Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation


Abstract
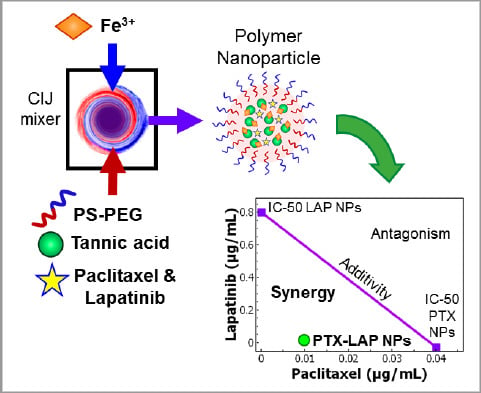
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Nanoparticle Formulation
2.4. Nanoparticle Characterization
2.5. Nanoparticle Drug Release In Vitro
2.6. Cytotoxicity
2.7. Cell Cycle Analysis by Flow Cytometry
3. Results and Discussion
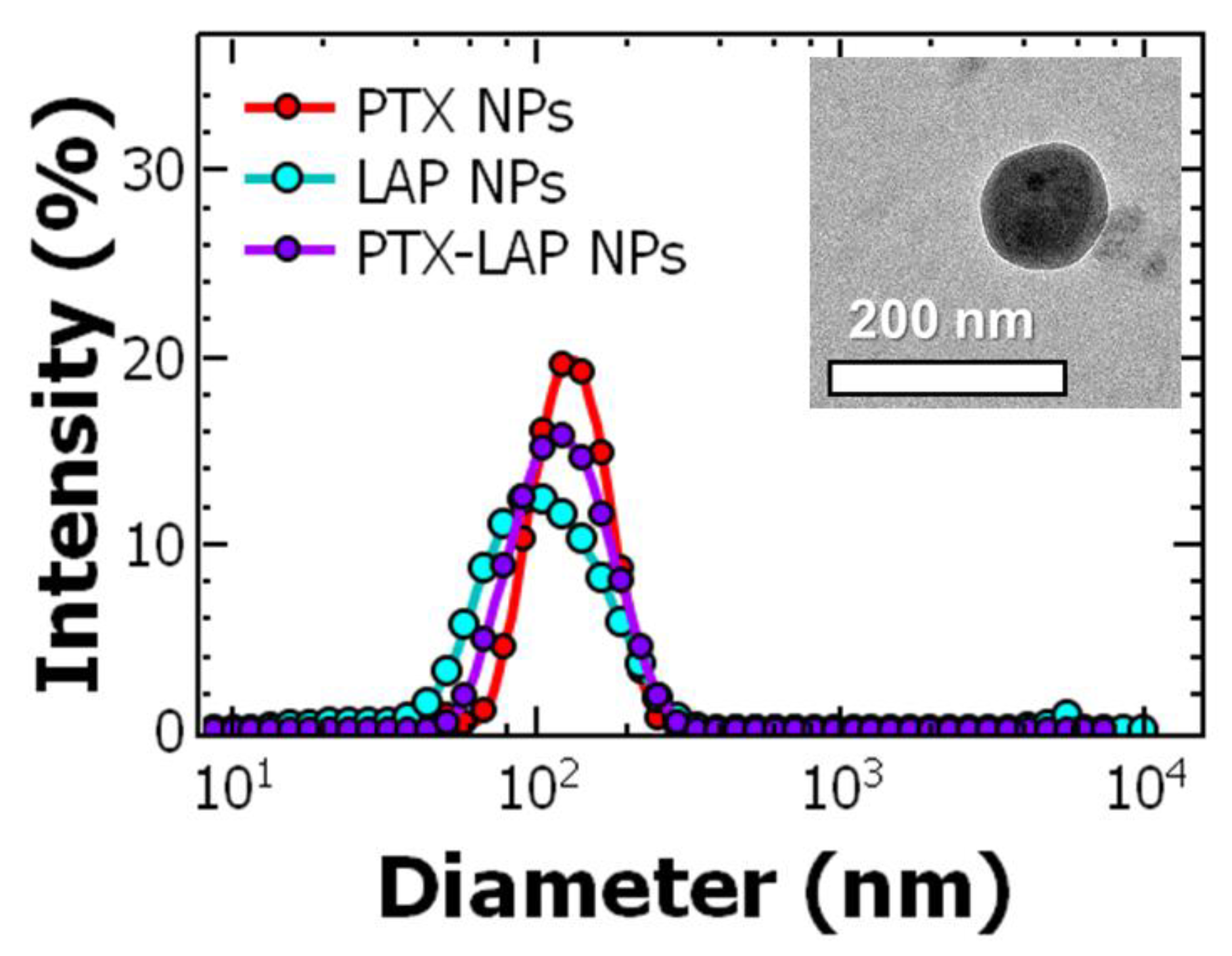
3.1. Nanoparticle Preparation and Characterization
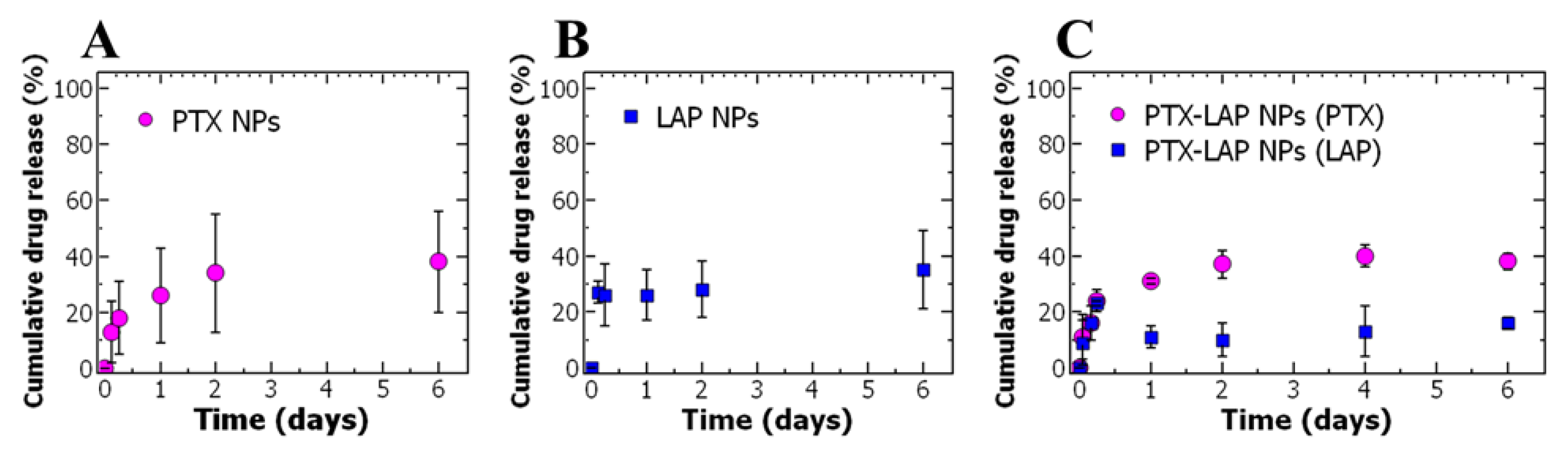
3.2. Drug Release
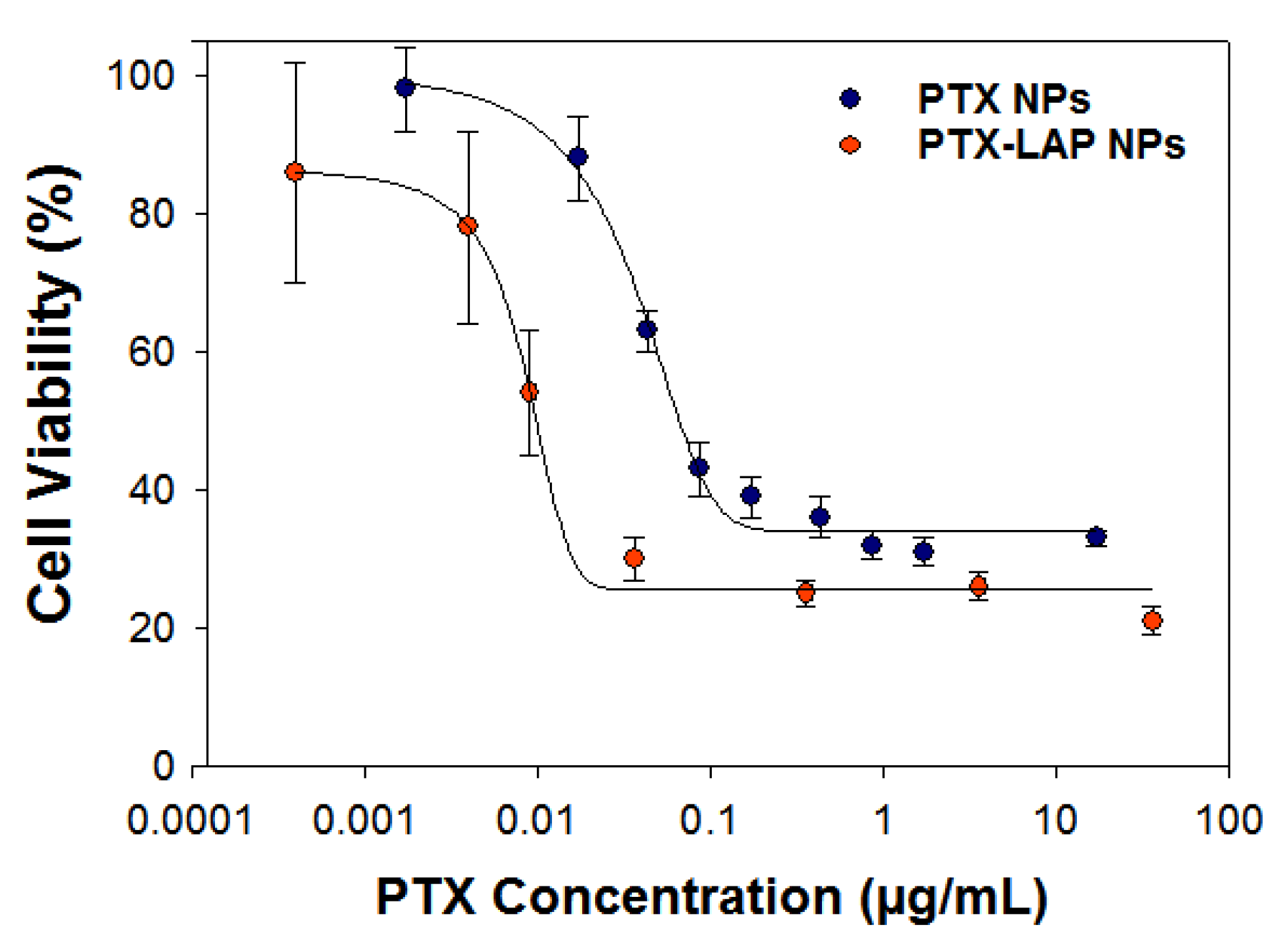
3.3. Assessing Drug Efficacy of Single-Drug Nanoparticles
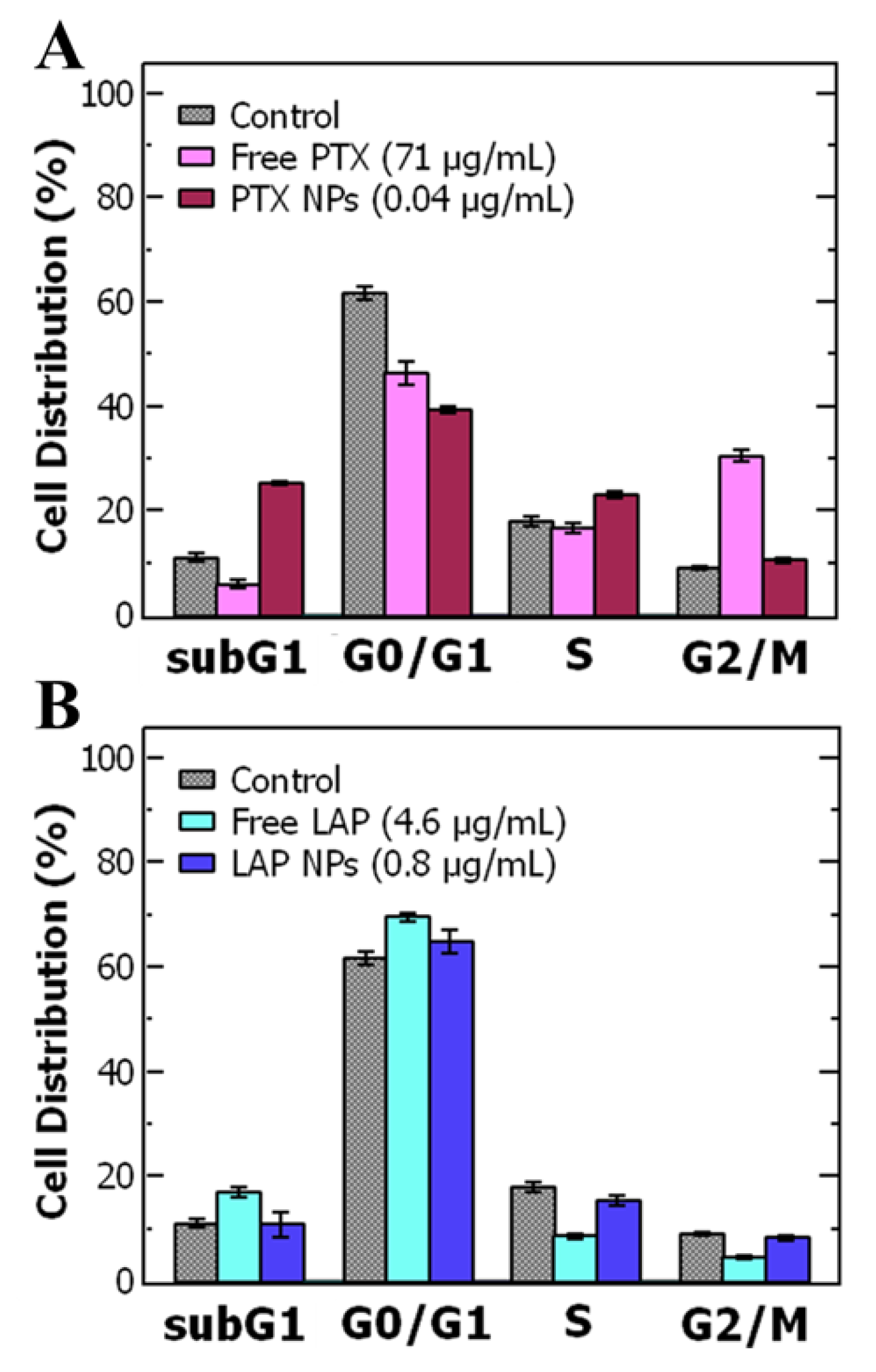
3.4. Cell Cycle Analysis
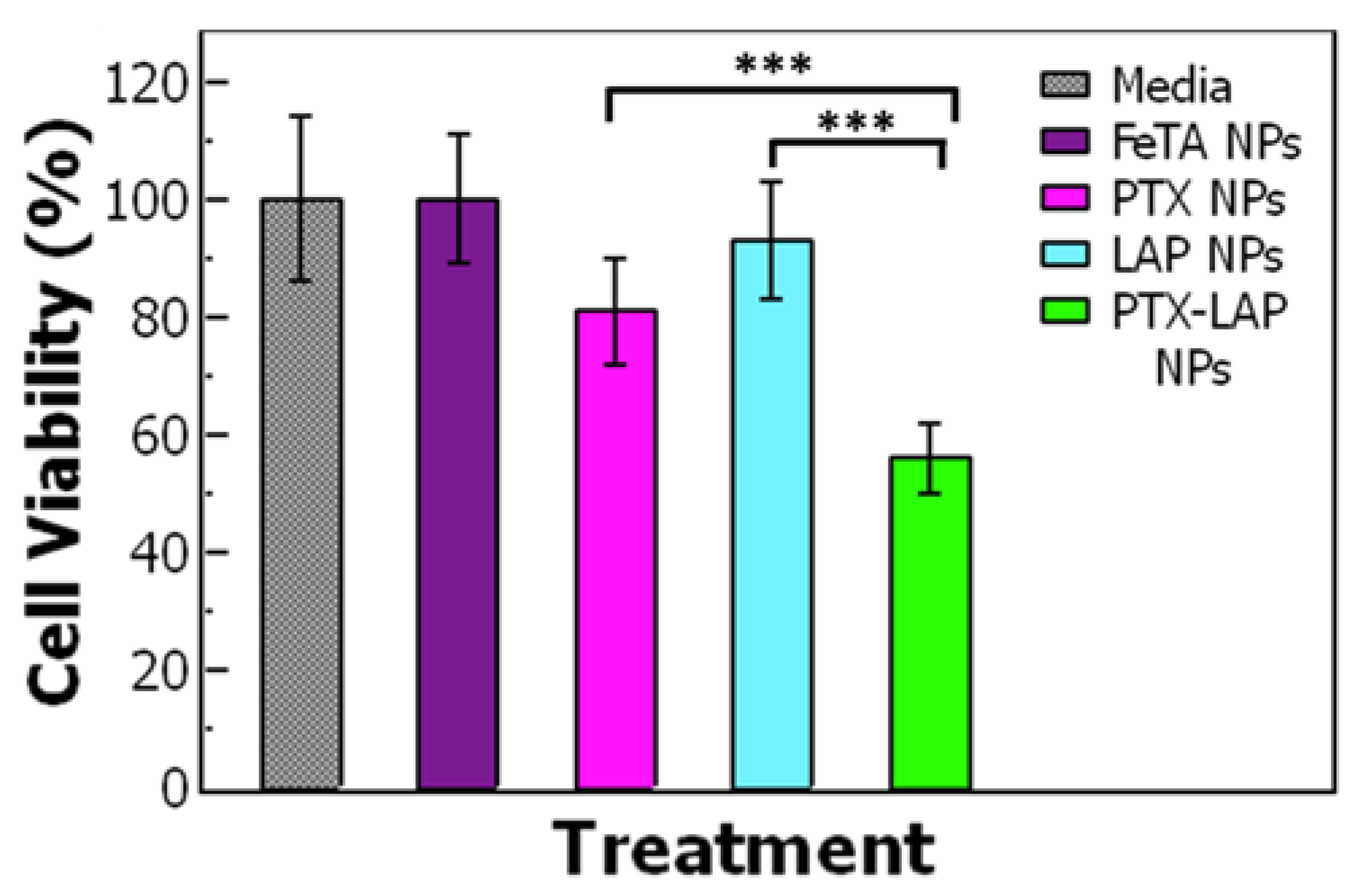
3.5. Drug Combination and Synergy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Ueno, N.T.; Mamounas, E.P. Neoadjuvant nab-paclitaxel in the treatment of breast cancer. Breast Cancer Res. Treat. 2016, 156, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.S.; Hotte, S.J.; Chin, J.L.; Hudes, G.R.; Gregg, R.; Trachtenberg, J.; Wang, L.; Tran-Thanh, D.; Pham, N.-A.; Tsao, M.-S.; et al. A multicenter phase II clinical trial of lapatinib (GW572016) in hormonally untreated advanced prostate cancer. Am. J. Clin. Oncol. 2010, 33, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Krauss, N.E.; Heerding, J.M.; Swindell, C.S.; Ringel, I.; Orr, G.A.; Horwitz, S.B. 3’-(p-azidobenzamido)taxol photolabels the N-terminal 31 amino acids of beta-tubulin. J. Biol. Chem. 1994, 269, 3132–3134. [Google Scholar] [PubMed]
- Rao, S.; He, L.; Chakravarty, S.; Ojima, I.; Orr, G.A.; Horwitz, S.B. Characterization of the Taxol binding site on the microtubule. Identification of Arg(282) in beta-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J. Biol. Chem. 1999, 274, 37990–37994. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef]
- Vergara, D.; Bellomo, C.; Zhang, X.; Vergaro, V.; Tinelli, A.; Lorusso, V.; Rinaldi, R.; Lvov, Y.M.; Leporatti, S.; Maffia, M. Lapatinib/Paclitaxel polyelectrolyte nanocapsules for overcoming multidrug resistance in ovarian cancer. Nanomedicine 2012, 8, 891–899. [Google Scholar] [CrossRef]
- Van Zuylen, L.; Verweij, J.; Sparreboom, A. Role of formulation vehicles in taxane pharmacology. Investig. New Drugs 2001, 19, 125–141. [Google Scholar] [CrossRef]
