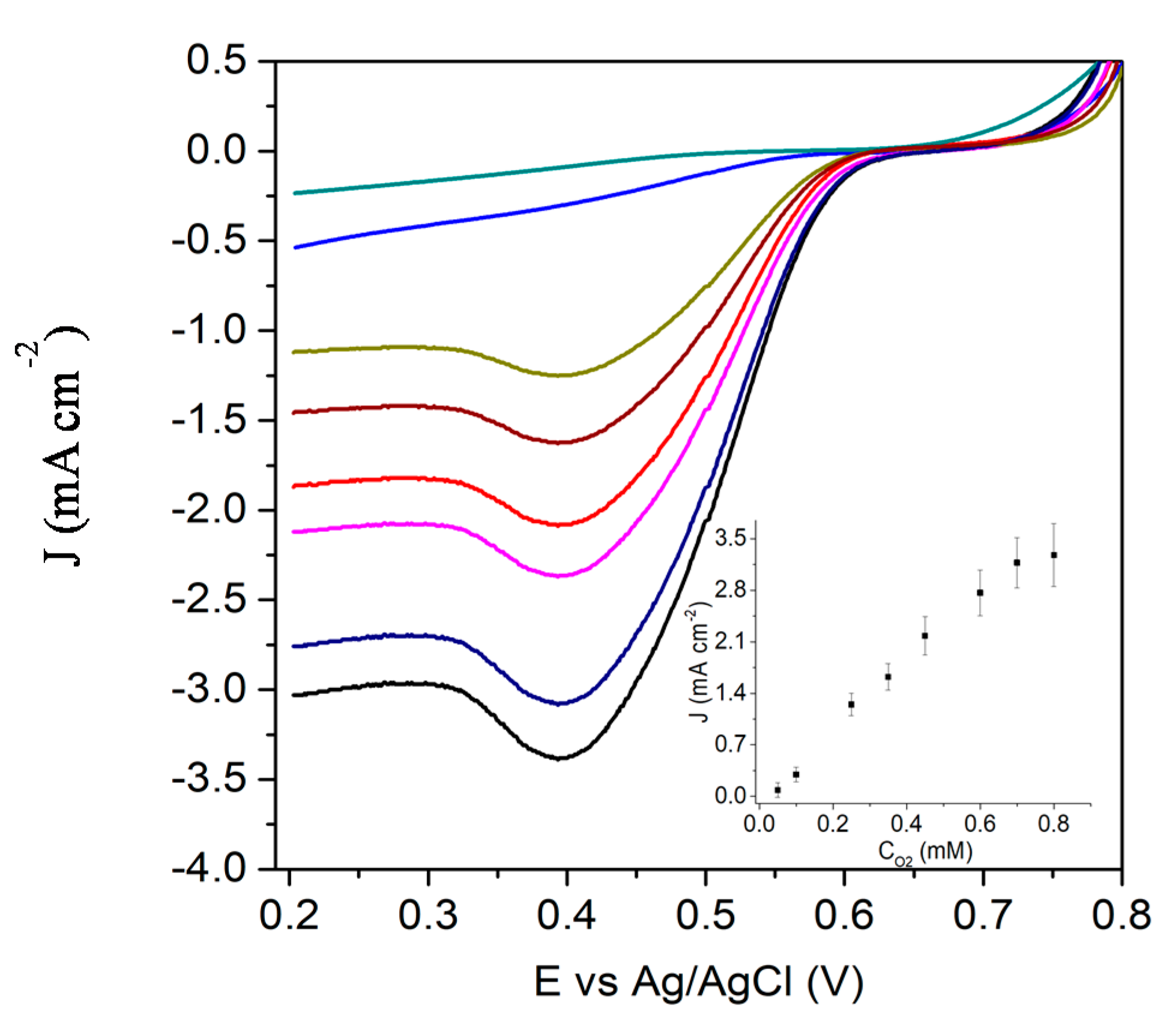
3.2. Characterization of the Biocathode and the Catalytic Process
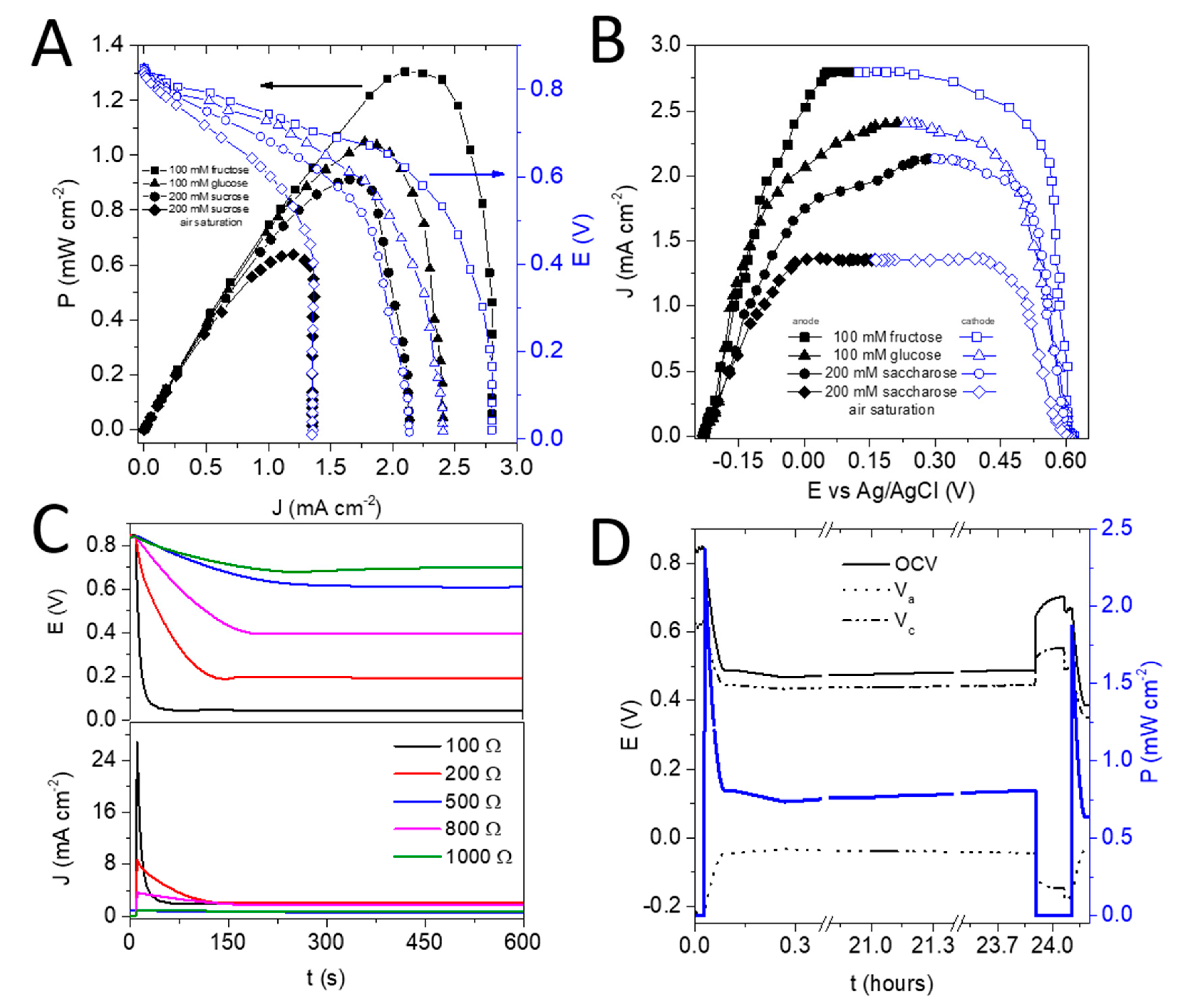
3.3. The Charge Generation Properties of the EFC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Falk, M.; Blum, Z.; Shleev, S. Direct electron transfer based enzymatic fuel cells. Electrochim. Acta 2012, 82, 191–202. [Google Scholar] [CrossRef]
- Stolarczyk, K.; Kizling, M.; Majdecka, D.; Zelechowska, K.; Biernat, J.F.; Rogalski, J.; Bilewicz, R. Biobatteries and biofuel cells with biphenylated carbon nanotubes. J. Power Sources 2014, 249, 263–269. [Google Scholar] [CrossRef]
- Bollella, P.; Fusco, G.; Stevar, D.; Gorton, L.; Ludwig, R.; Ma, S.; Boer, H.; Koivula, A.; Tortolini, C.; Favero, G.; et al. A Glucose/Oxygen Enzymatic Fuel Cell based on Gold Nanoparticles modified Graphene Screen-Printed Electrode. Proof-of-Concept in Human Saliva. Sens. Actuators B Chem. 2018, 256, 921–930. [Google Scholar] [CrossRef]
- Lau, C.; Moehlenbrock, M.J.; Arechederra, R.L.; Falase, A.; Garcia, K.; Rincon, R.; Minteer, S.D.; Banta, S.; Gupta, G.; Babanova, S.; et al. Paper based biofuel cells: Incorporating enzymatic cascades for ethanol and methanol oxidation. Int. J. Hydrogen Energy 2015, 40, 14661–14666. [Google Scholar] [CrossRef]
- Hickey, D.P.; Giroud, F.; Schmidtke, D.W.; Glatzhofer, D.T.; Minteer, S.D. Enzyme cascade for catalyzing sucrose oxidation in a biofuel cell. ACS Catal. 2013, 3, 2729–2737. [Google Scholar] [CrossRef]
- Shoji, K.; Akiyama, Y.; Suzuki, M.; Nakamura, N.; Ohno, H.; Morishima, K. Biofuel cell backpacked insect and its application to wireless sensing. Biosens. Bioelectron. 2016, 78, 390–395. [Google Scholar] [CrossRef]
- Zhu, Z.; Tam, T.K.; Sun, F.; You, C.; Zhang, Y.H.P. A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat. Commun. 2014, 5, 3026. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, Y.H.P. In vitro metabolic engineering of bioelectricity generation by the complete oxidation of glucose. Metab. Eng. 2017, 39, 110–116. [Google Scholar] [CrossRef]
- Sokic-Lazic, D.; Minteer, S.D. Pyruvate/air enzymatic biofuel cell capable of complete oxidation. Electrochem. Solid-State Lett. 2009, 12, 26–29. [Google Scholar] [CrossRef]
- Sokic-Lazic, D.; De Andrade, A.R.; Minteer, S.D. Utilization of enzyme cascades for complete oxidation of lactate in an enzymatic biofuel cell. Electrochim. Acta 2011, 56, 10772–10775. [Google Scholar] [CrossRef]
