3.1. TGA Analysis of Unmodified ZnO NPs
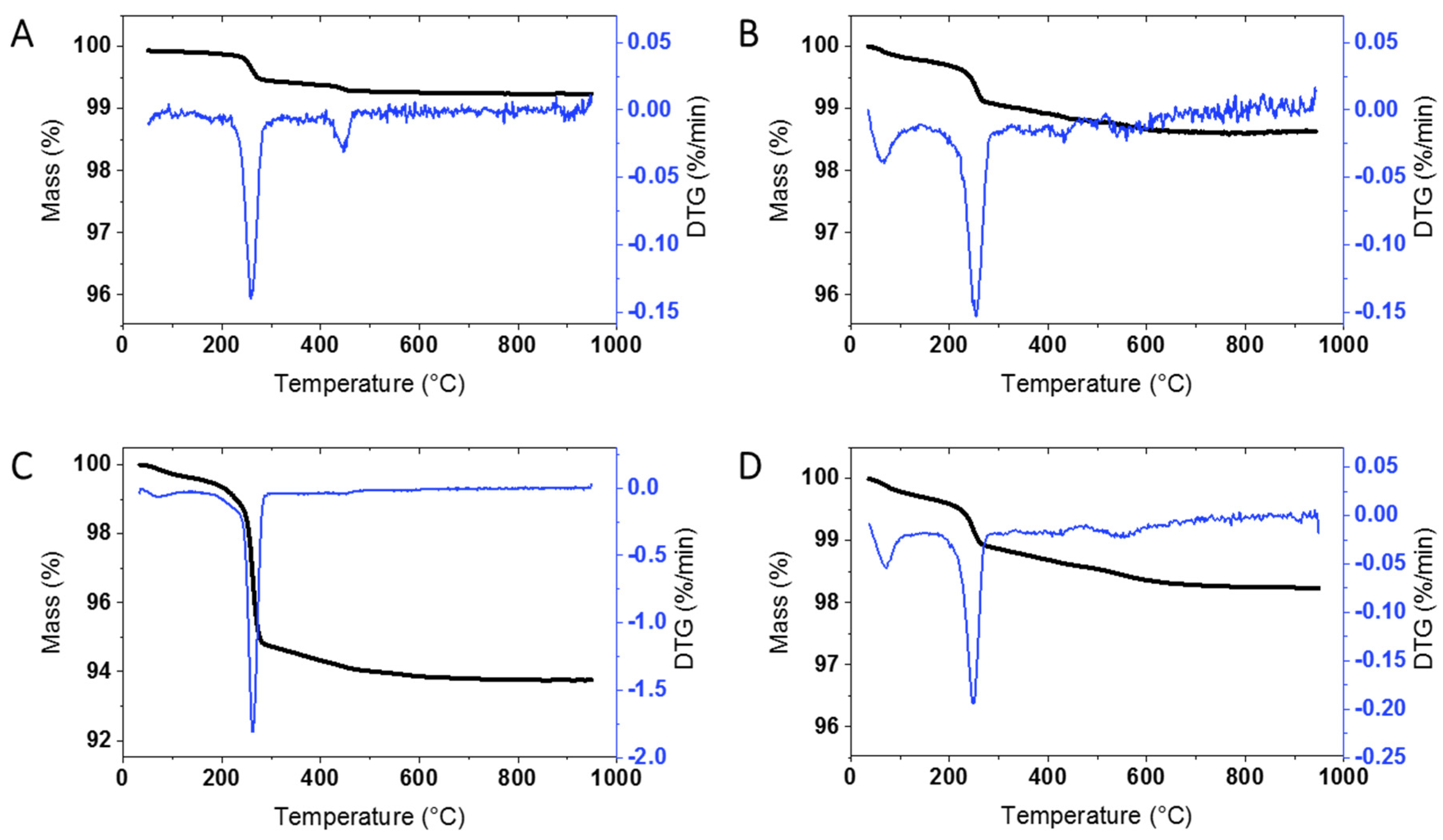
An initial survey of TGA results for at least one unmodified sample from each supplier was carried out in order to determine whether there is significant mass loss that will have to be taken into account for the surface modified samples. Representative results for ZnO from the four suppliers, as well as the JRC, are shown in
Figure 1 and
Figure S1; the sample codes are provided in the Materials and Methods. JRC-01 shows significant mass loss at ~260 °C (0.5%) and a smaller mass loss at higher temperatures. This can be contrasted to the behavior of BASF-01 sourced from the same supplier as JRC-01 but purchased at the beginning of this study. Here the overall mass loss is considerably lower (
Figure 1A vs
Figure S1A) without a clearly defined maximum at 260 °C. The original characterization report for JRC-01 reported only a small mass increase for this sample upon heating [
21]. However a later study that examined the JRC-01 sample and three batches of unmodified ZnO from the same supplier found that the total mass loss varied from 0.76 (JRC) to 0.93%. An inflection point at ~ 260 °C was also observed and FT-IR spectra showed loss of both carbon dioxide and water; carbon was detected in XPS spectra and was postulated to be from an adventitious carbon source [
25]. Overall these results indicate that the organic content of the BASF (JRC) sample may vary with time or sample handling.
Most of the samples from the three other commercial suppliers (USRN, NAM, Sky) give similar results with a clearly defined mass loss at ~ 260 °C (
Figure 1 and
Figure S1) and varying levels of mass loss at higher temperatures.
Table 1 summarizes the mass loss for each of the bare samples; the thermograms are divided into 3 temperature regions (200–300 °C, 300–600 °C and >600 °C; see Materials and Methods Section) for ease of comparison with the results for the surface modified materials discussed in the following sections. The mass loss from 200–300 °C, with a maximum at 260 °C, varied significantly for the bare samples, from a high of 4.61% for USRN-02 (
Figure 1C) to 0.24 for Sky-01 (
Figure S1B) and was low with lack of a clear inflection point in USRN-01 and NAM-02. The mass loss between 300–600 °C also varied across the samples, from a low of 0.13% for BASF-01 to a high of 1.6% for USRN-01. The mass loss in this region is expected to overlap with that for most organic coatings or modifications, as outlined below. With the exception of USRN-01, there is minimal mass loss (≤0.21%) above 600 °C. Note that in a few cases there is a small mass loss below 120 °C which is assigned to water but is not included in the Table. Multiple replicates were measured for 4 of the samples with good agreement between runs, with the exception of the high temperature mass loss which had a larger standard deviation for USRN-01 (
Table 1).