- Markman, M.; Kennedy, A.; Webster, K.; Kulp, B.; Peterson, G.; Belinson, J. Phase I Trial of Paclitaxel Plus Megestrol Acetate in Patients with Paclitaxel-refractory Ovarian Cancer. Clin. Cancer Res. 2000, 6, 4201–4204. [Google Scholar]
- Varma, M.V.S.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef]
- Yagi, H.; Yotsumoto, F.; Sonoda, K.; Kuroki, M.; Mekada, E.; Miyamoto, S. Synergistic anti-tumor effect of paclitaxel with CRM197, an inhibitor of HB-EGF, in ovarian cancer. Int. J. Cancer 2009, 124, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; An, S.-J.; Zhang, X.-C.; Dong, S.; Zhang, Y.-F.; Chen, Z.-H.; Chen, H.-J.; Guo, A.-L.; Lin, Q.-X.; Wu, Y.-L. In vitro sequence-dependent synergism between paclitaxel and gefitinib in human lung cancer cell lines. Cancer Chemother. Pharmacol. 2011, 67, 637–646. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, T. Alteration in the Balance of Prosurvival and Proapoptotic Signalling Pathways Leads to Sequence-Dependent Synergism Between Docetaxel and Sorafenib in Human Non-small Cell Lung Cancer Cell Lines. Cell Biochem. Biophys. 2014, 68, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Gomez, H.L.; Aziz, Z.; Zvirbule, Z.; Bines, J.; Arbushites, M.C.; Guerrera, S.F.; Koehler, M.; Oliva, C.; Stein, S.H.; et al. Phase III, Double-Blind, Randomized Study Comparing Lapatinib Plus Paclitaxel With Placebo Plus Paclitaxel As First-Line Treatment for Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Xu, R.-H.; Chung, H.C.; Sun, G.-P.; Doi, T.; Xu, J.-M.; Tsuji, A.; Omuro, Y.; Li, J.; Wang, J.-W.; et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN—A randomized, phase III study. J. Clin. Oncol. 2014, 32, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Schilder, R. Novel Approaches in Advancing the Treatment of Epithelial Ovarian Cancer: The Role of Angiogenesis Inhibition. J. Clin. Oncol. 2007, 25, 2894–2901. [Google Scholar] [CrossRef]
- Teplinsky, E.; Muggia, F. Targeting HER2 in ovarian and uterine cancers: Challenges and future directions. Gynecol. Oncol. 2014, 135, 364–370. [Google Scholar] [CrossRef]
