Salinomycin-Loaded Iron Oxide Nanoparticles for Glioblastoma Therapy



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of IONPs
2.3. Drug Loading on IONPs
2.4. Biocompatibility Evaluation of IONPs
2.5. Drug Release from IONPs
2.6. Cellular Uptake of IONPs
2.7. Cytotoxicity of Sali-IONPs in GBM Cell Line
2.8. Reactive Oxygen Species Determination
2.9. Quantitative RT-PCR
2.10. In Vitro BBB-GBM Model
2.11. Statistical Analysis
3. Results and Discussion
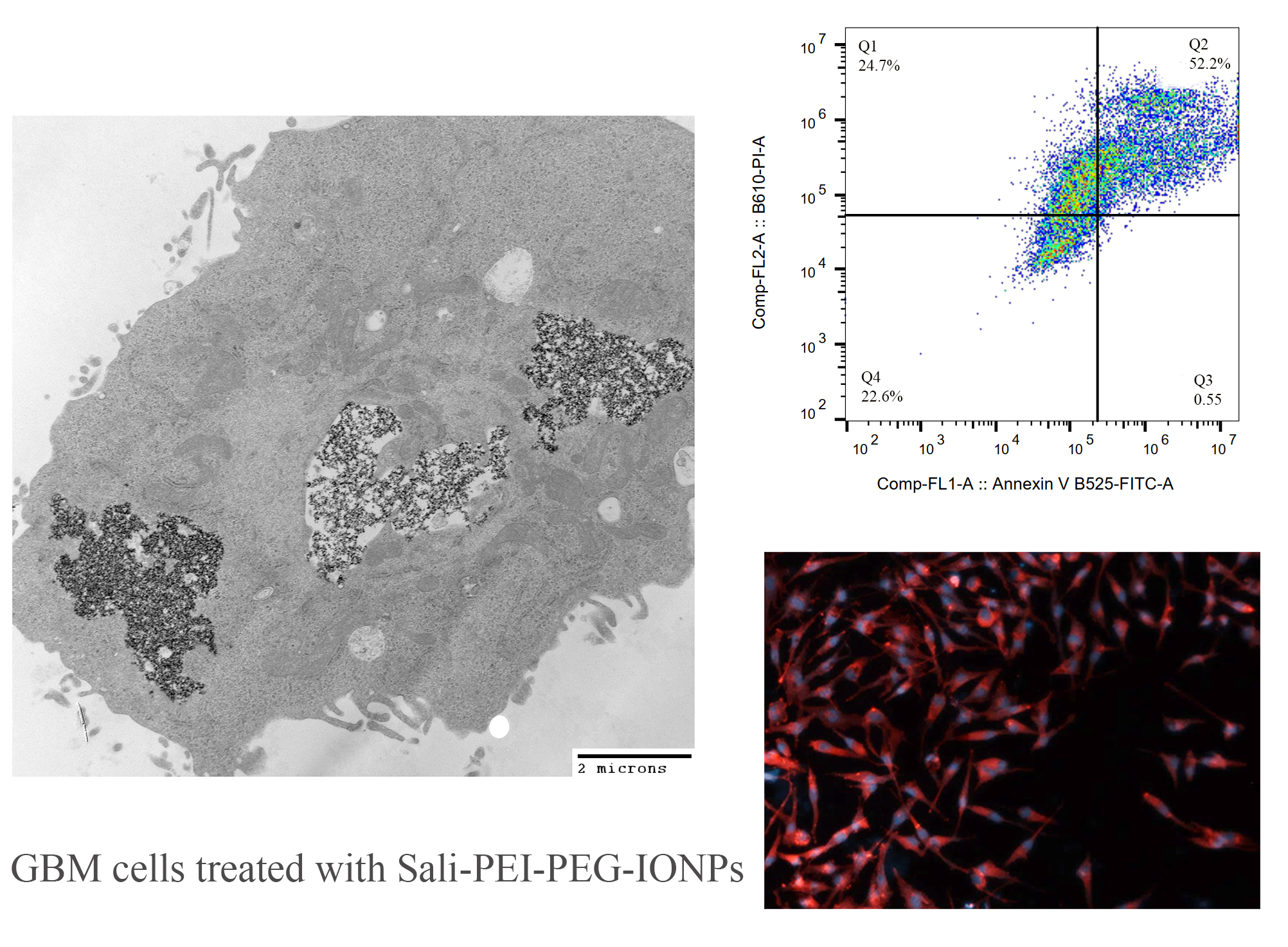
3.1. Characterization of IONPs
3.2. Characterization of the Sali-IONPs
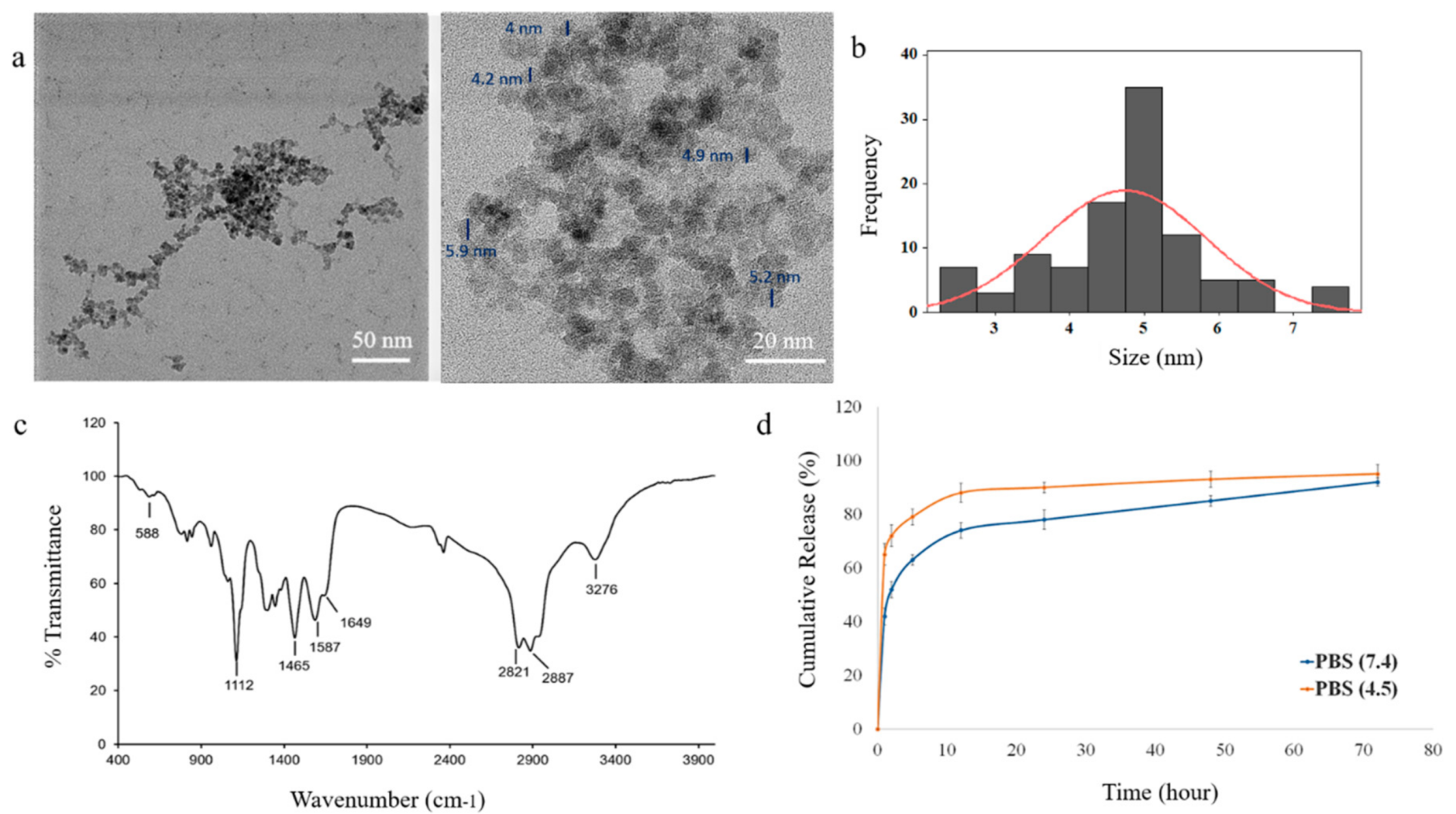
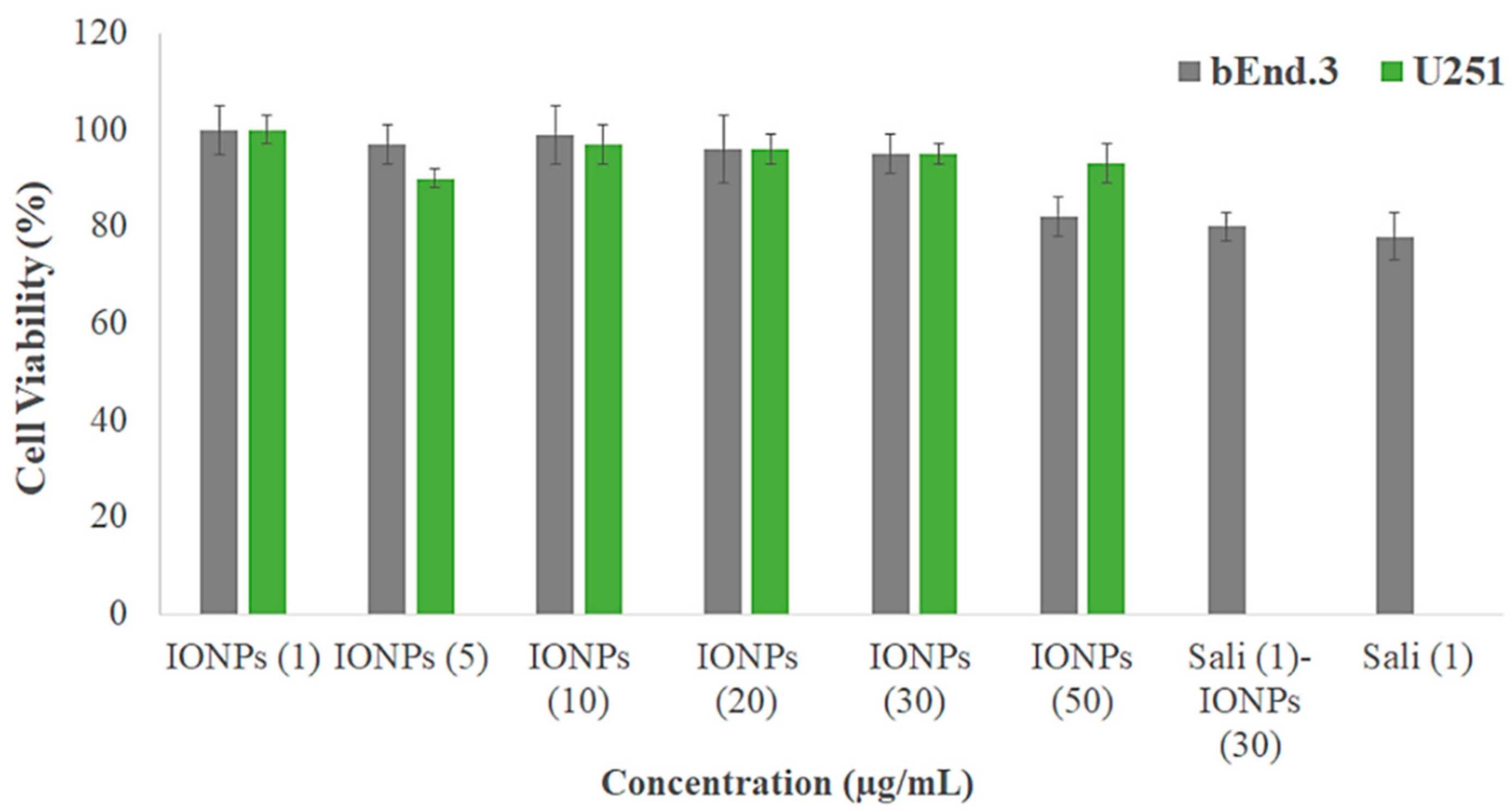
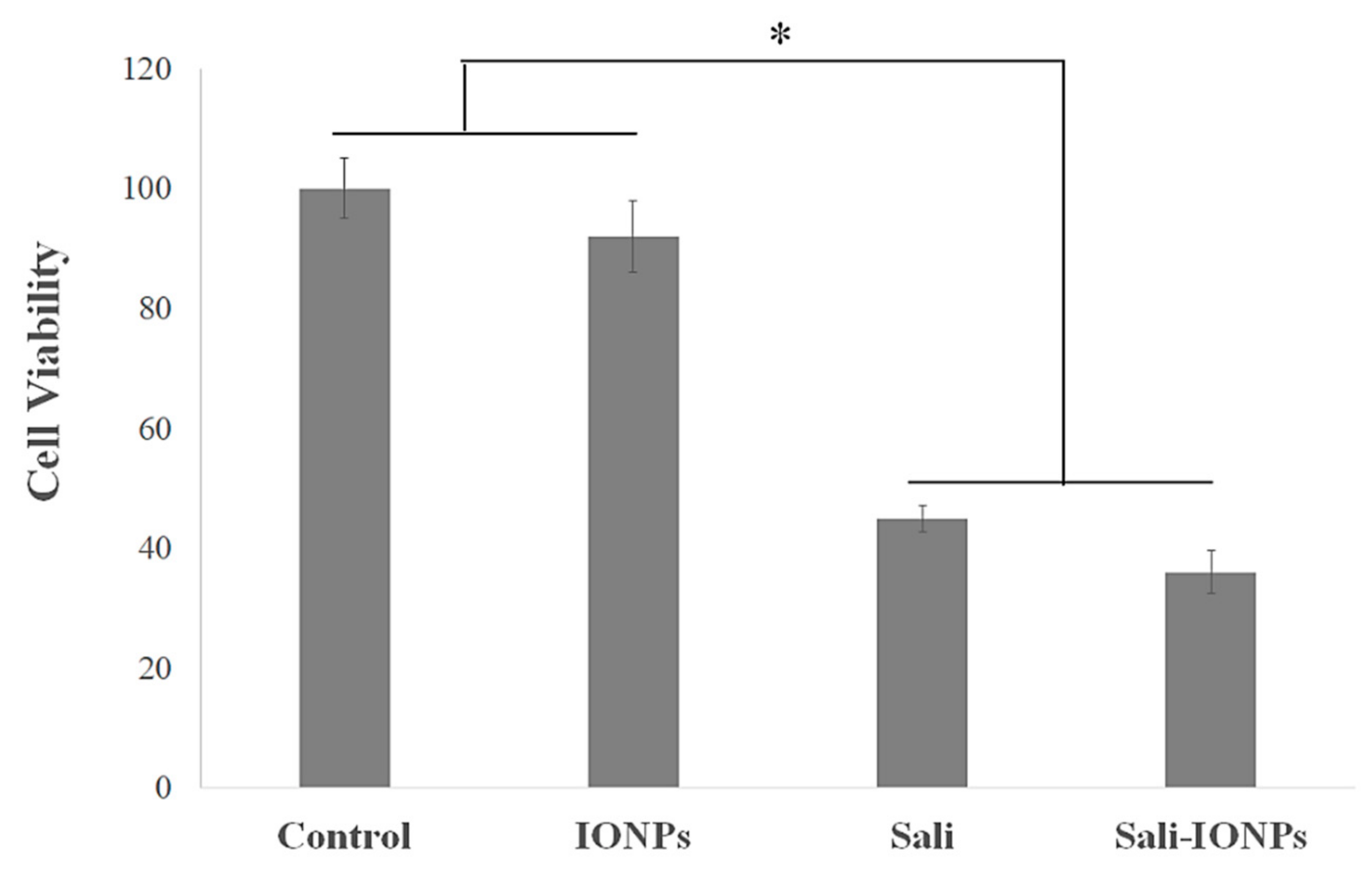
3.3. Biocompatibility of the IONPs
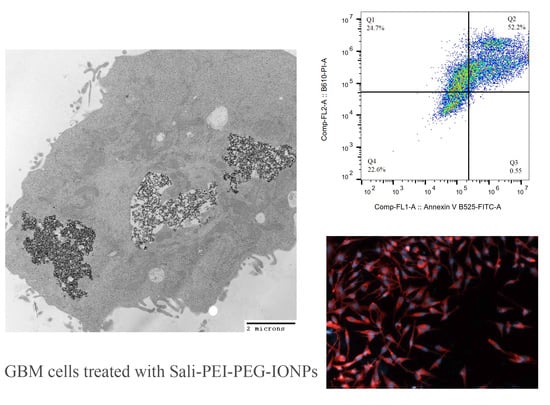
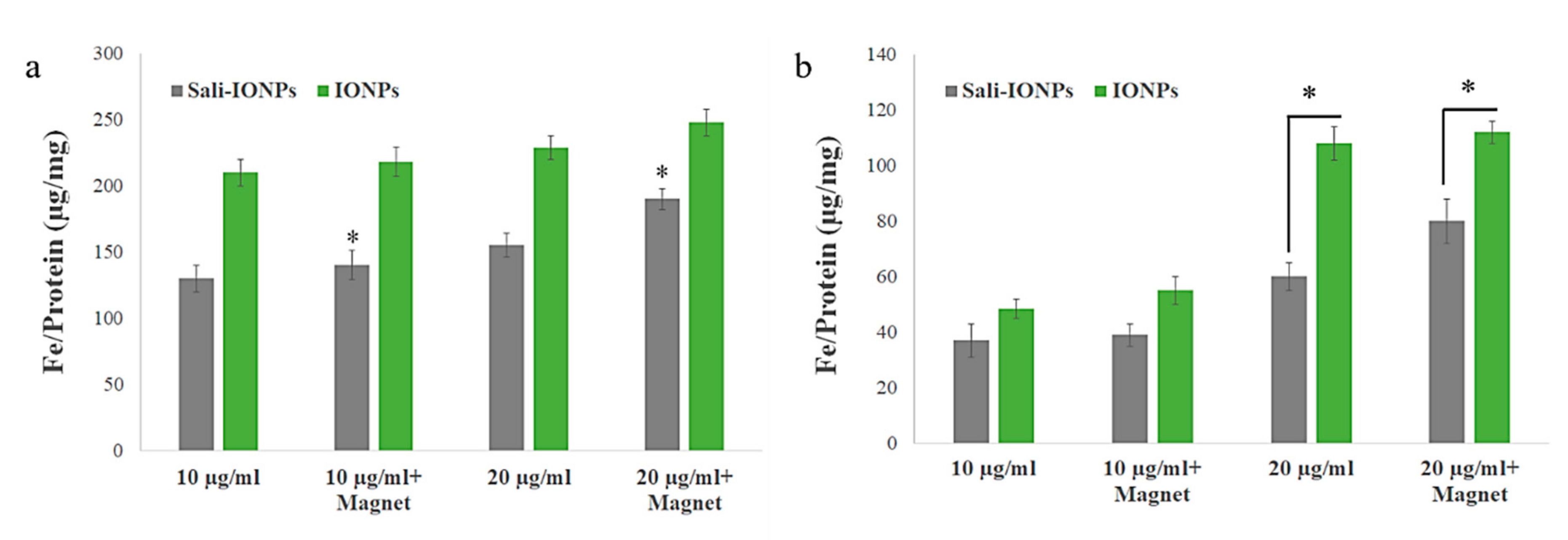
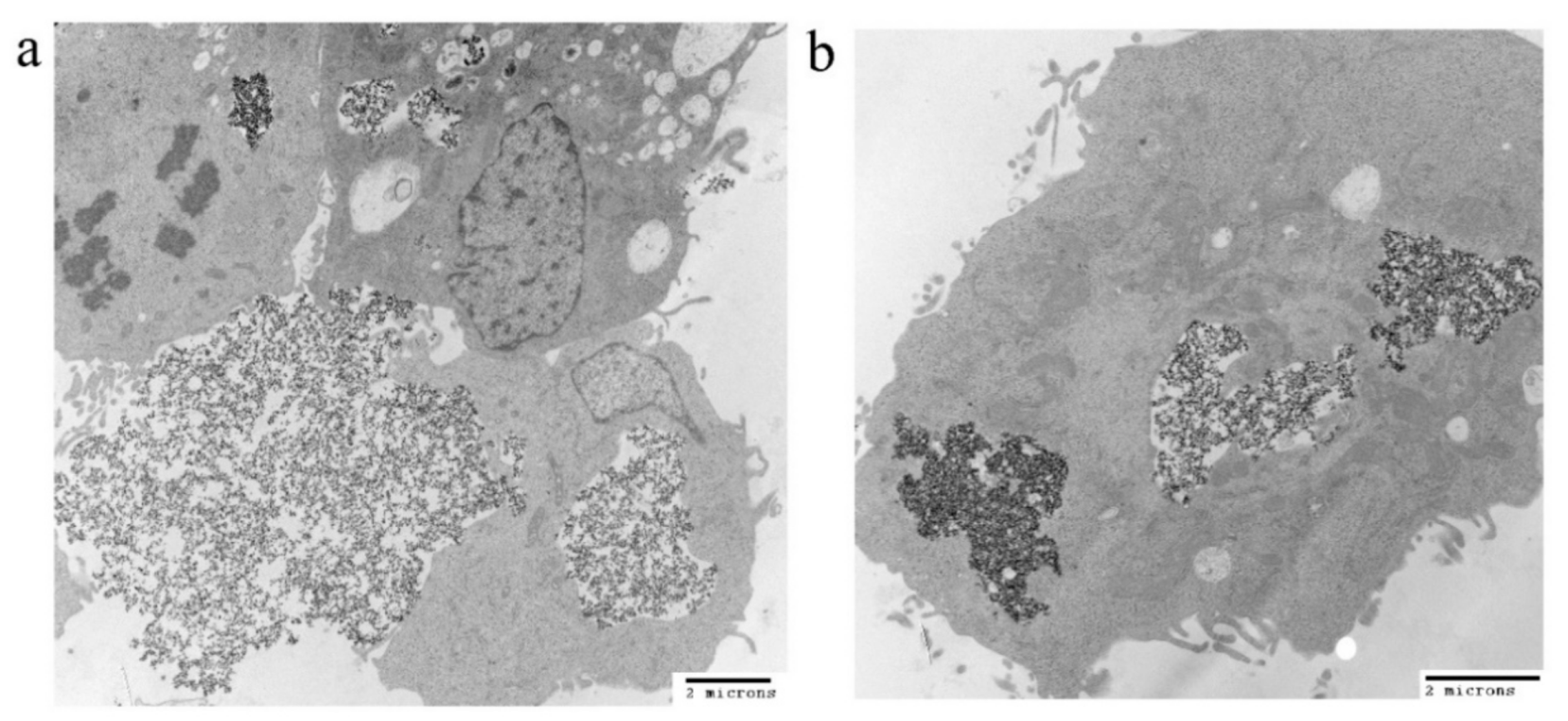
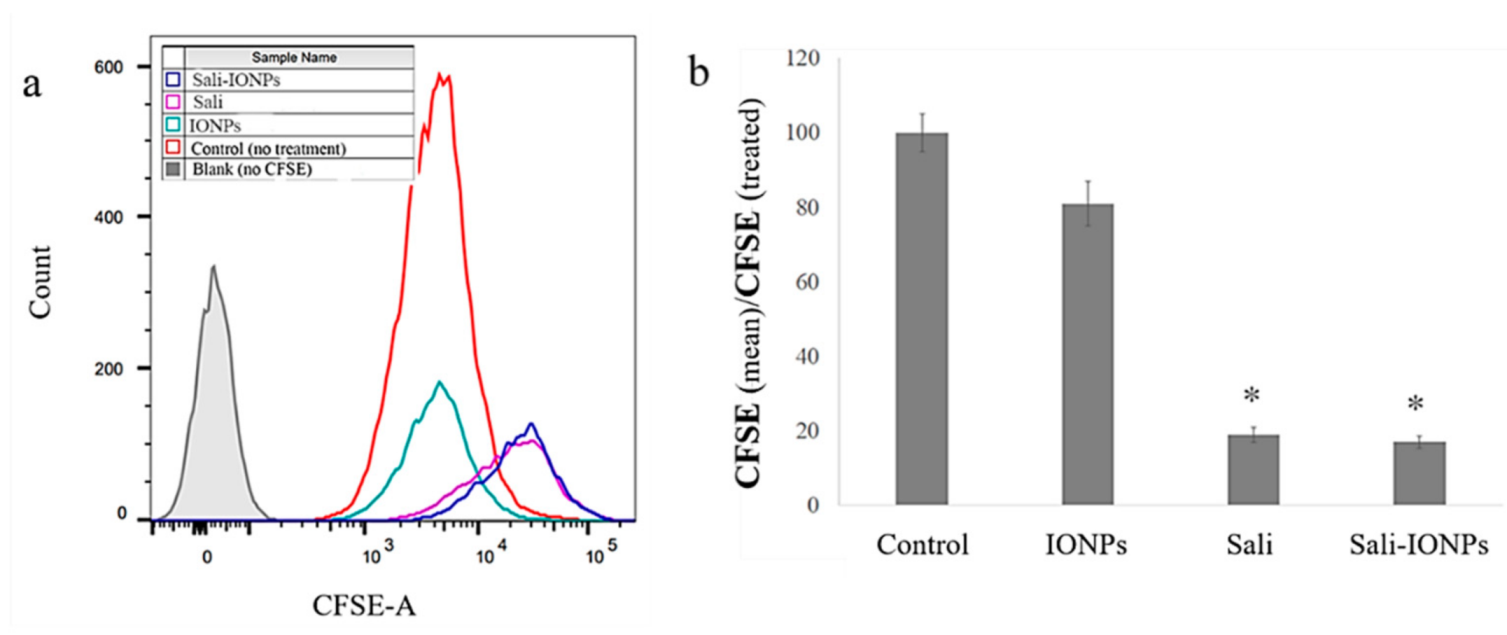
3.4. Cellular Uptake of the IONPs
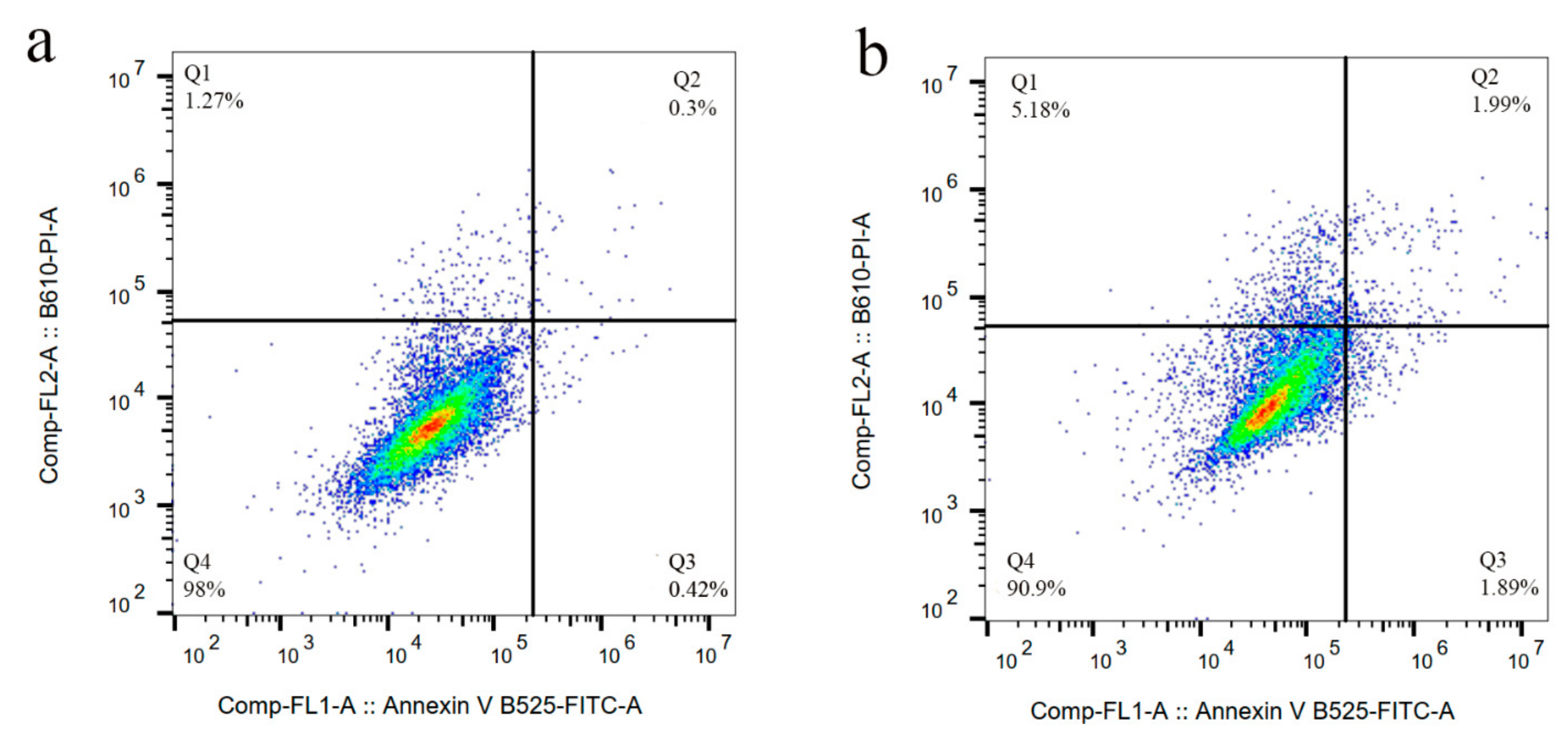
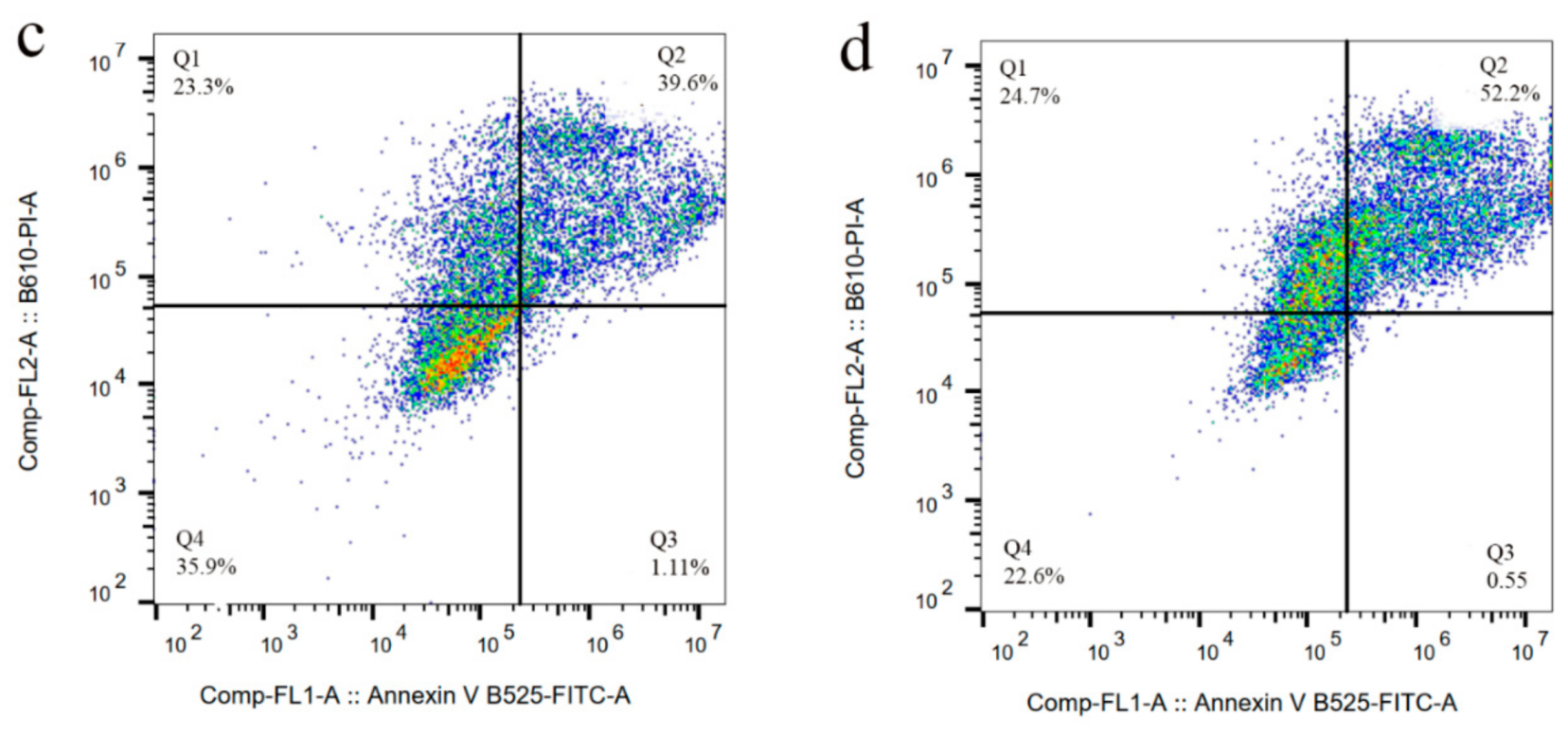
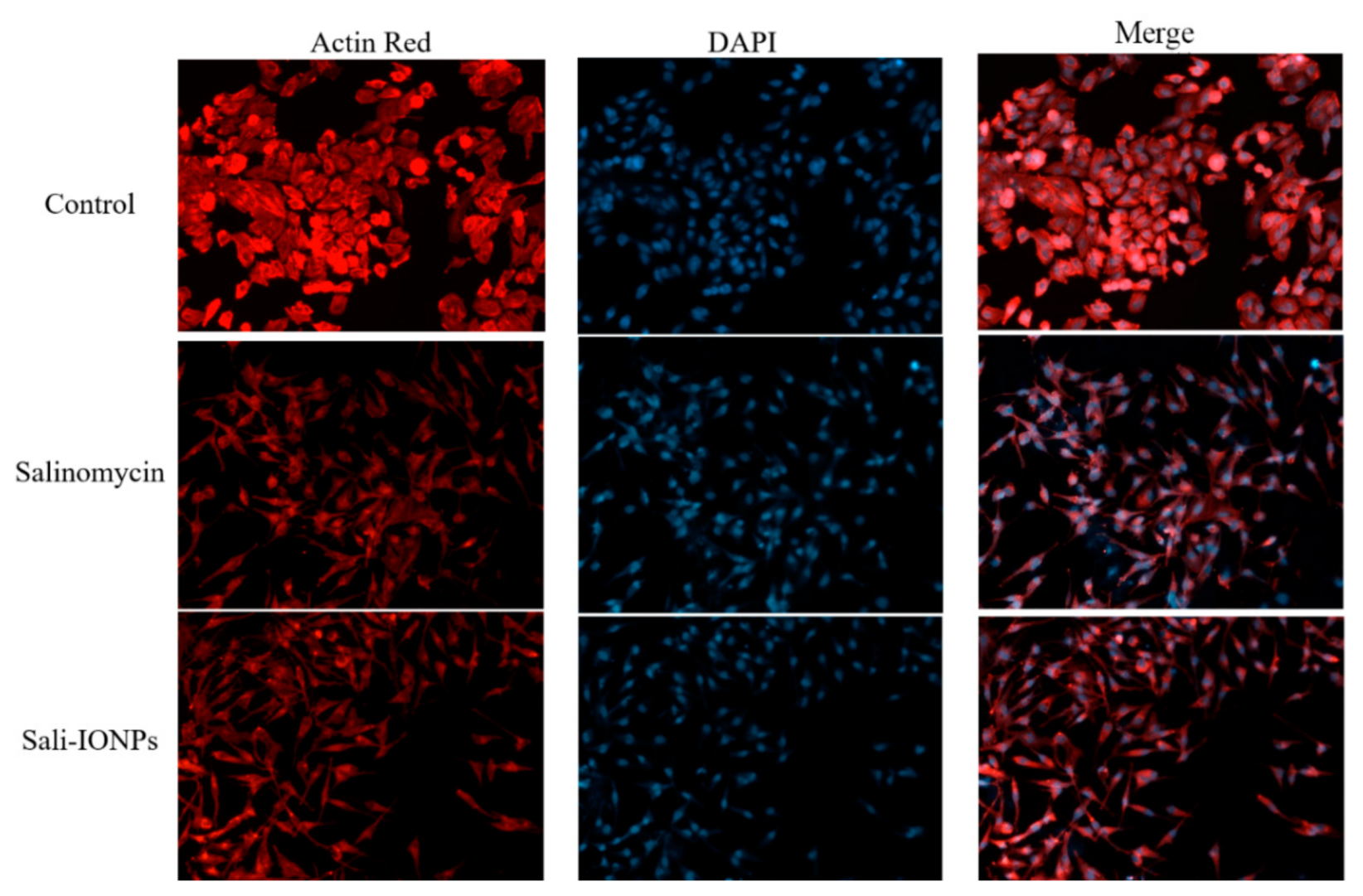
3.5. Cytotoxicity of Sali-IONPs on Cancer Cell
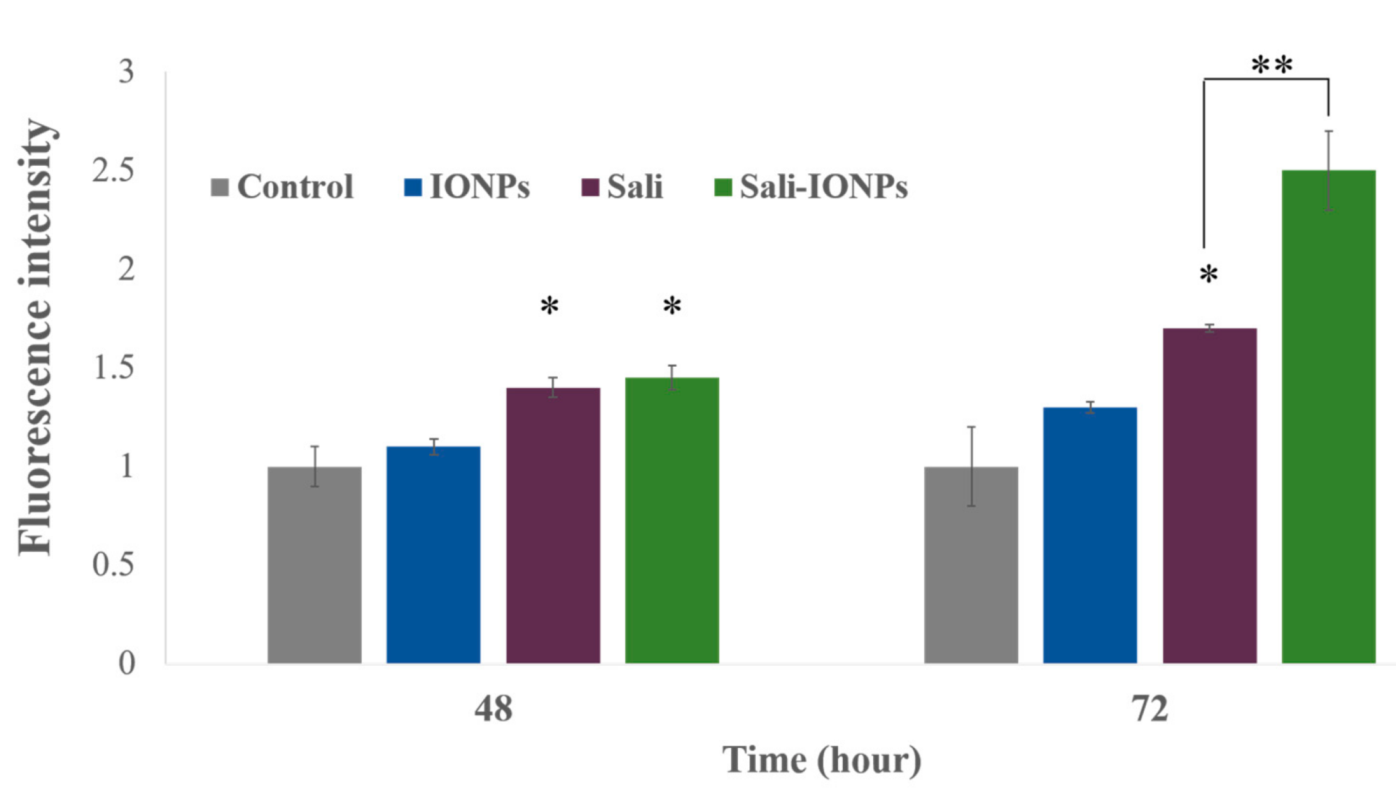
3.6. ROS Generation
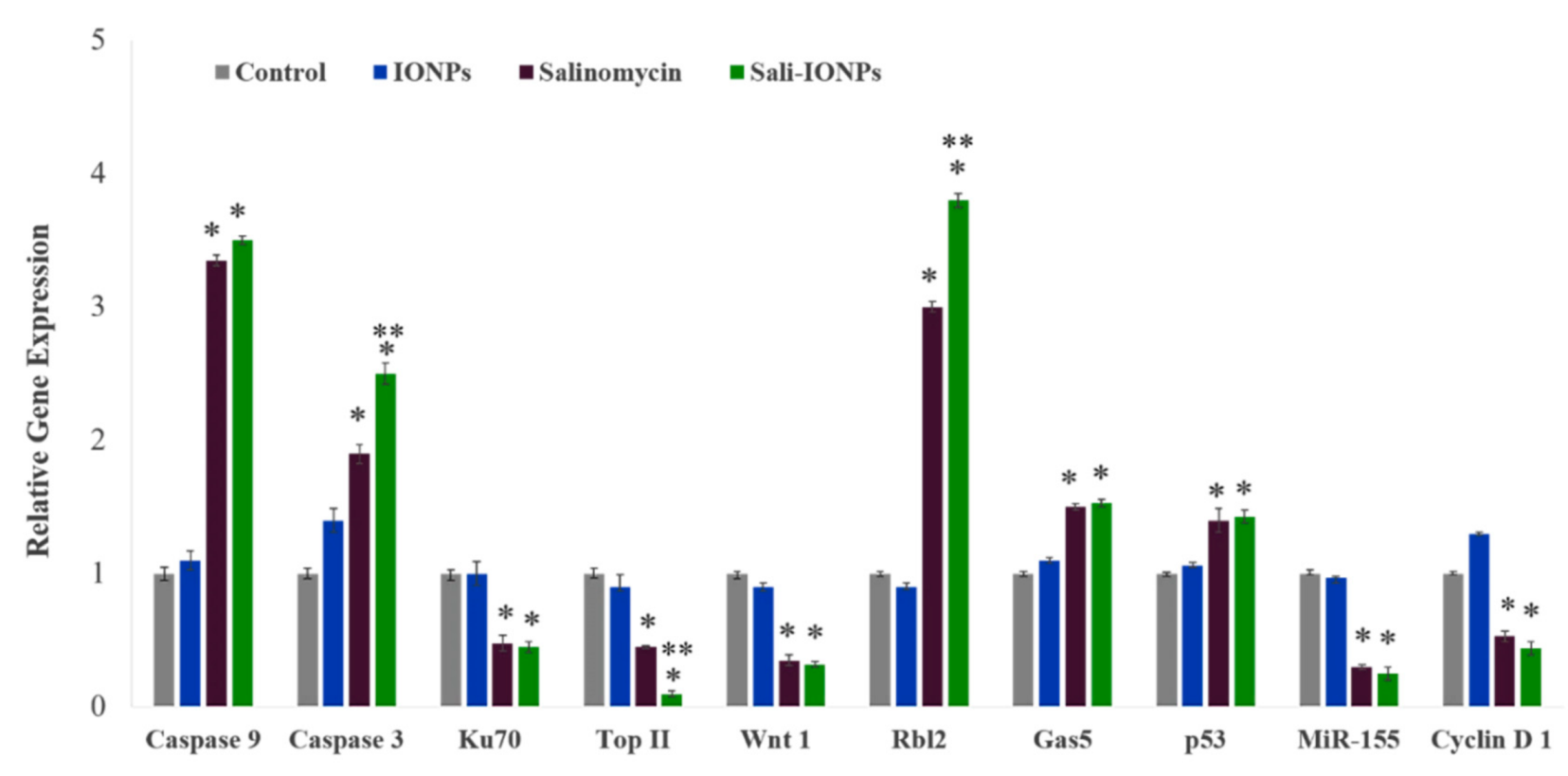
3.7. Quantitative RT-PCR