- Sokic-Lazic, D.; Minteer, S.D. Citric acid cycle biomimic on a carbon electrode. Biosens. Bioelectron. 2008, 24, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Handa, Y.; Yamagiwa, K.; Ikeda, Y.; Yanagisawa, Y.; Watanabe, S.; Yabuuchi, N.; Komaba, S. Fabrication of carbon-felt-based multi-enzyme immobilized anodes to oxidize sucrose for biofuel cells. ChemPhysChem 2014, 15, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Kajiya, K.; Nakamura, N.; Ohno, H. Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell. Energy Environ. Sci. 2009, 2, 1280–1285. [Google Scholar] [CrossRef]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Edembe, L. Bilirubin oxidases in bioelectrochemistry: Features and recent findings. Biosens. Bioelectron. 2013, 50, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, A.; Stemmer, A.; Tel-Vered, R. Enzymatic self-wiring in nanopores and its application in direct electron transfer biofuel cells. Nanoscale Adv. 2019, 1, 347–356. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The Immobilization of Fructose Dehydrogenase on Nanoporous Gold Electrodes for the Detection of Fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Nanostructured porous electrodes by the anodization of gold for an application as scaffolds in direct-electron-transfer-type bioelectrocatalysis. Anal. Sci. 2018, 34, 1317–1322. [Google Scholar] [CrossRef]
- Takahashi, Y.; Wanibuchi, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using porous gold electrodes. J. Electroanal. Chem. 2019, 843, 47–53. [Google Scholar] [CrossRef]
- Adachi, T.; Fujii, T.; Honda, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct electron transfer-type bioelectrocatalysis of FAD-dependent glucose dehydrogenase using porous gold electrodes and enzymatically implanted platinum nanoclusters. Bioelectrochemistry 2020, 133, 107457. [Google Scholar] [CrossRef]
- Kizling, M.; Dzwonek, M.; Więckowska, A.; Bilewicz, R. Size Does Matter—Mediation of Electron Transfer by Gold Clusters in Bioelectrocatalysis. ChemCatChem 2018, 10, 1988–1992. [Google Scholar] [CrossRef]
- Slaughter, G.; Kulkarni, T. Enzymatic Glucose Biofuel Cell and its Application. J. Biochips Tissue Chips 2015, 05, 1–10. [Google Scholar] [CrossRef]
- Xiao, X.; Xia, H.Q.; Wu, R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.; Liu, A. Tackling the Challenges of Enzymatic (Bio)Fuel Cells. Chem. Rev. 2019, 119, 9509–9558. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Urrios, A.; Kang, S.; Folch, A. The upcoming 3D-printing revolution in microfluidics. Lab Chip 2016, 16, 1720–1742. [Google Scholar] [CrossRef]