- Kondo, N.; Tsukuda, M.; Ishiguro, Y.; Kimura, M.; Fujita, K.; Sakakibara, A.; Takahashi, H.; Toth, G.; Matsuda, H. Antitumor effects of lapatinib (GW572016), a dual inhibitor of EGFR and HER-2, in combination with cisplatin or paclitaxel on head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 957–963. [Google Scholar] [CrossRef][Green Version]
- Guo, X.; Li, S.; Zhu, X.; Dou, Q.; Liu, D. Lapatinib in combination with paclitaxel plays synergistic antitumor effects on esophageal squamous cancer. Cancer Chemother. Pharmacol. 2018, 82, 383–394. [Google Scholar] [CrossRef]
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Baker, J.R.; Van Echo, D.A.; Von Hoff, D.D.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.K.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Formulating Paclitaxel in Nanoparticles Alters Its Disposition. Pharm. Res. 2005, 22, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Danquah, M.; Singh, S.; Wu, H.; Mahato, R.I. Paclitaxel- and lapatinib-loaded lipopolymer micelles overcome multidrug resistance in prostate cancer. Drug Deliv. Transl. Res. 2011, 1, 420–428. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, S.; Wang, F.; Zou, A.; Zhang, S.; Xiong, Y.; Cao, S.; Zhang, Q.; Wang, Y.; Jiang, X. A Novel Combined Micellar System of Lapatinib and Paclitaxel with Enhanced Antineoplastic Effect Against Human Epidermal Growth Factor Receptor-2 Positive Breast Tumor In Vitro. J. Pharm. Sci. 2015, 104, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Ravar, F.; Saadat, E.; Kelishadi, P.D.; Dorkoosh, F.A. Liposomal formulation for co-delivery of paclitaxel and lapatinib, preparation, characterization and optimization. J. Liposome Res. 2016, 26, 175–187. [Google Scholar] [CrossRef]
- Dehghankelishadi, P.; Saadat, E.; Ravar, F.; Safavi, M.; Pordeli, M.; Gholami, M.; Dorkoosh, F.A. In vitro and in vivo evaluation of paclitaxel–lapatinib-loaded F127 pluronic micelles. Drug Dev. Ind. Pharm. 2017, 43, 390–398. [Google Scholar] [CrossRef]
- Pinkerton, N.M.; Grandeury, A.; Fisch, A.; Brozio, J.; Riebesehl, B.U.; Prud’homme, R.K. Formation of Stable Nanocarriers by in Situ Ion Pairing during Block-Copolymer-Directed Rapid Precipitation. Mol. Pharm. 2013, 10, 319–328. [Google Scholar] [CrossRef]