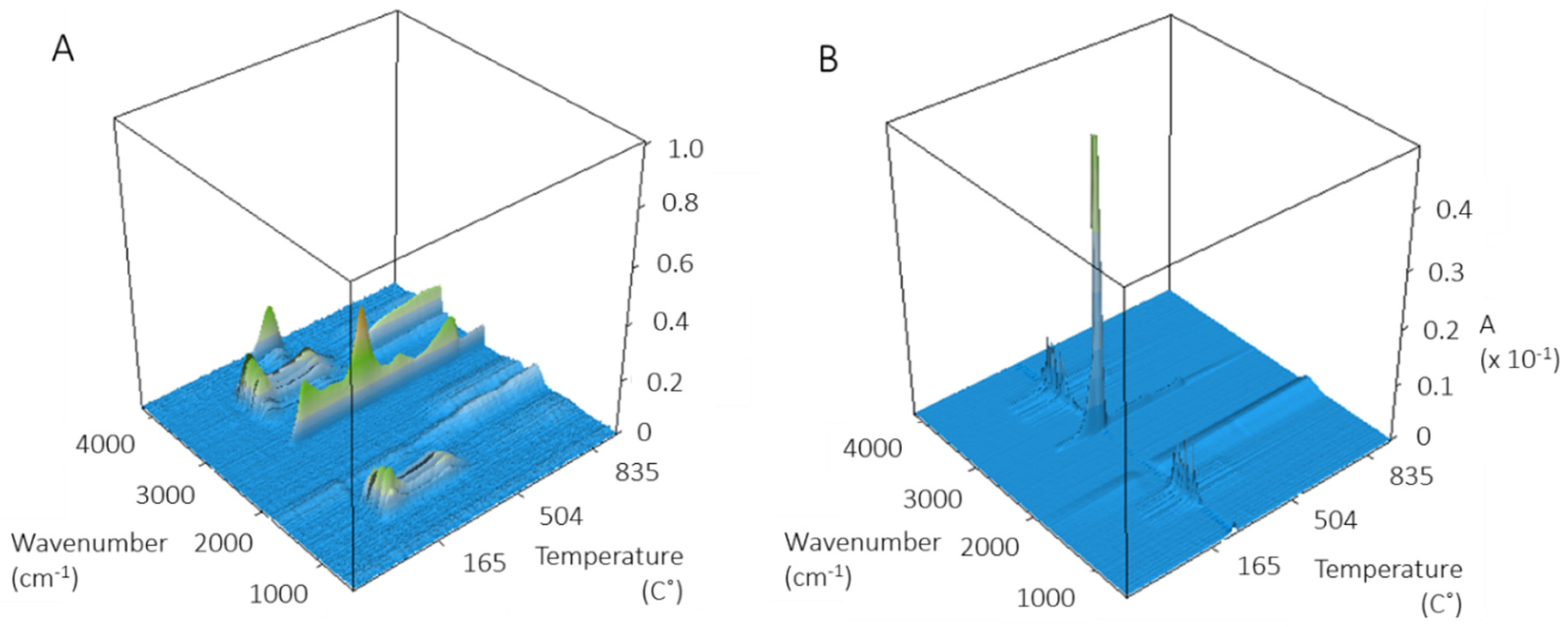
FT-IR spectra of evolved gases were measured for several ZnO NPs in an attempt to identify the source of the prominent component at 260 °C. The 3D spectra for USRN-01 and USRN-02 (
Figure 2) are dominated by loss of CO
2 at 260 °C with a minor amount of water; only water and CO
2 are detected across the temperature range examined. USRN-01 which had a larger mass loss between 300–600 °C than the other samples gave more complex 3D FT-IR spectra with loss of both water and CO
2 across a range of temperatures and the appearance of a peak assigned to ammonia at 965 cm
-1 at ~340 °C. Ammonia is not detected for USRN-02 or NAM-01 (
Figure S5A), suggesting that its presence in USRN-01 at least partly accounts for the large mass loss between 300–600 °C for this sample. Additional FT-IR signals in the 1100–1800 cm
-1 and 2800–3400 cm
-1 regions at temperatures above 600 °C for USRN-01 could not be conclusively identified, but are consistent with spectra for polyaromatic hydrocarbons. BASF-01 gave very weak FT-IR signals (not shown) consistent with the very low mass loss for this sample.
A control experiment using the procedure summarized for extraction of hydrophobic moieties (methanol-d
4/trifluoroacetic acid; see section below for stearic acid) was carried out for USRN-01 to test for the presence of hydrolysable or extractable material that could be identified by solution NMR. No signals were detected by
1H NMR, but there was an additional signal at ~ 160 ppm in the
13C-NMR spectrum. Based on the results in
Table 1, we hypothesized that the mass loss occurring in the intermediate temperature region (300–600 °C) was most likely to interfere with loss of functional groups from the modified ZnO samples. The loss of material below 300 °C and above 600 °C are likely to be below and above the temperature for functional group loss, based on previous studies.
3.2. qNMR and TGA Analysis of APTES-Functionalized ZnO NPs
The surface loading for APTES-functionalized NPs was first quantified by qNMR. Initial experiments focused on determining appropriate conditions to remove the covalently attached surface groups for qNMR analysis (
Tables S1–S3). Our previous studies using NMR for quantification of functional groups on silica were based on the dissolution of the silica matrix in basic D
2O (NaOD 0.4 M) which released the functional groups for solution NMR analysis and quantification by comparison to an internal standard. Since ZnO is insoluble in basic solution [
26] we tested a comparable approach based on dissolving APTES-functionalized zinc oxide in acidic D
2O (DCl 0.4 M). This did not always lead to the full dissolution of the powder; for example NAM-03-NH
2 produced a cloudy sample from which solids could be recovered by centrifugation (
Table S1). In another case (USRN-05-NH
2) the NMR spectra showed broad resonances (
Figure 3A) which we previously explained as an indication of insufficient dissolution with the 3-aminopropylsiloxane moiety existing in polymerised form [
19]. Note that the signals are much weaker for the other APTES-functionalized NPs, making it difficult to assess whether the NMR signals are also broadened.
