Anisotropic Thermal Conductivity in Few-Layer and Bulk Titanium Trisulphide from First Principles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
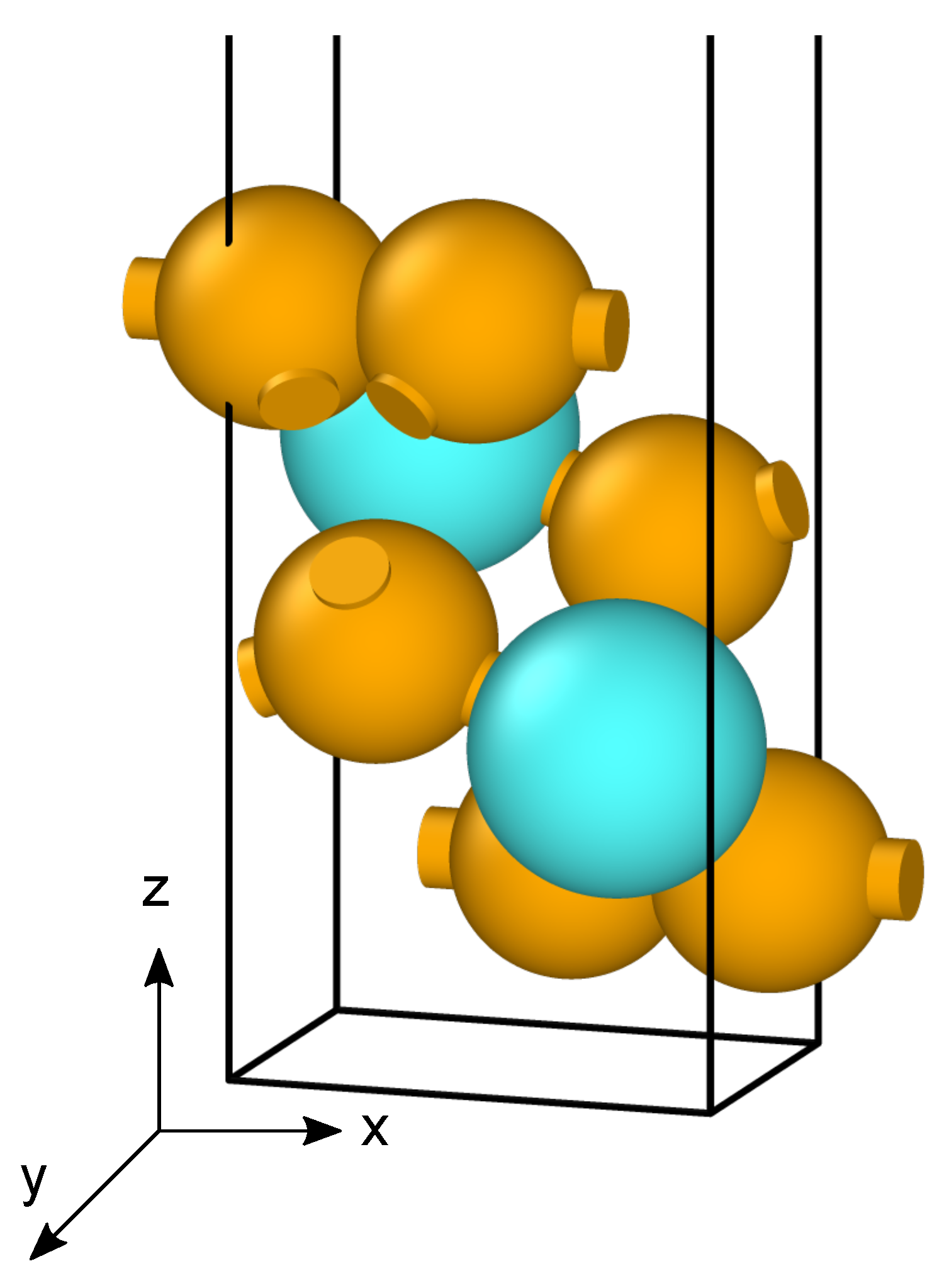
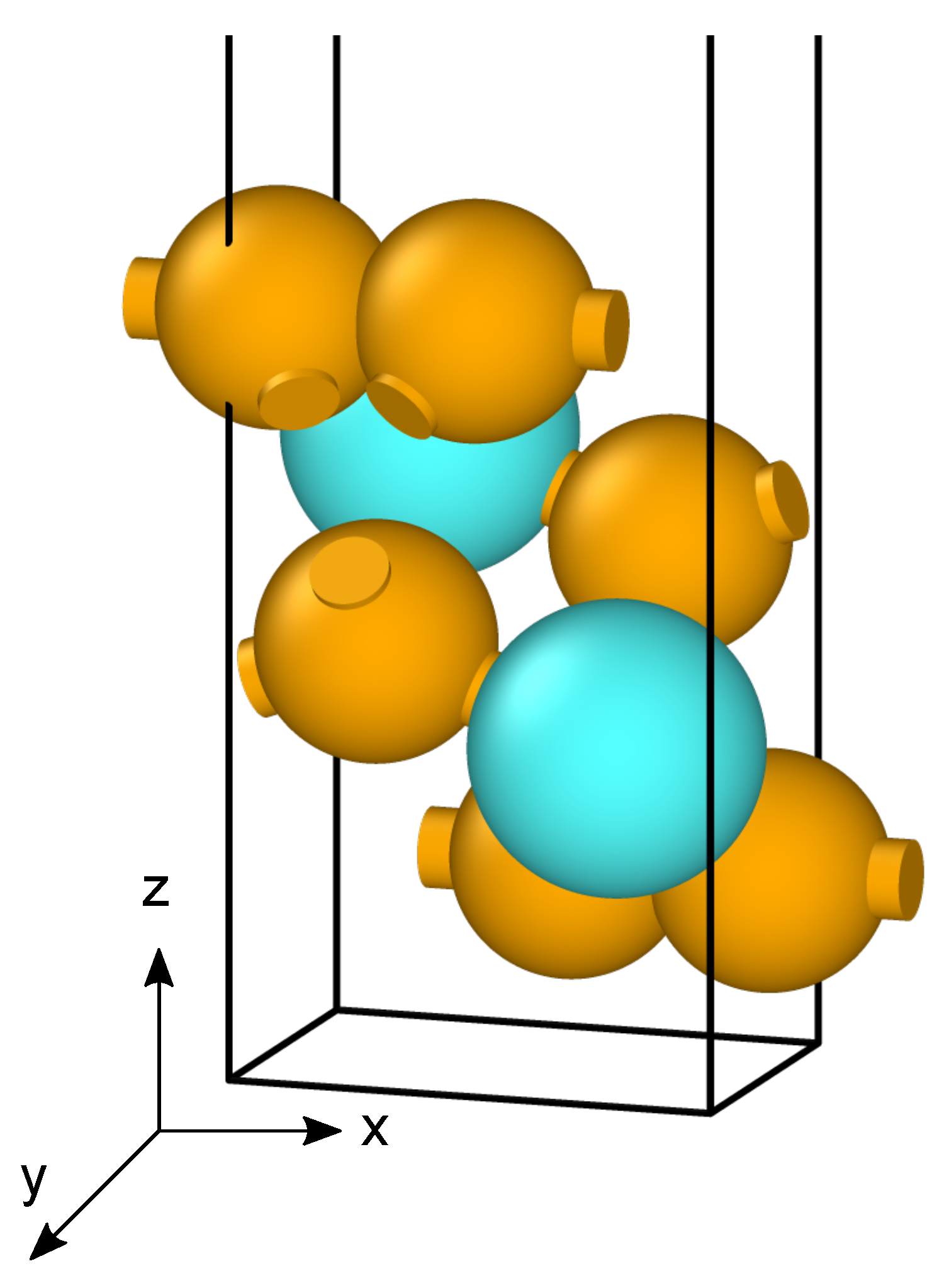
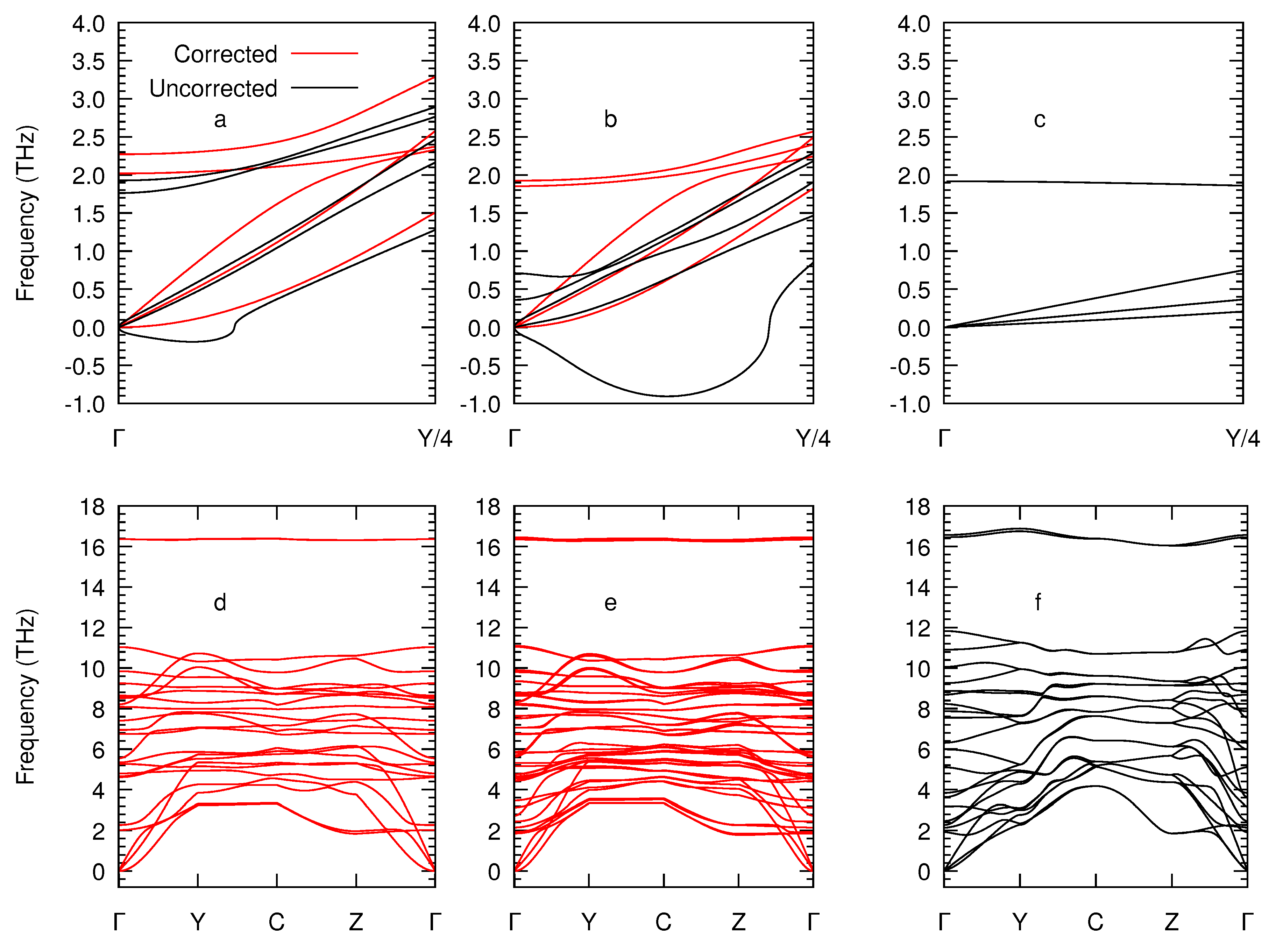
2. Theoretical Methods
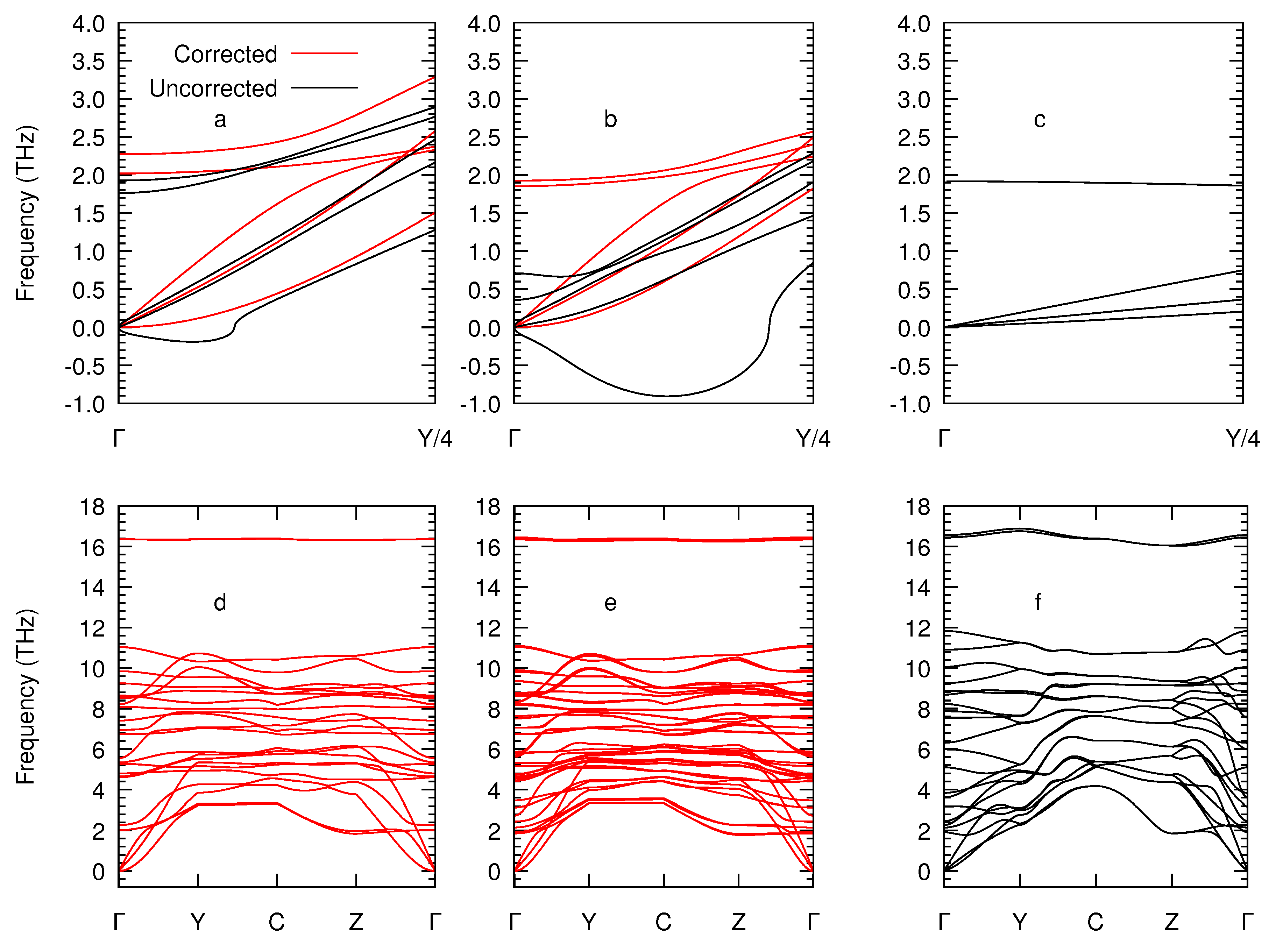
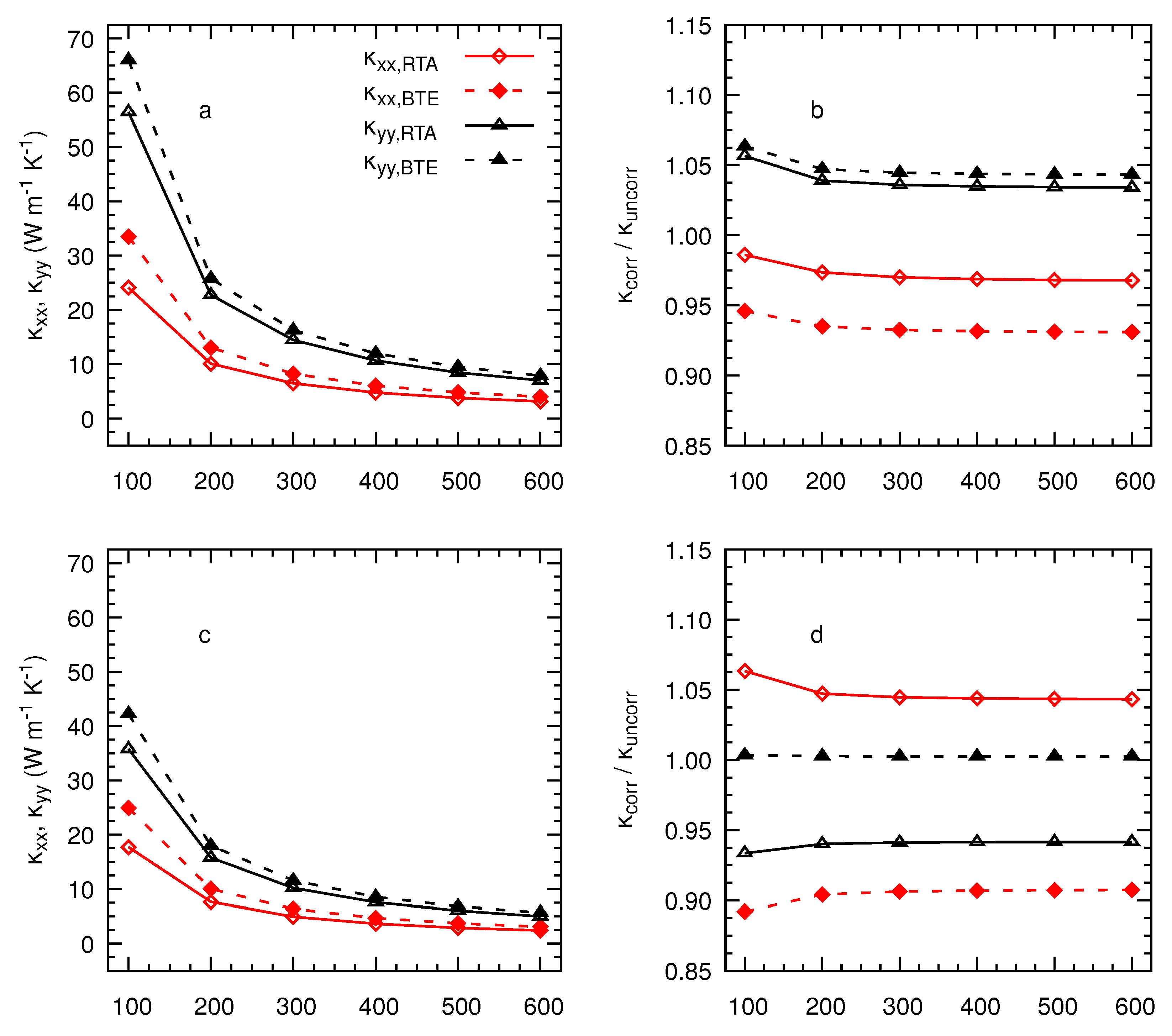
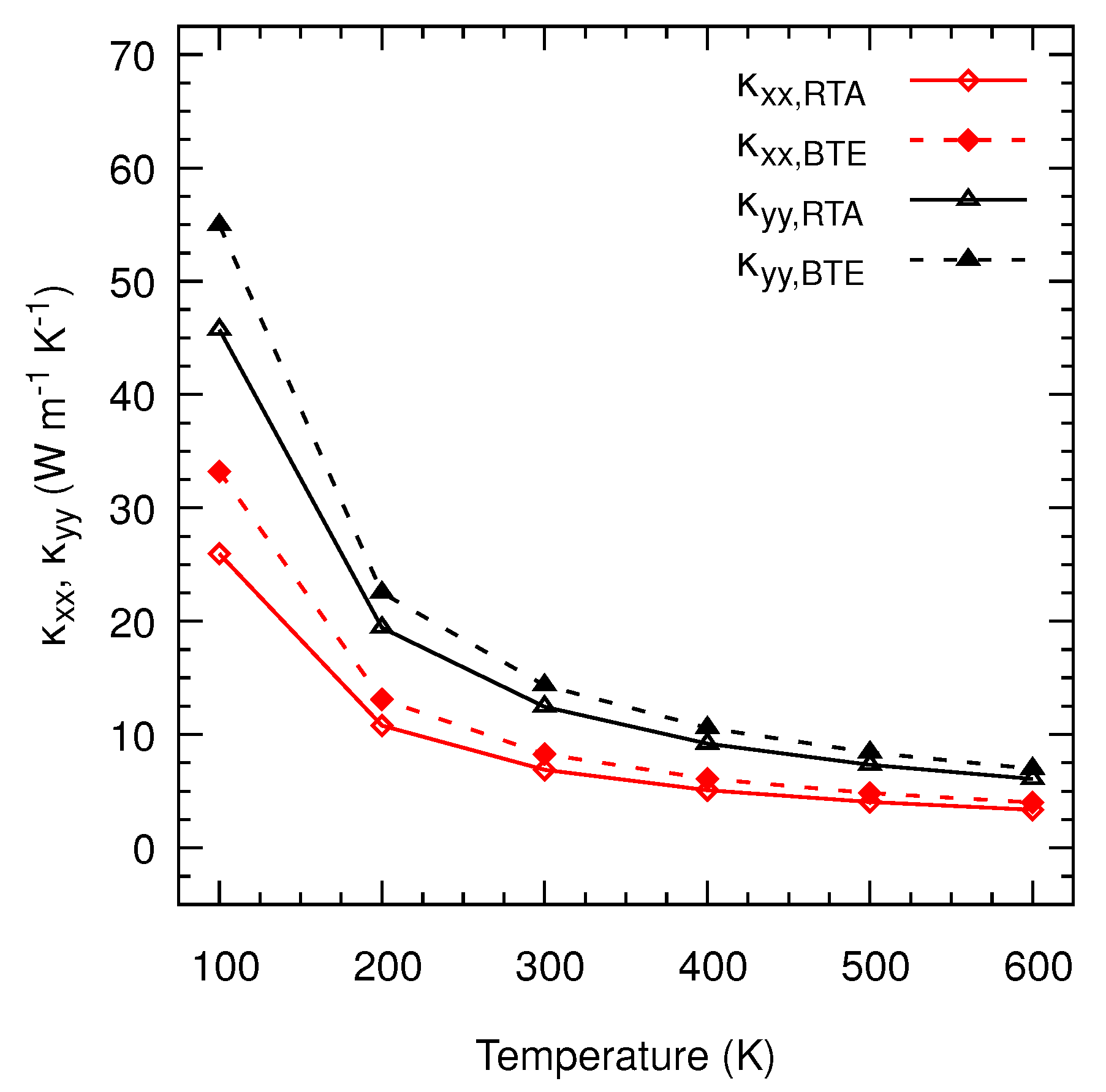
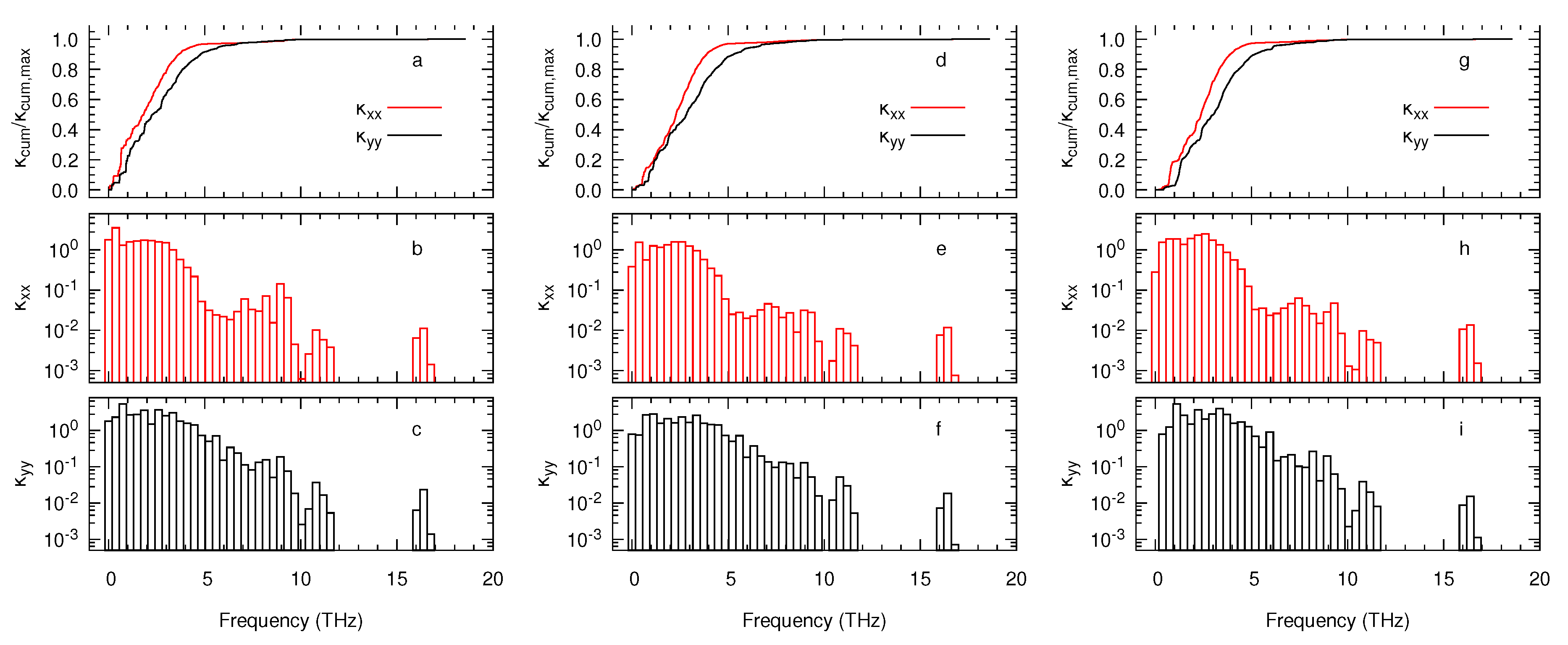
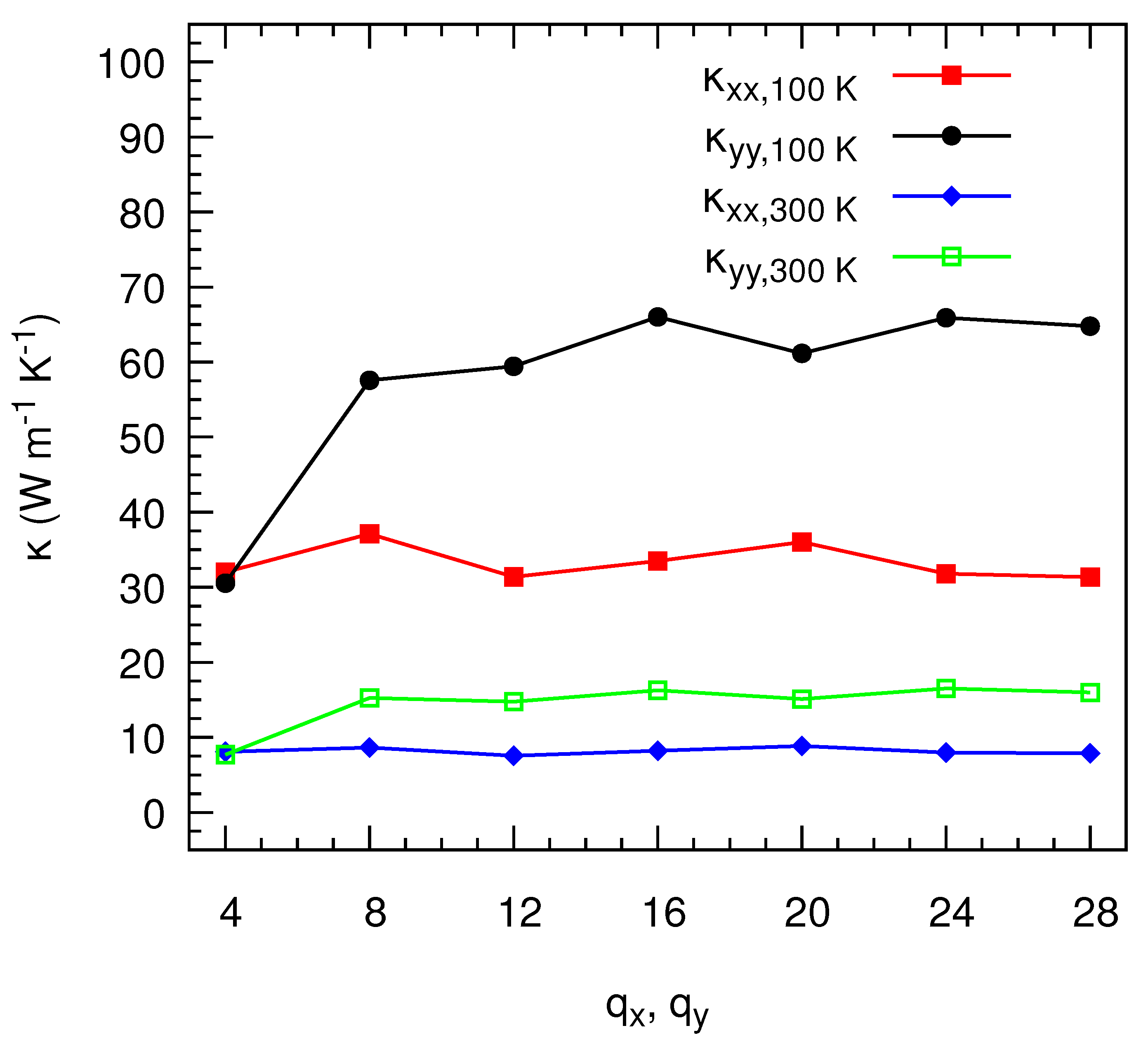
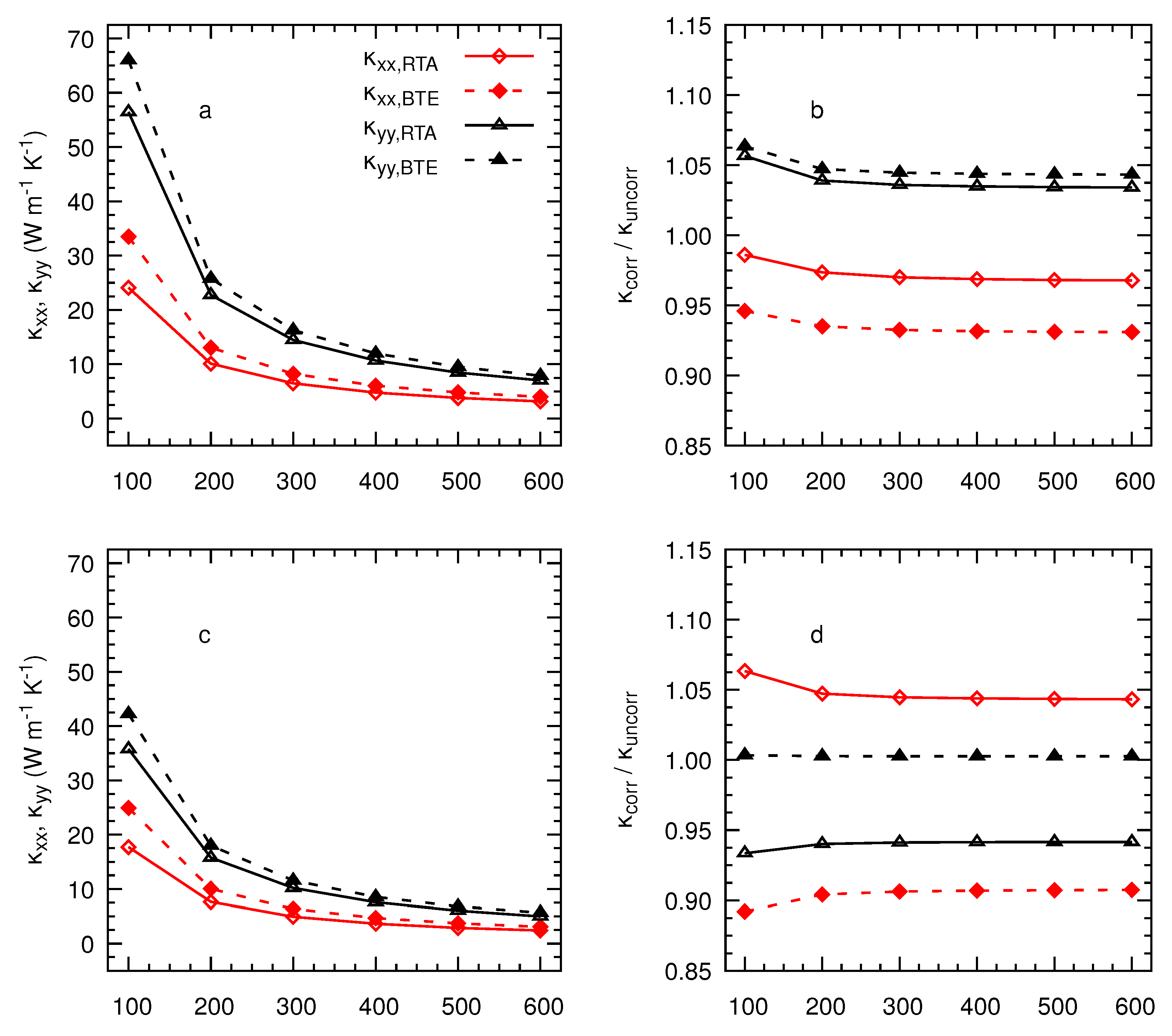
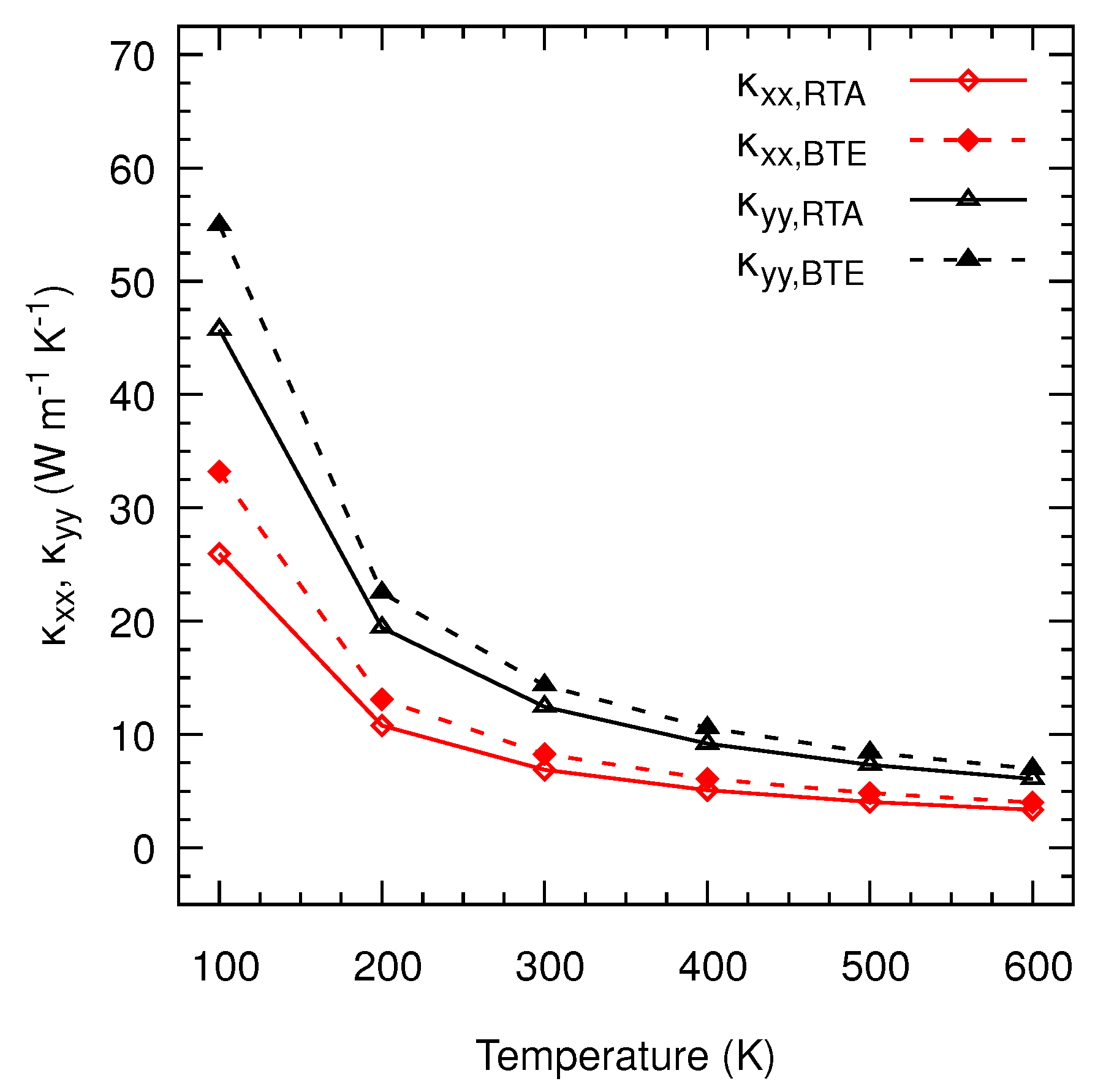
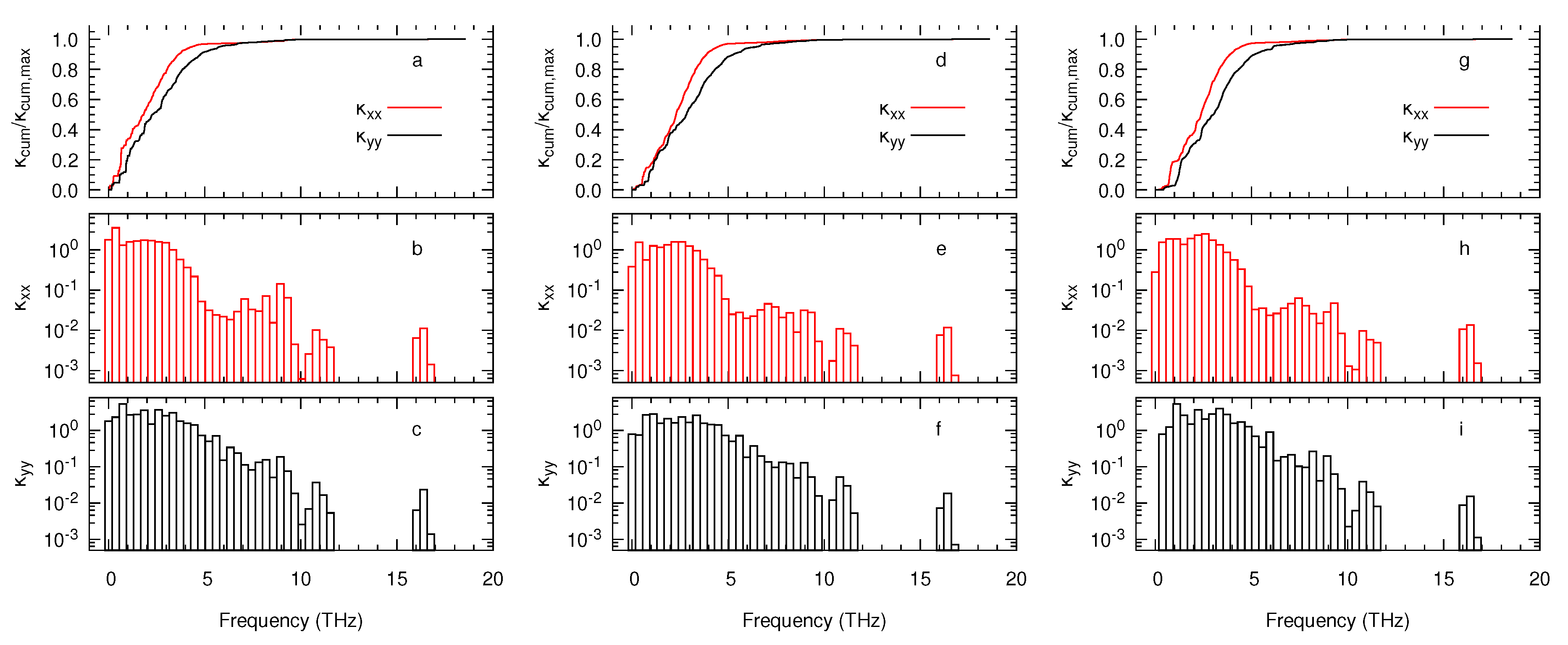
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.; Ares, J.R.; Ferrer, I.J.; Sánchez, C. Synthesis and characterization of a family of layered trichalcogenides for assisted hydrogen photogeneration. Phys. Status Solidi-Rapid Res. Lett. 2016, 10, 802–806. [Google Scholar] [CrossRef]