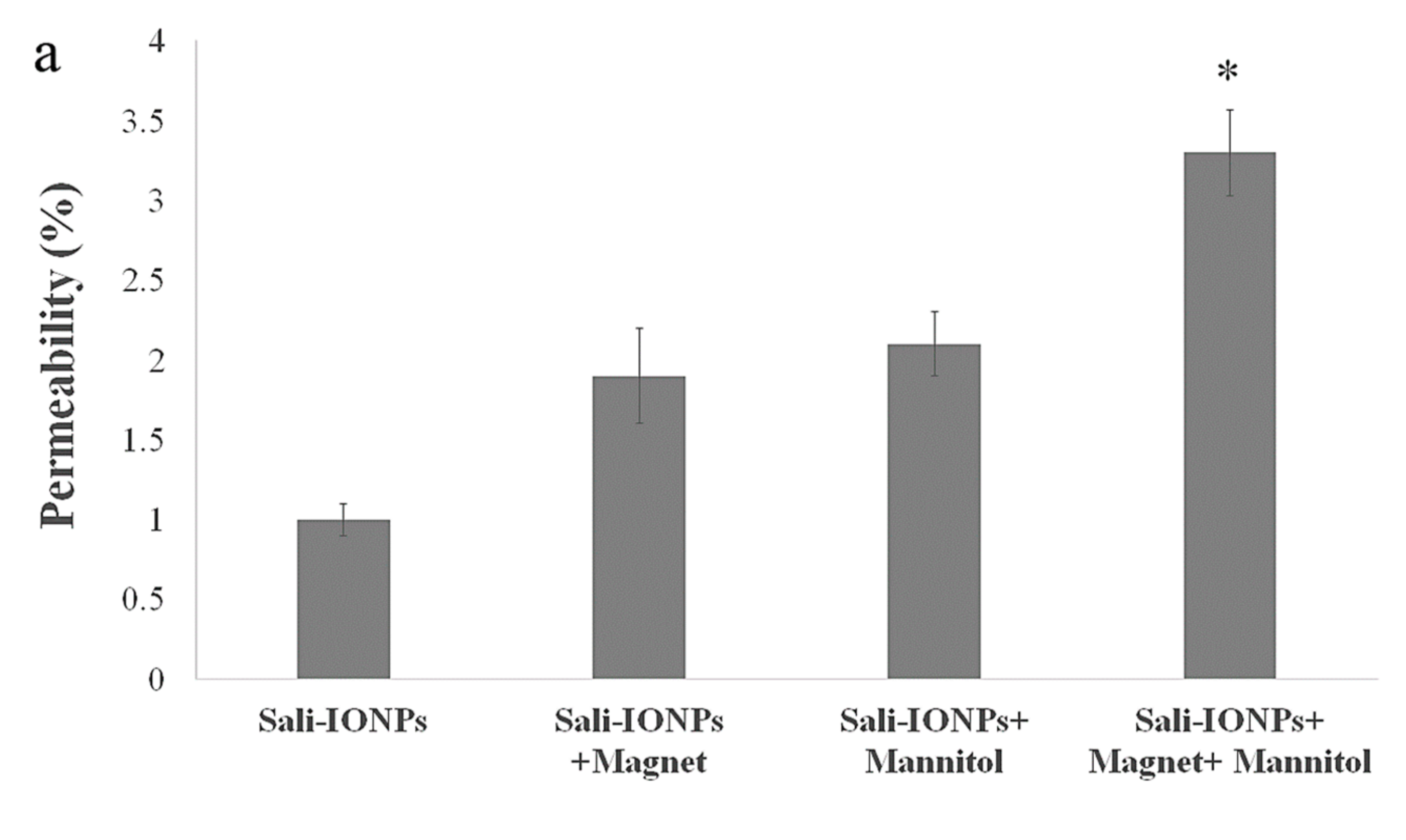
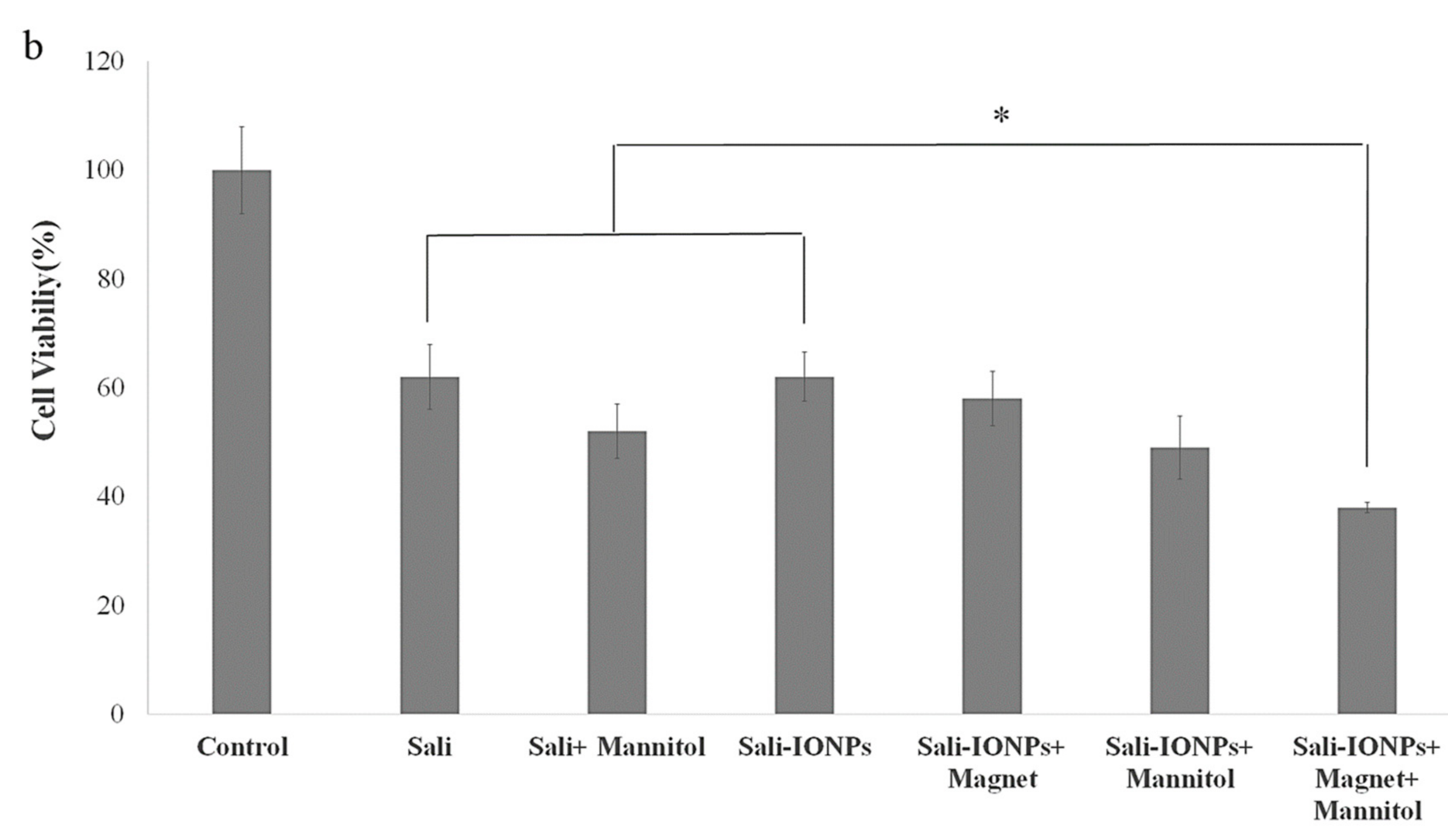
3.8. Evaluation of Sali-IONPs in BBB-GBM Co-Culture Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hathout, L.; Ellingson, B.; Pope, W. Modeling the efficacy of the extent of surgical resection in the setting of radiation therapy for glioblastoma. Cancer Sci. 2016, 107, 1110–1116. [Google Scholar] [CrossRef]
- Shi, W.; Bryan, M.S.; Gilbert, M.R.; Mehta, M.P.; Blumenthal, D.T.; Brown, P.D.; Valeinis, E.; Hopkins, K.; Souhami, L.; Andrews, D.W. Investigating the Effect of Reirradiation or Systemic Therapy in Patients With Glioblastoma After Tumor Progression: A Secondary Analysis of NRG Oncology/Radiation Therapy Oncology Group Trial 0525. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 38–44. [Google Scholar] [CrossRef]
- Norouzi, M.; Amerian, M.; Amerian, M.; Atyabi, F. Clinical applications of nanomedicine in cancer therapy. Drug Discov. Today 2019, 25, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Kenny, G.D.; Bienemann, A.S.; Tagalakis, A.D.; Pugh, J.A.; Welser, K.; Campbell, F.; Tabor, A.B.; Hailes, H.C.; Gill, S.S.; Lythgoe, M.F. Multifunctional receptor-targeted nanocomplexes for the delivery of therapeutic nucleic acids to the brain. Biomaterials 2013, 34, 9190–9200. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M. Recent advances in brain tumor therapy: Application of electrospun nanofibers. Drug Discov. Today 2018, 23, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 21, 1835–1849. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Chen, X.; Du, X.-S.; Zhang, J.-L.; Liu, G.; Zhang, W.-G. Application of iron oxide nanoparticles in glioma imaging and therapy: From bench to bedside. Nanoscale 2016, 8, 7808–7826. [Google Scholar] [CrossRef]
- Kulczar, C.; Lubin, K.E.; Lefebvre, S.; Miller, D.W.; Knipp, G.T. Development of a direct contact astrocyte-human cerebral microvessel endothelial cells blood–brain barrier coculture model. J. Pharm. Pharmacol. 2017, 69, 1684–1696. [Google Scholar] [CrossRef]
- Cheng, Y.; Dai, Q.; Morshed, R.A.; Fan, X.; Wegscheid, M.L.; Wainwright, D.A.; Han, Y.; Zhang, L.; Auffinger, B.; Tobias, A.L. Blood-brain barrier permeable gold nanoparticles: An efficient delivery platform for enhanced malignant glioma therapy and imaging. Small 2014, 10, 5137–5150. [Google Scholar] [CrossRef]
- Gao, H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharmaceut. Sin. B 2016, 6, 268–286. [Google Scholar] [CrossRef]