To explore the viability of the previous protocol using basic hydrolysis of siloxane bonds, the samples were treated with 0.4 M NaOD for various periods of time at 45 °C (data summarized in
Tables S1–S3). The aim was to release 3-aminopropylsiloxane from the NP surface and separate the supernatant from the remaining ZnO solids. This approach provided NMR spectra with well-defined signals (
Figure 3B). In order to test whether a single extraction was quantitative some samples were extracted with base a second time or extracted for a longer period of time. For USRN-05-NH
2 (which has a high APTES loading) this led to recovery of an additional minor amount of 3-aminopropylsiloxane. The quantification of APTES for the various ZnO samples using the basic hydrolysis method is summarized in
Table 2. The comparison of data for the acidic hydrolysis and the basic hydrolysis, followed by NMR of the supernatant, are summarized in
Tables S1–S3. The amount of recovered functional group is similar for the two methods for the two USRN samples, despite the evidence for incomplete dissolution for the acidic hydrolysis. However, acidic hydrolysis recovers 28% less functional group for NAM-03-NH
2. Based on the various tests, we conclude that basic hydrolysis and qNMR of the supernatant is the preferred method, although it does require a second hydrolysis step to ensure complete recovery of the functional group, particularly for samples with high amine loading.
As shown in
Table 2, the amine loading is relatively low for two of the APTES-modified samples, with 15 and 35 μmol/g for USRN-04-NH
2 and NAM-03-NH
2, respectively. A ten-fold higher loading of 454 μmol/g was obtained for USRN-05-NH
2. The variability in loading level for samples from a single supplier is similar to our previous results for silica nanoparticles [
17,
19] and literature results for other nanomaterials [
18,
25,
27]. Note that the more than twenty-fold difference in 3-aminopropyl loading for USRN-05-NH
2 and USRN-04-NH
2 cannot be explained by differences in surface area since these samples have nominal diameters of 10–30 and 20 nm, respectively.
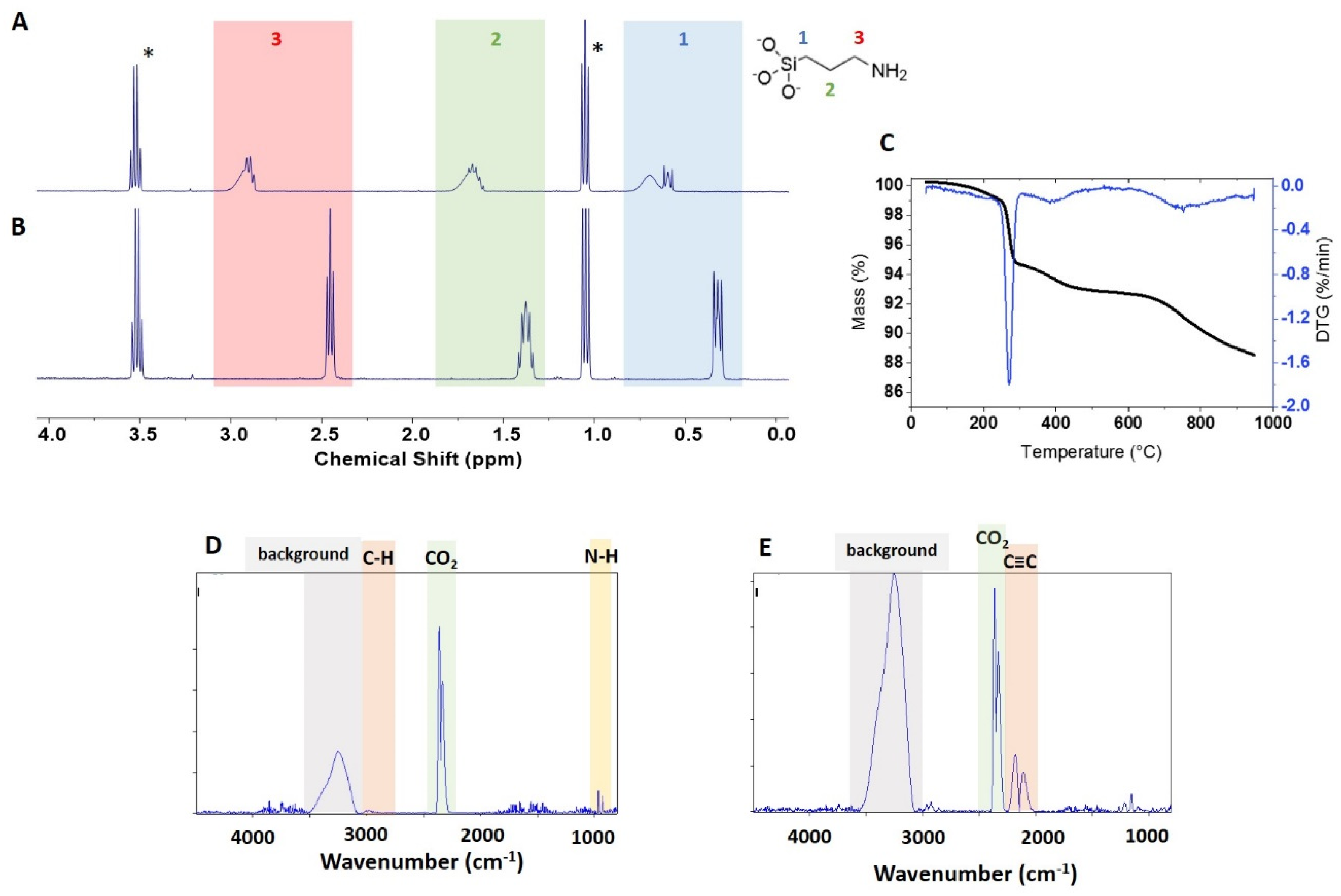
The APTES-functionalized samples were examined by TGA with the expectation that the sample with the highest loading should have a sufficient mass loss for reliable quantification. As shown in
Figure 3C, the mass loss for USRN-05-NH
2 occurs in three distinct regions, corresponding to 200–300 °C, 300–600 °C and >600 °C. The FT-IR spectra of evolved gases showed the presence of CO
2 and water (
Figure S5B), but the C–H bands expected for the loss of functional group were not detected in the expected region (
Figure 3D,E), probably due to low abundance. This is consistent with our previous experience with APTES-modified silicas [
20].