- Biele, R.; Flores, E.; Ares, J.R.; Sanchez, C.; Ferrer, I.J.; Rubio-Bollinger, G.; Castellanos-Gomez, A.; D’Agosta, R. Strain-induced band gap engineering in layered TiS3. Nano Res. 2018, 11, 225–232. [Google Scholar] [CrossRef]
- Jin, Y.; Li, X.; Yang, J. Single layer of MX3 (M = Ti, Zr; X = S, Se, Te): A new platform for nano-electronics and optics. Phys. Chem. Chem. Phys. 2015, 17, 18665–18669. [Google Scholar] [CrossRef] [Green Version]
- Saeed, Y.; Kachmar, A.; Carignano, M.A. First-principles study of the transport properties in bulk and monolayer MX3 (M = Ti, Zr, Hf and X = S, Se) compounds. J. Phys. Chem. C 2017, 121, 1399–1403. [Google Scholar] [CrossRef]
- Abudukelimu, A.; Kakushima, K.; Ahmet, P.; Geni, M.; Tsutsui, K.; Nishiyama, A.; Sugii, N.; Natori, K.; Hattori, T.; Iwai, H. The effect of isotropic and anisotropic scattering in drain region of ballistic channel diode. In Proceedings of the 2010 10th IEEE International Conference on Solid-State and Integrated Circuit Technology, Shanghai, China, 1–4 November 2010; pp. 1247–1249. [Google Scholar] [CrossRef]
- Wu, J.; Wang, D.; Liu, H.; Lau, W.M.; Liu, L.M. An ab initio study of TiS3: A promising electrode material for rechargeable Li and Na ion batteries. RSC Adv. 2015, 5, 21455–21463. [Google Scholar] [CrossRef]
- Barawi, M.; Flores, E.; Ferrer, I.J.; Ares, J.R.; Sánchez, C. Titanium trisulphide (TiS3) nanoribbons for easy hydrogen photogeneration under visible light. J. Mater. Chem. A 2015, 3, 7959–7965. [Google Scholar] [CrossRef]
- Island, J.O.; Buscema, M.; Barawi, M.; Clamagirand, J.M.; Ares, J.R.; Sánchez, C.; Ferrer, I.J.; Steele, G.A.; van der Zant, H.S.J.; Castellanos-Gomez, A. Ultrahigh Photoresponse of Few-Layer TiS3 Nanoribbon Transistors. Adv. Opt. Mater. 