- Cocovi-Solberg, D.J.; Rosende, M.; Michalec, M.; Miró, M. 3D Printing: The Second Dawn of Lab-On-Valve Fluidic Platforms for Automatic (Bio)Chemical Assays. Anal. Chem. 2019, 91, 1140–1149. [Google Scholar] [CrossRef]
- Mihranyan, A.; Nyholm, L.; Garcia Bennett, A.E.; Strømme, M. A novel high specific surface area conducting paper material composed of polypyrrole and Cladophora cellulose. J. Phys. Chem. B 2008, 112, 12249–12255. [Google Scholar] [CrossRef]
- Nyström, G.; Razaq, A.; Strømme, M.; Nyholm, L.; Mihranyan, A. Ultrafast all-polymer paper-based batteries. Nano Lett. 2009, 9, 3635–3639. [Google Scholar] [CrossRef]
- Ammam, M.; Fransaer, J. Microbiofuel cell powered by glucose/O2 based on electrodeposition of enzyme, conducting polymer and redox mediators. Part II: Influence of the electropolymerized monomer on the output power density and stability. Electrochim. Acta 2014, 121, 83–92. [Google Scholar] [CrossRef]
- Tominaga, M.; Shirakihara, C.; Taniguchi, I. Direct heterogeneous electron transfer reactions and molecular orientation of fructose dehydrogenase adsorbed onto pyrolytic graphite electrodes. J. Electroanal. Chem. 2007, 610, 1–8. [Google Scholar] [CrossRef]
- Kawai, S.; Yakushi, T.; Matsushita, K.; Kitazumi, Y.; Shirai, O.; Kano, K. The electron transfer pathway in direct electrochemical communication of fructose dehydrogenase with electrodes. Electrochem. Commun. 2014, 38, 28–31. [Google Scholar] [CrossRef]
- Ratautas, D.; Laurynenas, A.; Dagys, M.; Marcinkevičiene, L.; Meškys, R.; Kulys, J. High current, low redox potential mediatorless bioanode based on gold nanoparticles and glucose dehydrogenase from Ewingella americana. Electrochim. Acta 2016, 199, 254–260. [Google Scholar] [CrossRef]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. The influence of pH and divalent/monovalent cations on the internal electron transfer (IET), enzymatic activity, and structure of fructose dehydrogenase. Anal. Bioanal. Chem. 2018, 410, 3253–3264. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Hickey, D.P.; Abdellaoui, S.; Lim, K.; Wu, F.; Tan, B.; Minteer, S.D. Rational design of quinones for high power density biofuel cells. Chem. Sci. 2015, 6, 4867–4875. [Google Scholar] [CrossRef] [PubMed]
- Kizling, M.; Bilewicz, R. Fructose Dehydrogenase Electron Transfer Pathway in Bioelectrocatalytic Reactions. ChemElectroChem 2018, 5, 166–174. [Google Scholar] [CrossRef]