Based on the observation of the 260 °C mass loss for unmodified ZnO samples and the observation of a weak signal consistent with an amine (
Figure 3C), we hypothesized that the 300–600 °C mass loss was most likely to be due to to loss of APTES from the surface. The mass loss in this region was estimated to correspond to 381 μmol/g of APTES, in reasonable agreement with the qNMR data (454 μmol/g). Correction of the APTES mass loss using data for NPs of the same size gives a value of 299 μmol/g, considerably lower than the qNMR value and suggesting that the bare sample does not provide a reliable correction. The mass loss at >600 °C is much larger for this sample than the modest changes (~ 0.2%) observed for most of the unmodified ZnO NPs. The FT-IR spectra in this region show typical CO
2 signals and a minor 2000–2300 cm
-1 band which occurs in this temperature region in other functionalized samples (
Figure 3E). Although it was not possible to identify the probable chemical structure of this component by comparison to the spectral library, a number of molecules with unsaturated C≡C bonds show signals in this region. Note that the large background signal between 3000–3500 cm
-1 is due to ice condensation in the detector and makes it impossible to detect other signals in this region.
TGA curves for the remaining APTES-functionalized samples show smaller mass loss in the 300–600 °C range (
Figure S2). For USRN-04-NH
2 the mass loss in this region is 0.9%, the same as that for the mass loss in the equivalent-size bare sample (USRN-02,
Table 1). This suggests that that amine loading is low, as observed by NMR (~15 μmol/g); the entire mass loss of 0.9% would correspond to an amine content of ~154 μmol/g). Similarly for NAM-03, the mass loss is 0.36%, similar to the value of 0.41 for a bare sample of the same size from the same supplier. Note that one could possibly improve on the correction if the precursor of the modified sample was available. Both of these samples show only a small mass loss above 600 °C that is similar to that in unmodified samples, suggesting that the mass loss at high temperatures for USRN-04-NH
2 is somewhat anomalous.
3.3. qNMR and TGA Analysis of Caprylsilane Modified ZnO NPs
Two caprylsilane-functionalized samples were examined using a method similar to the one optimized for APTES-modified ZnO, but with methanol-d
4 as solvent in order to solubilize the more hydrophobic alkyl group.