- Ganta, S.; Amiji, M. Coadministration of Paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol. Pharm. 2009, 6, 928–939. [Google Scholar] [CrossRef]
- Kelishady, P.D.; Saadat, E.; Ravar, F.; Akbari, H.; Dorkoosh, F. Pluronic F127 polymeric micelles for co-delivery of paclitaxel and lapatinib against metastatic breast cancer: Preparation, optimization and in vitro evaluation. Pharm. Dev. Technol. 2015, 20, 1009–1017. [Google Scholar] [CrossRef]
- Duong, A.D.; Ruan, G.; Mahajan, K.; Winter, J.O.; Wyslouzil, B.E. Scalable, semicontinuous production of micelles encapsulating nanoparticles via electrospray. Langmuir 2014, 30, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Prud’homme, R.K. Targeted Theragnostic Nanoparticles via Flash Nanoprecipitation: Principles of Material Selection. In Polymer Nanoparticles for Nanomedicines; Springer: Cham, Switzerland, 2016; pp. 55–85. ISBN 978-3-319-41419-5. [Google Scholar]
- Pustulka, K.M.; Wohl, A.R.; Lee, H.S.; Michel, A.R.; Han, J.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C.W. Flash nanoprecipitation: Particle structure and stability. Mol. Pharm. 2013, 10, 4367–4377. [Google Scholar] [CrossRef] [PubMed]
- Levit, S.L.; Stwodah, R.M.; Tang, C. Rapid, Room Temperature Nanoparticle Drying and Low-Energy Reconstitution via Electrospinning. J. Pharm. Sci. 2018, 107, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Amin, D.; Messersmith, P.B.; Anthony, J.E.; Prud’homme, R.K. Polymer Directed Self-Assembly of pH-Responsive Antioxidant Nanoparticles. Langmuir 2015, 31, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhu, Z.; Qian, H.; Wohl, A.R.; Beaman, C.J.; Hoye, T.R.; Macosko, C.W. A simple confined impingement jets mixer for flash nanoprecipitation. J. Pharm. Sci. 2012, 101, 4018–4023. [Google Scholar] [CrossRef] [PubMed]
- Understanding the Colloidal Stability of Protein Therapeutics Using Dynamic Light Scattering; Malvern Instruments Limited: Malvern, UK, 2014.
- Han, J.; Michel, A.R.; Lee, H.; Kalscheuer, S.; Wohl, A.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C. Nanoparticles Containing High Loads of Paclitaxel-Silicate Prodrugs: Formulation, Drug Release, and Anticancer Efficacy. Curr. Heart Fail. Rep. 2015, 12, 4329–4335. [Google Scholar] [CrossRef]