2014, 2, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Island, J.O.; Barawi, M.; Biele, R.; Almazán, A.; Clamagirand, J.M.; Ares, J.R.; Sánchez, C.; van der Zant, H.S.J.; Álvarez, J.V.; D’Agosta, R.; et al. TiS3 Transistors with Tailored Morphology and Electrical Properties. Adv. Mater. 2015, 27, 2595–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Mendoza, A.J.; Barawi, M.; Biele, R.; Flores, E.; Ares, J.R.; Sánchez, C.; Rubio-Bollinger, G.; Agraït, N.; D’Agosta, R.; Ferrer, I.J.; et al. Electronic Bandgap and Exciton Binding Energy of Layered Semiconductor TiS3. Adv. Electron. Mater. 2015, 1, 1500126. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Ares, J.; Clamagirand, J.; Barawi, M.; Sánchez, C. Optical properties of titanium trisulphide (TiS3) thin films. Thin Sol. Films 2013, 535, 398–401. [Google Scholar] [CrossRef]
- Guilmeau, E.; Berthebaud, D.; Misse, P.R.; Hebert, S.; Lebedev, O.I.; Chateigner, D.; Martin, C.; Maignan, A. ZrSe3-Type variant of TiS3: Structure and thermoelectric properties. Chem. Mat. 2014, 26, 5585–5591. [Google Scholar] [CrossRef]
- Hsieh, P.L.; Jackson, C.; Grüner, G. Disorder effects in the linear chain compound TiS3. Sol. Stat. Commun. 1983, 46, 505–507. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Wen, Y.; Shi, L.; Chen, R.; Liu, H.; Shan, B. Titanium Trisulfide Monolayer as a Potential Thermoelectric Material: A First-Principles-Based Boltzmann Transport Study. ACS Appl. Mater. Interfaces 2017, 9, 2509–2515. [Google Scholar] [CrossRef]
- Carrete, J.; Li, W.; Lindsay, L.; Broido, D.A.; Gallego, L.J.; Mingo, N. Physically founded phonon dispersions of few-layer materials and the case of borophene. Mater. Res. Lett. 2016, 4, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Furuseth, S.; Brattas, L.; Kjekshus, A. Crystal Structures of TiS 3, ZrS 3, ZrSe 3, ZrTe 3, HfS 3 and HfSe 3. Acta Chem. Scand. A 1975, 29, 623. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar]