Tables S4 and S5 summarize the conditions that were investigated to optimize the hydrolysis and release the caprylsilane moiety into the solution. Multiple concentrations of NaOD were tested in order to achieve quantitative hydrolysis while maintaining a sufficiently hydrophobic solvent to solubilize the released caprylsilane. NaOD concentrations of 0.2, 0.4 and 0.8 M were tested with reaction times of 24 and 48 h at 45 °C. A second and third hydrolysis step were also tested in some experiments. Overall the highest recovery of functional group was for samples with 2 hydrolysis steps of 24 h in 0.4 M NaOD, with an additional third hydrolysis step yielding only traces of caprylsilane (
Table S4). The recovery obtained for a single 78-h extraction with either 0.4 M or 0.8 M NaOD was ~10% lower, whereas the use of 0.2 M NaOD gave 50–60% of the maximum recovery. This indicates that base concentration rather than time is the more important factor for quantitative release of the caprylsilane. Higher temperatures (85 °C) led to deposition of material on the walls of the glass ampoule and low recovery of the caprylsilane.
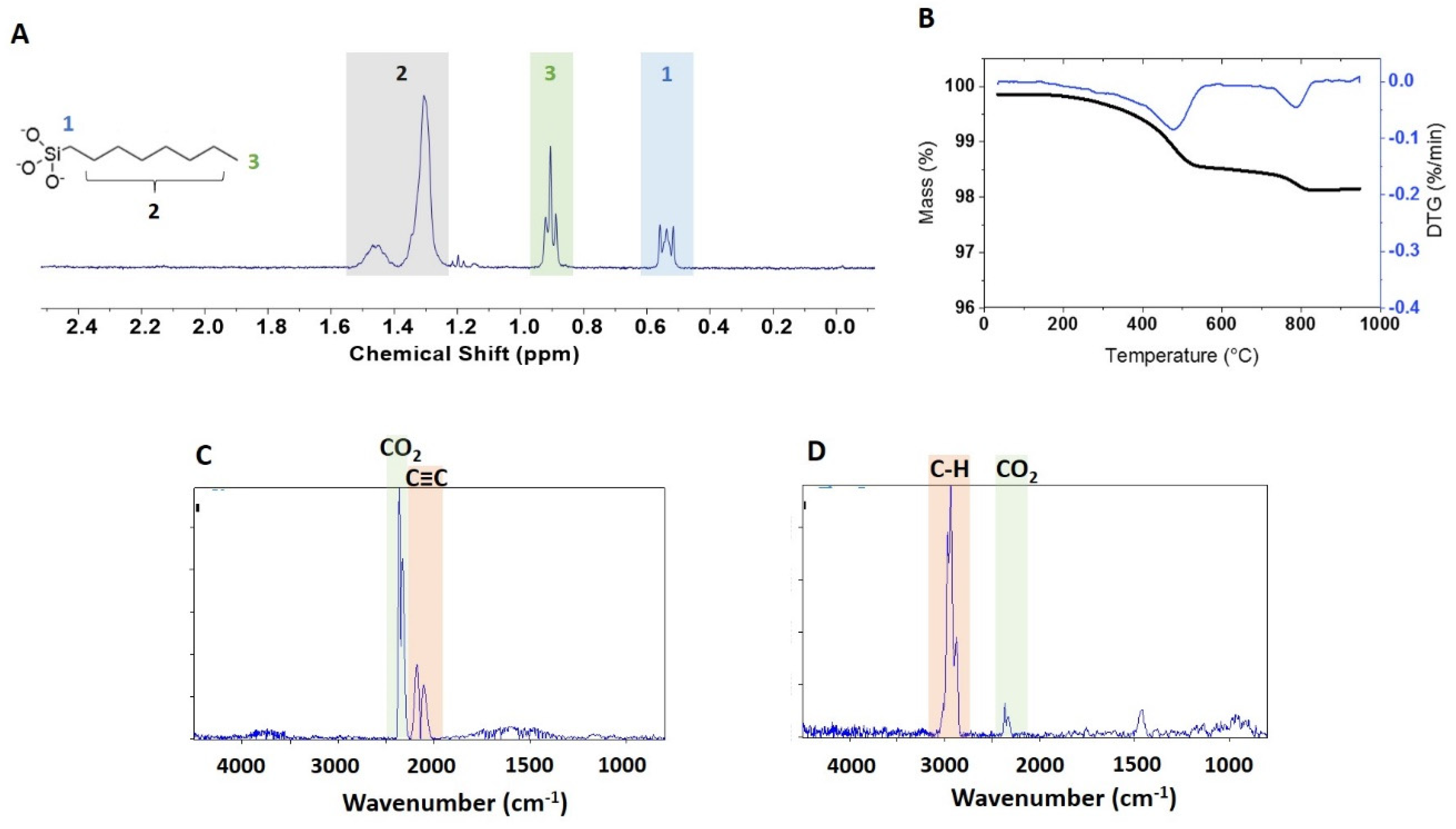
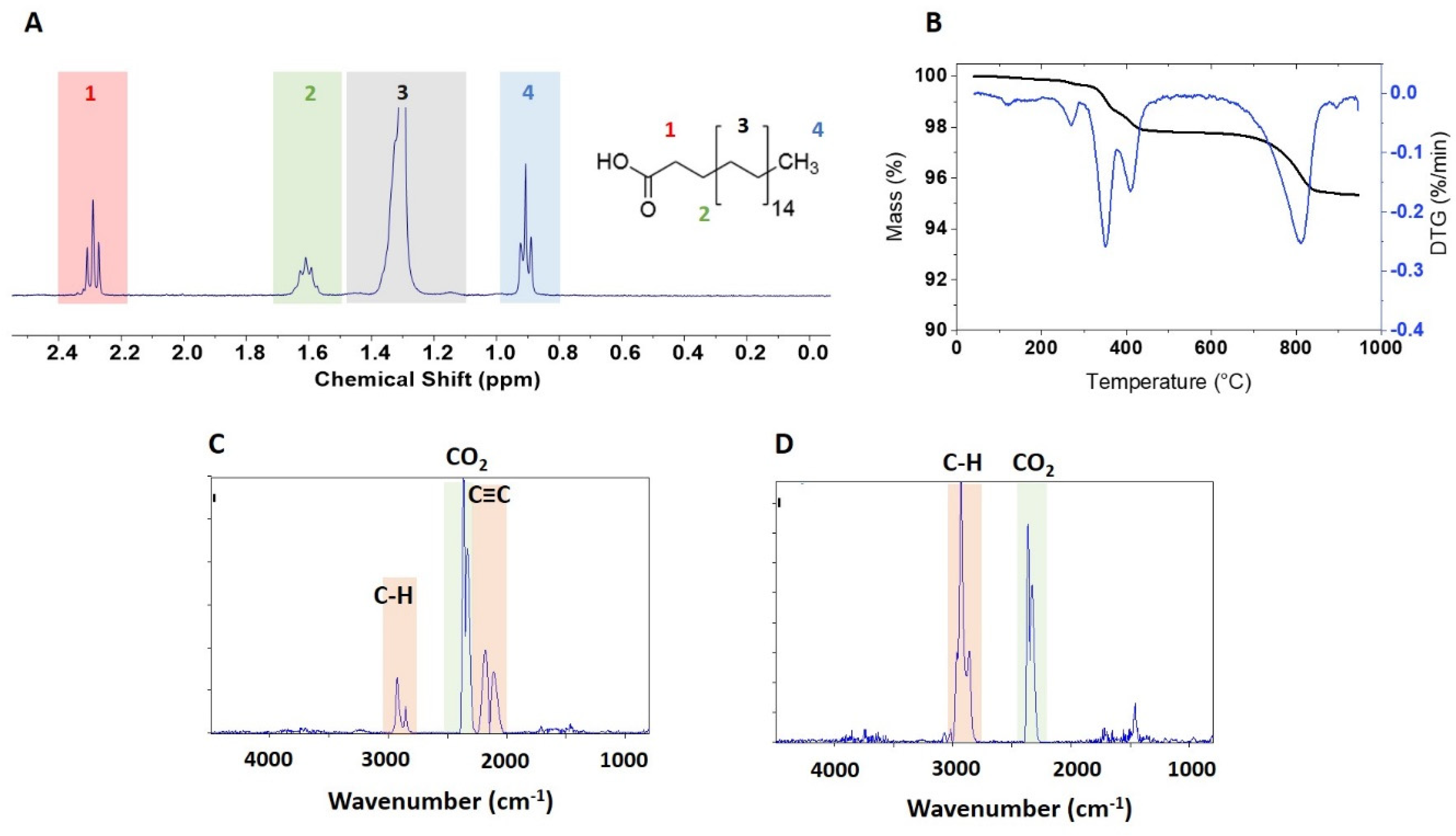
A qNMR spectrum of the extracted functional group is shown in
Figure 4A for BASF-02-C8. The JRC-02-C8 and BASF-02-C8 samples (both from supplier BASF) gave the same caprylsilane loading (101, 102 μmol/g),
Table 2, suggesting that the surface modification reaction is reproducible for these samples. TGA for these two samples showed mass losses between 300 and 600 °C with an inflection point at ~ 475 °C (
Figure 4B and
Figrue S3). Making the assumption that this corresponds to loss of the caprylsilane, the mass losses correspond to 98 and 93 umol/g for JRC-02-C8 and BASF-02-C8, after correction for the small mass loss in this region for the corresponding bare samples. The agreement with the qNMR data supports our hypothesis that the mass loss between 300 and 600 °C can be assigned to the caprylsilane. FT-IR spectra for BASF-02-C8 showed C–H signals at 2700–3000 cm
-1 at temperatures between 300–600 °C (
Figure 4C and
Figrue S5C), consistent with the loss of alkyl groups, further confirmation that this component is due to loss of the caprylsilane. Neither caprylsilane-modified ZnO sample shows the clear signature for mass loss at 260 °C that was observed for some other samples and there was no detectable loss of CO
2 at this temperature in the FT-IR spectra (
Figure S5C). However, there is additional mass loss at high temperatures that is of variable intensity for the samples examined and that varies from run to run for the same sample. The FT-IR spectra show a large signal due to CO
2 and the previously mentioned bands at 2000–2300 cm
-1 (
Figure 4D). Note that the original characterization data reported a mass loss of ~ 1% for JRC-02-C8 [
21], whereas a later study of the JRC sample and two other batches of the same material reported 2-step mass losses between 2.1 and 2.4% [
25].