- Pagels, R.F.; Edelstein, J.; Tang, C.; Prud’homme, R.K. Controlling and Predicting Nanoparticle Formation by Block Copolymer Directed Rapid Precipitations. Nano Lett. 2018, 18, 1139–1144. [Google Scholar] [CrossRef]
- Tang, C.; Sosa, C.L.; Pagels, R.F.; Priestley, R.D.; Prud’homme, R.K. Efficient preparation of size tunable PEGylated gold nanoparticles. J. Mater. Chem. B 2016, 4, 4813–4817. [Google Scholar] [CrossRef]
- Tang, C.; Levit, S.; Zeevi, M.; Vasey, C.; Fromen, C. Chapter 12: Polymer Colloids Enable Medical Applications. In Polymer Colloids: Formulation, Characterization, and Applications; Royal Society of Chemistry: Cambridge, UK, 2019; pp. 358–398. [Google Scholar]
- Johnson, B.K.; Prud homme, R.K. Flash NanoPrecipitation of Organic Actives and Block Copolymers using a Confined Impinging Jets Mixer. Aust. J. Chem. 2003, 56, 1021–1024. [Google Scholar] [CrossRef]
- Zajdel, A.; Wilczok, A.; Jelonek, K.; Musiał-Kulik, M.; Foryś, A.; Li, S.; Kasperczyk, J. Cytotoxic Effect of Paclitaxel and Lapatinib Co-Delivered in Polylactide-co-Poly(ethylene glycol) Micelles on HER-2-Negative Breast Cancer Cells. Pharmaceutics 2019, 11, 169. [Google Scholar] [CrossRef]
- Jia, L.; Shen, J.; Li, Z.; Zhang, D.; Zhang, Q.; Liu, G.; Zheng, D.; Tian, X. In vitro and in vivo evaluation of paclitaxel-loaded mesoporous silica nanoparticles with three pore sizes. Int. J. Pharm. 2013, 445, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chavanpatil, M.D.; Patil, Y.; Panyam, J. Susceptibility of nanoparticle-encapsulated paclitaxel to P-glycoprotein-mediated drug efflux. Int. J. Pharm. 2006, 320, 150–156. [Google Scholar] [CrossRef]
- Modi, S.; Anderson, B.D. Determination of Drug Release Kinetics from Nanoparticles: Overcoming Pitfalls of the Dynamic Dialysis Method. Mol. Pharm. 2013, 10, 3076–3089. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.S.; DeLuca, P.P. Methods to Assess in Vitro Drug Release from Injectable Polymeric Particulate Systems. Pharm. Res. 2006, 23, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Gillespie, D.T.; Seitaridou, E. Simple Brownian Diffusion: An Introduction to the Standard Theoretical Models; Oxford University Press: Oxford, NY, USA, 2012; ISBN 978-0-19-966450-4. [Google Scholar]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; di Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