- Xu, H.-L.; Mao, K.-L.; Huang, Y.-P.; Yang, J.-J.; Xu, J.; Chen, P.-P.; Fan, Z.-L.; Zou, S.; Gao, Z.-Z.; Yin, J.-Y. Glioma-targeted superparamagnetic iron oxide nanoparticles as drug-carrying vehicles for theranostic effects. Nanoscale 2016, 8, 14222–14236. [Google Scholar] [CrossRef] [PubMed]
- Mujokoro, B.; Adabi, M.; Sadroddiny, E.; Adabi, M.; Khosravani, M. Nano-structures mediated co-delivery of therapeutic agents for glioblastoma treatment: A review. Mater. Sci. Eng. C 2016, 69, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Nazari, B.; Miller, D.W. Electrospun-based systems in cancer therapy. In Electrospun Materials for Tissue Engineering and Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2017; pp. 337–356. [Google Scholar]
- Hola, K.; Markova, Z.; Zoppellaro, G.; Tucek, J.; Zboril, R. Tailored functionalization of iron oxide nanoparticles for MRI, drug delivery, magnetic separation and immobilization of biosubstances. Biotechnol. Adv. 2015, 33, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wu, Z.; Yu, T.; Jiang, C.; Kim, W.-S. Recent progress on magnetic iron oxide nanoparticles: Synthesis, surface functional strategies and biomedical applications. Sci. Technol. Adv. Mater. 2015, 16, 023501. [Google Scholar] [CrossRef]