- Hou, C.; Lang, Q.; Liu, A. Tailoring 1,4-naphthoquinone with electron-withdrawing group: Toward developing redox polymer and FAD-GDH based hydrogel bioanode for efficient electrocatalytic glucose oxidation. Electrochim. Acta 2016, 211, 663–670. [Google Scholar] [CrossRef]
- Kizling, M.; Dzwonek, M.; Wieckowska, A.; Bilewicz, R. Gold nanoparticles in bioelectrocatalysis—The role of nanoparticle size. Curr. Opin. Electrochem. 2018, 12, 113–120. [Google Scholar] [CrossRef]
- Kizling, M.; Stolarczyk, K.; Tammela, P.; Wang, Z.; Nyholm, L.; Golimowski, J.; Bilewicz, R. Bioelectrodes based on pseudocapacitive cellulose/polypyrrole composite improve performance of biofuel cell. Bioelectrochemistry 2016, 112, 184–190. [Google Scholar] [CrossRef]
- Wolfenden, R.; Yuan, Y. Rates of spontaneous cleavage of glucose, fructose, sucrose, and trehalose in water, and the catalytic proficiencies of invertase and trehalas. J. Am. Chem. Soc. 2008, 130, 7548–7549. [Google Scholar] [CrossRef]
- Karaskiewicz, M.; Majdecka, D.; Wieckowska, A.; Biernat, J.F.; Rogalski, J.; Bilewicz, R. Induced-fit binding of laccase to gold and carbon electrodes for the biological fuel cell applications. Electrochim. Acta 2014, 126, 132–138. [Google Scholar] [CrossRef]
- Giroud, F.; Milton, R.D.; Tan, B.-X.; Minteer, S. Simplifying Enzymatic Biofuel Cells: Immobilized Naphthoquinone as a Biocathodic Orientational Moiety and Bioanodic Electron Mediator. ACS Catal. 2015, 5, 1240–1244. [Google Scholar] [CrossRef]
- Zhang, Y.; Arugula, M.A.; Williams, S.T.; Minteer, S.D.; Simonian, A.L. Layer-by-Layer Assembly of Carbon Nanotubes Modified with Invertase/Glucose Dehydrogenase Cascade for Sucrose/O 2 Biofuel Cell. J. Electrochem. Soc. 2016, 163, F449–F454. [Google Scholar] [CrossRef]
- Herkendell, K.; Tel-Vered, R.; Stemmer, A. Switchable aerobic/anaerobic multi-substrate biofuel cell operating on anodic and cathodic enzymatic cascade assemblies. Nanoscale 2017, 9, 14118–14126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ma, C.; Percival Zhang, Y.H. Co-utilization of mixed sugars in an enzymatic fuel cell based on an in vitro enzymatic pathway. Electrochim. Acta 2018, 263, 184–191. [Google Scholar] [CrossRef]
- Trifonov, A.; Stemmer, A.; Tel-Vered, R. Power generation by selective self-assembly of biocatalysts. ACS Nano 2019, 13, 8630–8638. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INV | X | X | X | X | X | X | X | X | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MUT | X | X | X | X | X | X | X | X | |||||||