- Liu, P.; De Wulf, O.; Laru, J.; Heikkilä, T.; van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Peltonen, L.; Laaksonen, T. Dissolution Studies of Poorly Soluble Drug Nanosuspensions in Non-sink Conditions. AAPS PharmSciTech 2013, 14, 748–756. [Google Scholar] [CrossRef]
- Mobasseri, R.; Karimi, M.; Tian, L.; Naderi-Manesh, H.; Ramakrishna, S. Hydrophobic lapatinib encapsulated dextran-chitosan nanoparticles using a toxic solvent free method: Fabrication, release property & in vitro anti-cancer activity. Mater. Sci. Eng. C 2017, 74, 413–421. [Google Scholar]
- Bullock, J. Dissolution of Nanoparticle Drug Formulations. In Poorly Soluble Drugs: Dissolution and Drug Release; Taylor & Francis Group: Abingdon, UK, 2017; pp. 301–352. ISBN 9781315364537. [Google Scholar]
- D’Addio, S.M.; Bukari, A.A.; Dawoud, M.; Bunjes, H.; Rinaldi, C.; Prud’homme, R.K. Determining drug release rates of hydrophobic compounds from nanocarriers. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374. [Google Scholar] [CrossRef]
- Gülçin, İ.; Huyut, Z.; Elmastaş, M.; Aboul-Enein, H.Y. Radical scavenging and antioxidant activity of tannic acid. Arab. J. Chem. 2010, 3, 43–53. [Google Scholar] [CrossRef]
- Varbiro, G.; Veres, B.; Gallyas, F.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Rad. Biol. Med. 2001, 31, 548–558. [Google Scholar] [CrossRef]
- Fonseca, C.; Simões, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control. Release 2002, 83, 273–286. [Google Scholar] [CrossRef]
- Lee, Y.; Graeser, R.; Kratz, F.; Geckeler, K.E. Paclitaxel-Loaded Polymer Nanoparticles for the Reversal of Multidrug Resistance in Breast Cancer Cells. Adv. Funct. Mater. 2011, 21, 4211–4218. [Google Scholar] [CrossRef]
- Hirota, K.; Ter, H. Endocytosis of Particle Formulations by Macrophages and Its Application to Clinical Treatment. In Molecular Regulation of Endocytosis; Ceresa, B., Ed.; InTech: London, UK, 2012; ISBN 978-953-51-0662-3. [Google Scholar]
- Swanson, J.A.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef]