3.4. qNMR and TGA Analysis of Stearic Acid Modified ZnO NPs
A number of conditions were tested to remove the surface coating from stearic acid modified ZnO NPs. Initial experiments tested multiple extraction steps (each 24 h at 45 °C) using DMSO and methanol-d
4 but, ≤10 μmol/g stearic acid was recovered (
Table S6). A modified extraction procedure using pyridine-d
4 in methanol-d
4 gave a higher recovery of stearic acid from ZnO NPs (43 µmol/g), although this value was still lower than the estimate from TGA (see below) [
28]. This may be caused by the proposed strong chemical interaction between ZnO and stearic acid, since some literature reports indicate that stearic acid is covalently bonded to ZnO [
29]. Therefore, we investigated a two-phase method in which stearic acid modified ZnO was first dispersed in an organic solvent (toluene, cyclohexane, and chloroform tested) and then combined with HCl (0.4 M) followed by 24 h reflux. After this, both layers appeared transparent, indicating the disintegration of starting material. Due to its high partition coefficient (log P
O/W = 8.23), stearic acid should be concentrated in the organic layer. However, after solvent evaporation, NMR analysis revealed the presence of multiple impurities that interfered with the diagnostic signals of stearic acid. These impurities may have been introduced during the reflux or be due to traces of organic solvent and therefore this method was not suitable for quantitative analysis.
An alternate method based on dissolution of the ZnO matrix in mixtures of methanol-d
4 and trifluoroacetic acid (TFA-d) was tested (
Tables S7–S9). This limited the sample manipulation and excluded non-deuterated solvents. The initially cloudy dispersion became transparent which suggests that the core nanomaterial had dissolved. The use of an organic acid is necessary because stearic acid is insoluble in methanol containing even low fractions of water (>5%). Integration of the acquired NMR spectra confirmed the chemical structure of stearic acid and gave values for stearic acid content that were similar to the TGA results (see below). The values obtained by dissolution of three commercial stearic acid-modified ZnO NPs in TFA- methanol-d
4 are provided in
Table 2. This method gave values with relatively high standard deviation for two USRN samples, although the repeatability was reasonably good for Sky-02-C18. In order to further confirm the suitability of the method, a bare ZnO sample (BASF-01) was modified with stearic acid (NRC-01-C18) and analyzed by qNMR (
Figure 5A,
Table 2,
Table S10). This demonstrated recovery of ~90% of the initial stearic acid used for surface modification at several TFA concentrations. Based on this it appears that the TFA concentration used for the experiment is not critical as long as it facilitates dissolution of ZnO. The relatively high standard deviation in the two USRN samples is independent of TFA concentration, and appears to be specifically related to samples from this manufacturer.
TGA of the four samples modified with stearic acid showed mass loss in 3 regions, 200–300 °C, 300–600 °C and >600 °C,
Figure 5B and
Figure S4, although with intensities that differed from sample to sample. The low temperature region has a clear maximum at 260 °C for all samples, with a mass loss that varies from ~0.2% (NRC-01-C18) to ~5% (USRN-07-C18, Sky-02-C18); USRN-06-C18 has a small shoulder at ~170 °C that may be due to water or solvent (
Figure S4A). As for other samples described above, the FT-IR spectra of NRC-01-C18 and USRN-07-C18 are dominated by loss of CO
2 and water at low temperatures (
Figure S5D,E). Interestingly the unmodified sample BASF-01 does not show a maximum at 260 °C, but there is a clear, although small, mass loss in this region for the stearic acid-modified sample. A control sample prepared by treating BASF-01 with the stearic acid modification conditions in the absence of stearic acid had a mass loss of ~ 0.1% between 200–300 °C.
The middle temperature region (300–600 °C) has maxima at 355 and 405 °C that are assigned to loss of stearic acid, based on the loss of other organic groups in this region and on the observation of bands consistent with alkyl groups between 2700–3000 cm
-1 at a temperature of 403 °C in the FT-IR spectra (
Figure 5C,
Figures S5D,E and S6A). The magnitude of the mass loss above 600 °C is variable for multiple runs of the same sample. Consistent with other functionalized samples, signals at 2000–2300 cm
-1 start to emerge in the FT-IR spectra for both NRC-01-C18 and USRN-07-C18 above 600 °C (
Figure 5D, S6B). These signals are of higher intensity for NRC-01-C18 than for the commercial samples and there is a low intensity alkyl band at 2700–3000 cm
-1 at temperatures above 600 °C (
Figure 5D). However, the continuous release of high boiling point components in the FT-IR may not be related to their desorption from the analysed material; this is more likely to be due to the condensation of these compounds in the TGA-FT-IR transfer line and their slow release at later times during the TGA run. The control sample of BASF-01 that was subjected to the same surface modification conditions in the absence of stearic acid showed only a modest mass loss of 0.34% between 200 and 500 °C.
3.5. X-Ray Photoelectron Spectroscopy
XPS data was obtained for selected samples to provide further confirmation of the conclusions from NMR and TGA and to seek additional insight on the source of the mass loss at 260 °C for both bare and modified ZnO and the high temperature mass loss that was significant for several samples. Survey XPS scans for bare ZnO powders (see
Table S11) indicated that the main elements present are Zn, O and C, with a small Cl signal (~1–3% atomic composition). Chlorine impurities have previously been reported in XPS of ZnO NPs [
25] and are likely due to the procedure used for NP synthesis. While survey scans are useful to rapidly obtain information on the atomic composition of the sample and identify the presence of impurities, high resolution scans of the strongest core levels associated with the major constituents were used for further quantitative analysis as summarized in
Table 3. The O/Zn atomic composition ratios are close to 1 (0.99 to 1.06), as expected; a notable exception is USRN-02 which had a significantly higher O/Zn ratio (1.18). All samples showed the presence of a substantial amount of carbon with most of the samples exhibiting C/Zn ratios between 0.39 and 0.67 with no clear correlation between this ratio and the total mass loss measured by TGA for these samples. However, the largest carbon content was for USRN-02 which also had a high O/Zn ratio and a large mass loss at 260 °C, suggesting that this mass loss corresponds to a species containing both C and O. As XPS is a surface sensitive technique and sensitive to adventitious C contamination, as observed previously for silica NPs [
17], we assume that the observed C/Zn ratios reflect a mixture of C contamination and carbon-containing impurities in these ZnO samples.