- Li, W.; Mingo, N.; Lindsay, L.; Broido, D.A.; Stewart, D.A.; Katcho, N.A. Thermal conductivity of diamond nanowires from first principles. Phys. Rev. B 2012, 85, 195436. [Google Scholar]
- Li, W.; Carrete, J.; Katcho, N.A.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comp. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar]
- Carrete, J.; Vermeersch, B.; Katre, A.; van Roekeghem, A.; Wang, T.; Madsen, G.K.; Mingo, N. almaBTE: A solver of the space-time dependent Boltzmann transport equation for phonons in structured materials. Comput. Phys. Commun. 2017, 220, 351–362. [Google Scholar]
- Li, W.; Lindsay, L.; Broido, D.A.; Stewart, D.A.; Mingo, N. Thermal conductivity of bulk and nanowire Mg2SixSn1−x alloys from first principles. Phys. Rev. B 2012, 86, 174307. [Google Scholar]
- Torres, P.; Torelló, A.; Bafaluy, J.; Camacho, J.; Cartoixà, X.; Alvarez, F.X. First principles kinetic-collective thermal conductivity of semiconductors. Phys. Rev. B 2017, 95, 165407. [Google Scholar]
- Tamura, S. Isotope scattering of dispersive phonons in Ge. Phys. Rev. B 1983, 27, 858–866. [Google Scholar]
- Torres, P.; Alvarez, F.X.; Cartoixà, X.; Rurali, R. Thermal conductivity and phonon hydrodynamics in transition metal dichalcogenides from first-principles. 2D Mater. 2019, 6, 035002. [Google Scholar]
- Kong, B.D.; Paul, S.; Nardelli, M.B.; Kim, K.W. First-principles analysis of lattice thermal conductivity in monolayer and bilayer graphene. Phys. Rev. B 2009, 80, 033406. [Google Scholar]
- Li, W.; Carrete, J.; Mingo, N. Thermal conductivity and phonon linewidths of monolayer MoS2 from first principles. Appl. Phys. Lett. 2013, 103, 253103. [Google Scholar]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saiz, F.; Carrete, J.; Rurali, R. Anisotropic Thermal Conductivity in Few-Layer and Bulk Titanium Trisulphide from First Principles. Nanomaterials 2020, 10, 704. https://doi.org/10.3390/nano10040704
Saiz F, Carrete J, Rurali R. Anisotropic Thermal Conductivity in Few-Layer and Bulk Titanium Trisulphide from First Principles. Nanomaterials. 2020; 10(4):704. https://doi.org/10.3390/nano10040704
Chicago/Turabian StyleSaiz, Fernan, Jesus Carrete, and Riccardo Rurali. 2020. "Anisotropic Thermal Conductivity in Few-Layer and Bulk Titanium Trisulphide from First Principles" Nanomaterials 10, no. 4: 704. https://doi.org/10.3390/nano10040704
APA StyleSaiz, F., Carrete, J., & Rurali, R. (2020). Anisotropic Thermal Conductivity in Few-Layer and Bulk Titanium Trisulphide from First Principles. Nanomaterials, 10(4), 704. https://doi.org/10.3390/nano10040704