- Shu, C.-H.; Yang, W.K.; Shih, Y.-L.; Kuo, M.-L.; Huang, T.-S. Cell cycle G2/M arrest and activation of cyclin-dependent kinases associated with low-dose paclitaxel-induced sub-G1 apoptosis. Apoptosis 1997, 2, 463–470. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Y.; Strom, A.; Gustafsson, J.-Å.; Guan, X. Lapatinib induces p27Kip1-dependent G1 arrest through both transcriptional and post-translational mechanisms. Cell Cycle 2013, 12, 2665–2674. [Google Scholar] [CrossRef]
- Wang, T.-H.; Wang, H.-S.; Soong, Y.-K. Paclitaxel-induced cell death. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Kalurupalle, S.; Hanko, C.; Lim, C.; Broude, E.; Blagosklonny, M.V. Mechanism of G1-like arrest by low concentrations of paclitaxel: Next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene 2008, 27, 4402–4410. [Google Scholar] [CrossRef]
- Hsiao, J.-R.; Leu, S.-F.; Huang, B.-M. Apoptotic mechanism of paclitaxel-induced cell death in human head and neck tumor cell lines. J. Oral. Pathol. Med. 2009, 38, 188–197. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Azmi, A.S.; Banerjee, S.; Ali, S.; Wang, Z.; Bao, B.; Beck, F.W.J.; Maitah, M.; Choi, M.; Shields, T.F.; Philip, P.A.; et al. Network Modeling of MDM2 Inhibitor-Oxaliplatin Combination Reveals Biological Synergy in wt-p53 solid tumors. Oncotarget 2011, 2, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C. Drug Combination Studies and Their Synergy Quantification Using the Chou–Talalay Method—Letter. Cancer Res. 2015, 75, 2400. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (Taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef]
- Moy, B.; Kirkpatrick, P.; Kar, S.; Goss, P. Lapatinib. Nat. Rev. Drug Discov. 2007, 6, 431–432. [Google Scholar] [CrossRef]
- Dai, C.; Tiwari, A.K.; Wu, C.-P.; Su, X.; Wang, S.-R.; Liu, D.; Ashby, C.R.; Huang, Y.; Robey, R.W.; Liang, Y.; et al. Lapatinib (Tykerb, GW572016) Reverses Multidrug Resistance in Cancer Cells by Inhibiting the Activity of ATP-Binding Cassette Subfamily B Member 1 and G Member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef]
- Dai, C.; Ma, S.; Wang, F.; Zhao, H.; Wu, X.; Huang, Z.; Chen, Z.; To, K.; Fu, L. Lapatinib promotes the incidence of hepatotoxicity by increasing chemotherapeutic agent accumulation in hepatocytes. Oncotarget 2015, 6, 17738–17752. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Solids (mg/mL) | Ratio of BCP:Core | PTX Concentration (mg/mL) | Size 1 (nm) * | Size 2 (nm) * | PDI * |