The high-resolution scans of the C1s and O1s regions, shown in
Figure 6, also reveal additional information on the nature of the chemical species present in these samples. The C1s region is dominated by a peak with maximum intensity at 285 eV along with a smaller peak at 289 eV (
Figure 6). Standard curve fitting procedures were used to decompose the spectra into several components, reflecting the different bonding environments for carbon present on the sample. The main peak at 285 eV (C1) is characteristic of carbon-carbon or carbon-hydrogen bonding whereas the shifted peaks (C2 at 286eV, C3 at 289 eV and C4 at 290 eV) are assigned to carbon bound to oxygen. While all the samples exhibited C1, C2 and C3 components, the broad C4 feature was only observed in three of the samples, including NAM-01 in
Figure 6.
The fraction of total carbon assigned to each of the four signals is summarized in
Table 3. Based on literature precedent we hypothesize that the C3 and C4 signals are due to carbonate as a range of carbonates have XPS signals at ~ 289–290 eV [
30,
31]. Carbonate could be introduced by the synthetic procedure used (which is not divulged by the manufacturer) or by adsorption of carbon dioxide [
32,
33]. An earlier study had provided FT-IR evidence for the formation of surface ZnCO
3, showing that the amount increased with time [
32]. In that study an XPS signal at 286.5 eV was assigned to the carbonate, although this is inconsistent with XPS data for a range of carbonates. In the present work, the BASF-01 sample did exhibit a greater fraction of its carbon signal in the C3 and C4 states in a second measurement performed at a later time, indicating that C–O species may be accumulating on the sample. The O1s region (
Figure 6,
Table 3) can be fit with two components at 530.5 (O2) and 532 eV (O3) which are assigned to metal oxide and O–C containing moieties (such as carbonates), respectively. Whereas USRN-02 showed an almost equal fraction of O2 (52%) and O3 (48%) signals, for all the other bare particles O2 (58-62%) was significantly higher than O3. This is consistent with USRN-02 exhibiting a greater degree of carbonate (or other C–O containing impurity).
Surface-modified ZnO NPs showed changes that are consistent with introduction of the expected functional groups (
Table 4,
Table S12). Detection of Si in the silane modified samples was complicated by the presence of Zn plasmon loss features associated with Zn 3p and Zn 3s peaks that are adjacent to the Si 2p and Si2s peaks. Despite this complication Si signals are clearly observed for both APTES and caprylsilane functionalized samples in high resolution spectra (
Figure S7). The sharper Si features were separated from the broad Zn plasmon losses for quantification. As seen in
Table 4, Si/Zn ratios extracted using the Si 2s and Si 2p peaks are in reasonable agreement. The caprylsilane and APTES samples exhibit similar Si signals, indicative of similar degrees of functionalization. This is consistent with the qNMR and TGA data once the results are converted to surface coverage of functional groups on the particles. For the APTES samples a N1s signal is also observed with the N/Zn ratio comparable to the Si/Zn ratio as expected. O/Zn ratios were higher (1.12–1.45) than those measured for all bare samples, except USRN-02. Similarly, the C/Zn ratios were higher than for bare ZnO samples from the same manufacturer, with the highest values obtained for stearic acid modified samples. The presence of adventitious carbon contamination, as noted for the bare samples above as well as in our previous study of silica particles [
17], complicates the use of the C/Zn ratio for quantification of the surface functional groups. However, comparing the C/Zn ratios for the three different stearic acid modified particles shows a trend that mirrors the NMR and TGA results with USRN-06-C18 showing the highest degree of stearic acid loading. The C/Zn ratios were similar for the two caprylsilane modified samples, again consistent with the Si/Zn ratios and the NMR and TGA data, although the O/Zn ratio varied. For APTES, the C/Zn increased by a smaller amount and repeat measurements showed higher variability, presumably due to the lower carbon content in the functional group.
As for the bare particles, the high-resolution spectra in the C1s and O1s regions were analyzed to identify contributions of different chemical species to the overall carbon and oxygen signals. The spectra and fits are shown in
Figure 7 and
Figure S7 and the results are summarized in
Table 4. In the C1s region, the modified particles all exhibited a large C1 (285 eV) signal which was usually the majority contribution (50–70%). Most of the samples also exhibited C2 and C3 features, although the latter signal was not observed for either of the two caprylsilane-modified samples, even though it was evident in the bare samples from the same source. However, BASF-02-C8 had an additional shoulder at lower binding energy, C0 (283.5 eV), shown in
Figure S8. This feature is assigned to Zn-C [
34], but was not detected in the other caprylsilane-modified sample (JRC-02).
There is again no obvious correlation with the TGA results since BASF-02-C8 had the lower overall mass loss and C/Zn ratio, despite the presence of this new XPS signal. The BASF-02-C8 particles also exhibit unusual features in the Zn2p region. The Zn2p signal for ZnO exhibits two peaks separated by 23 eV due to spin-orbit splitting. For most of the particles studied here the Zn region can be fit with two components Zn2p3/2 and Zn2p1/2 with the latter having half the intensity of the former. For BASF-02-C8, this region is best fit with an additional spin-orbit pair shifted to lower binding energy by ~2 eV (see
Figure S9). This feature is not observed on JRC-02-C8 or any of the bare particles but has been seen for C doped ZnO produced by calcination [
34]. The observation of signals consistent with Zn-C bonds in both the Zn2p and C1s regions provides strong evidence for the formation of carbon-doped ZnO during TGA analysis although it is not clear why such species are only formed for BASF-02-C8. This sample does exhibit a new lower energy signal O1 (529 eV) in the O1s region. Less intense C0 and O1 features are also observed in the first measurement of USRN-05-NH
2 (but not in the 2
nd measurement) suggesting it may be due to extrinsic contamination.