| GDH | X | X | X | X | X | X | X | X | |||||||
| FDH | X | X | X | X | X | X | X | X | |||||||
| J (mA cm−2) | 1.93 ± 0.37 | 0.77 ± 0.18 | 1.03 ± 0.18 | 1.11 ± 0.25 | 0.05 ± 0.01 | 0.0 ± 0.0 | 0.04 ± 0.01 | 0.06 ± 0.01 | 1.01 ± 0.23 | 0.03 ± 0.01 | 0.48 ± 0.06 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.02 ± 0.01 | 0.03 ± 0.01 |
| Enzymes | Electrode Materials | Fuel Solution | OCV (V) | Pmax (µW cm−2) | Jmax (µA cm−2) | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| INV, FDH, glucose oxidase | Anode: Toray Paper ferrocene-based hydrogel; Cathode: Pt electrode | 100 mM sucrose; O2 saturated | 0.61 | 50 | 340 | [5] | |
| INV, glucose oxidase | Anode: carbon paper, DNA template prepared by rolling circle amplification; Cathode: Pt electrode | 50 mM sucrose; O2 saturated | 0.59 | 22.2 | 140 | [41] | |
| INV, MUT, FDH, glucose oxidase LAC, catalase | Mesoporous carbon nanoparticle matrix | 100 mM glucose, 100 mM fructose, 180 mM sucrose, O2 saturated, 140 mM H2O2 | 0.64 | 240 | 890 | [42] | |
| sucrose phosphorylase, xylose isomerase, polyphosphate glucokinase, glucose-6-phosphate DH, 6-phosphogluconate DH, PPP enzymes | Carbon paper, enzymes and mediator dissolved in anolyte; Cathode: Pt electrode | 0.64 mM sucrose, O3 saturated | 0.80 | 690 | 1330 | 27% of initial power after 6 days | [43] |
| INV, MUT, FDH, GDH; LAC | CPPy, AuNQ modified with naphthoquinone | 200 mM sucrose; O2 saturated | 0.82 | 810 | 2230 | 36% of initial power after 8 days | This paper |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizling, M.; Dzwonek, M.; Nowak, A.; Tymecki, Ł.; Stolarczyk, K.; Więckowska, A.; Bilewicz, R. Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels. Nanomaterials 2020, 10, 1534. https://doi.org/10.3390/nano10081534
Kizling M, Dzwonek M, Nowak A, Tymecki Ł, Stolarczyk K, Więckowska A, Bilewicz R. Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels. Nanomaterials. 2020; 10(8):1534. https://doi.org/10.3390/nano10081534
Chicago/Turabian StyleKizling, Michał, Maciej Dzwonek, Anna Nowak, Łukasz Tymecki, Krzysztof Stolarczyk, Agnieszka Więckowska, and Renata Bilewicz. 2020. "Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels" Nanomaterials 10, no. 8: 1534. https://doi.org/10.3390/nano10081534
APA StyleKizling, M., Dzwonek, M., Nowak, A., Tymecki, Ł., Stolarczyk, K., Więckowska, A., & Bilewicz, R. (2020). Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels. Nanomaterials, 10(8), 1534. https://doi.org/10.3390/nano10081534