- Lee, N.; Yoo, D.; Ling, D.; Cho, M.H.; Hyeon, T.; Cheon, J. Iron oxide based nanoparticles for multimodal imaging and magnetoresponsive therapy. Chem. Rev. 2015, 115, 10637–10689. [Google Scholar] [CrossRef]
- Arami, H.; Khandhar, A.; Liggitt, D.; Krishnan, K.M. In vivo delivery, pharmacokinetics, biodistribution and toxicity of iron oxide nanoparticles. Chem. Soc. Rev. 2015, 44, 8576–8607. [Google Scholar] [CrossRef]
- Kievit, F.M.; Wang, F.Y.; Fang, C.; Mok, H.; Wang, K.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J. Control. Release 2011, 152, 76–83. [Google Scholar] [CrossRef]
- Mu, Q.; Jeon, M.; Hsiao, M.H.; Patton, V.K.; Wang, K.; Press, O.W.; Zhang, M. Stable and efficient Paclitaxel nanoparticles for targeted glioblastoma therapy. Adv. Healthc. Mater. 2015, 4, 1236–1245. [Google Scholar] [CrossRef]
- Mu, Q.; Lin, G.; Patton, V.K.; Wang, K.; Press, O.W.; Zhang, M. Gemcitabine and chlorotoxin conjugated iron oxide nanoparticles for glioblastoma therapy. J. Mater. Chem. B 2016, 4, 32–36. [Google Scholar] [CrossRef]