Figure 7 shows the spectra for the three stearic acid modified samples. Interestingly, USRN-07-C18, the sample with the large 260 °C TGA mass loss has the largest C3 signal and the largest O3 signal in the C1s and O1s regions, respectively. This further supports our assignment of this species to a carbonate.
3.6. In Situ Quantification of Stearic Acid Using ssNMR
Although ssNMR is a time-consuming method, it is one of the few methods that provides both structural and quantitative information for the intact nanomaterial. Therefore, two of the stearic acid modified samples were also examined by
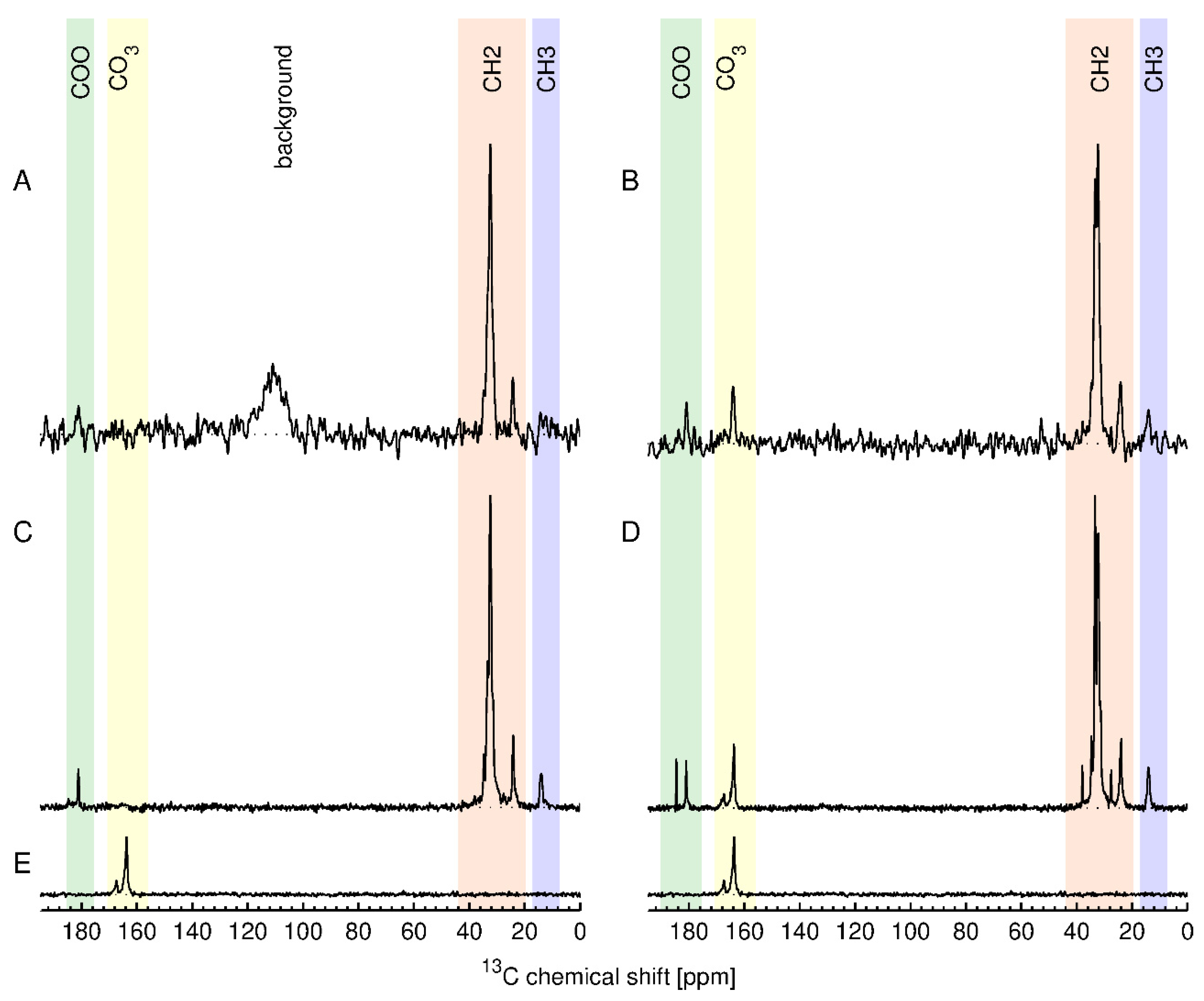
13C ssNMR, along with one unmodified ZnO. We reasoned that the high total carbon content for the stearic acid modified samples should give the maximum sensitivity for direct detection of the functional group. The results are shown in
Figure 8 together with their assignment. The top row (A, B) shows
13C ssNMR spectra obtained using the inherent direct polarization (DP) of the
13C nuclei in the external magnetic field. The obtained spectral signal intensity is directly proportional to the number of
13C nuclear spins contributing to the respective signal. This allows to absolutely quantify the different components in the sample by comparing the signal intensity to an external standard [
35]. The second and third rows (C, D, E) in
Figure 8 show
13C cross-polarization (CP) spectra, which are obtained by transferring nuclear polarization from the abundant protons to the
13C nuclei, significantly enhancing the sensitivity of the resulting
13C spectra. However, these spectra are in general not absolutely quantitative. The spectra of USRN-06-C18 show the expected signals for stearic acid whereas the spectra of USRN-07-C18 have additional signals in the COO and the CH
2 region. Furthermore, the spectra of both USRN-07-C18 and unmodified ZnO show signals at 164 and 167 ppm that were assigned to carbonate and/or bicarbonate ions on the ZnO surface. The presence of additional ssNMR signals is consistent with the large mass loss at 260 °C for this sample, indicating that the low temperature mass loss is likely to be due to the presence of carbonate. The concentration of the stearic acid groups in USRN-06-C18 and USRN-07-C18 was determined to be 172 µmol/g and 277 µmol/g, respectively, obtained by integration of the CH
2 signals in the direct polarization experiment. The estimate for USRN-06-C18 is slightly lower than the qNMR value (208 ± 18 µmol/g,), although that for USRN-07-C18 is significantly higher than that determined by qNMR (185 ± 34 µmol/g,). Note that the signal-to-noise ratio for the DP experiment will result in an error on the order of 7–10% (20–30 µmol/g) which may partially account for these differences.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}