|---|---|---|---|---|---|
| 18 | 2:1 | 1 | 107 ± 2 | 32 ± 1 | 0.275 ± 0.009 |
| 36 | 2:1 | 2 | 113 ± 6 | 31 ± 1 | 0.372 ± 0.012 |
| 11 | 1:1 | 1 | 170 ± 33 | 0 | 0.142 ± 0.053 |
| 13.5 | 1.5:1 | 1 | 128 ± 7 | 0 | 0.244 ± 0.034 |
| 16 | 2:1 | 1 | 111 ± 10 | 0 | 0.255 ± 0.021 |
| 19 | 2:1 | 2 | 117 ± 3 | 20 ± 1 | 0.268 ± 0.009 |
| 16 | 2:1 | 1 | 111 ± 10 | 0 | 0.255 ± 0.021 |
| 14.5 | 2:1 | 0.5 | 134 ± 42 | 0 | 0.232 ± 0.145 |
| Total Solids (mg/mL) | Ratio of BCP:Core | Size 1 (nm) * | Size 2 (nm) * | PDI * |
|---|---|---|---|---|
| 16 | 2:1 | 91 ± 10 | 0 | 0.214 ± 0.038 |
| 36 | 2:1 | 106 ± 5 | 26 ± 4 | 0.288 ± 0.004 |
| 10.5 | 1:1 | 134 ± 8 | 0 | 0.255 ± 0.012 |
| 15.3 | 2:1 | 126 ± 7 | 0 | 0.380 ± 0.037 |
| Samples | Encapsulation Efficiency (EE%) * | Drug Loading (DL%) * | ||
|---|---|---|---|---|
| PTX | LAP | PTX | LAP | |
| PTX NPs | 37.6 ± 14.4 | - | 3.11 ± 1.88 | - |
| LAP NPs | - | 25.0 ± 1.5 | - | 1.82 ± 0.71 |
| PTX-LAP NPs | 67.0 ± 2.2 | 25.9 ± 3.5 | 2.11 ± 0.50 | 0.79 ± 0.40 |
| Drug Treatment | IC-50 (µg/mL) ** | |
|---|---|---|
| PTX | LAP | |
| Free PTX * | 70.6 ± 5.1 | - |
| Free LAP * | - | 4.6 ± 1.3 |
| PTX NPs | 0.040 ± 0.003 | - |
| LAP NPs | - | 0.80 ± 0.26 |
| Drug Treatment | IC-50 (µg/mL) | Combination Index (CI) | |
|---|---|---|---|
| PTX | LAP | ||
| PTX NPs and LAP NPs | 0.015 ± 0.003 | 0.007 ± 0.001 | 0.40 |
| PTX-LAP NPs | 0.0090 ± 0.0009 | 0.0040 ± 0.0004 | 0.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levit, S.L.; Yang, H.; Tang, C. Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation. Nanomaterials 2020, 10, 561. https://doi.org/10.3390/nano10030561
Levit SL, Yang H, Tang C. Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation. Nanomaterials. 2020; 10(3):561. https://doi.org/10.3390/nano10030561
Chicago/Turabian StyleLevit, Shani L., Hu Yang, and Christina Tang. 2020. "Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation" Nanomaterials 10, no. 3: 561. https://doi.org/10.3390/nano10030561
APA StyleLevit, S. L., Yang, H., & Tang, C. (2020). Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation. Nanomaterials, 10(3), 561. https://doi.org/10.3390/nano10030561