- Kaluzova, M.; Bouras, A.; Machaidze, R.; Hadjipanayis, C.G. Targeted therapy of glioblastoma stem-like cells and tumor non-stem cells using cetuximab-conjugated iron-oxide nanoparticles. Oncotarget 2015, 6, 8788. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Machaidze, R.; Kaluzova, M.; Wang, L.; Schuette, A.J.; Chen, H.; Wu, X.; Mao, H. EGFRvIII antibody–conjugated iron oxide nanoparticles for magnetic resonance imaging–guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010, 70, 6303–6312. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Lin, B.; Li, D.; Ai, H. Superparamagnetic iron oxide nanoparticles for MR imaging and therapy: Design considerations and clinical applications. Curr. Opin. Pharmacol. 2014, 18, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.J. Current status of superparamagnetic iron oxide contrast agents for liver magnetic resonance imaging. World J. Gastroenterol. 2015, 21, 13400. [Google Scholar] [CrossRef] [PubMed]
- Chertok, B.; Moffat, B.A.; David, A.E.; Yu, F.; Bergemann, C.; Ross, B.D.; Yang, V.C. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials 2008, 29, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. BioMed Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Huczynski, A. Salinomycin–a new cancer drug candidate. Chem. Biol. Drug Design 2012, 79, 235–238. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Ghavami, S.; Grabarek, J.; Kratz, G.; Wiechec, E.; Fredriksson, B.-A.; Pariti, R.K.R.; Cieślar-Pobuda, A.; Panigrahi, S.; Łos, M.J. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: Differences between primary and cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 2057–2069. [Google Scholar] [CrossRef]
- Managò, A.; Leanza, L.; Carraretto, L.; Sassi, N.; Grancara, S.; Quintana-Cabrera, R.; Trimarco, V.; Toninello, A.; Scorrano, L.; Trentin, L. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 2015, 6, e1930. [Google Scholar] [CrossRef]
- Norouzi, M.; Abdali, Z.; Liu, S.; Miller, D.W. Salinomycin-loaded Nanofibers for Glioblastoma Therapy. Sci. Rep. 2018, 8, 9377. [Google Scholar] [CrossRef]
- Lagas, J.S.; Sparidans, R.W.; Van Waterschoot, R.A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein limits oral availability, brain penetration, and toxicity of an anionic drug, the antibiotic salinomycin. Antimicrob. Agents Chemother. 2008, 52, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Yathindranath, V.; Sun, Z.; Worden, M.; Donald, L.J.; Thliveris, J.A.; Miller, D.W.; Hegmann, T. One-pot synthesis of iron oxide nanoparticles with functional silane shells: A versatile general precursor for conjugations and biomedical applications. Langmuir 2013, 29, 10850–10858. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yathindranath, V.; Worden, M.; Thliveris, J.A.; Chu, S.; Parkinson, F.E.; Hegmann, T.; Miller, D.W. Characterization of cellular uptake and toxicity of aminosilane-coated iron oxide nanoparticles with different charges in central nervous system-relevant cell culture models. Int. J. Nanomed. 2013, 8, 961. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. In Advanced Protocols in Oxidative Stress II; Springer: Berlin, Germany, 2010; pp. 57–72. [Google Scholar]
- Lee, J.-Y.; Termsarasab, U.; Lee, M.Y.; Kim, D.-H.; Lee, S.Y.; Kim, J.S.; Cho, H.-J.; Kim, D.-D. Chemosensitizing indomethacin-conjugated chitosan oligosaccharide nanoparticles for tumor-targeted drug delivery. Acta Biomater. 2017, 57, 262–273. [Google Scholar] [CrossRef]
- Huang, P.; Liu, J.; Wang, W.; Zhang, Y.; Zhao, F.; Kong, D.; Liu, J.; Dong, A. Zwitterionic nanoparticles constructed from bioreducible RAFT–ROP double head agent for shell shedding triggered intracellular drug delivery. Acta Biomater. 2016, 40, 263–272. [Google Scholar] [CrossRef]
- Zhang, T.; Lip, H.; He, C.; Cai, P.; Wang, Z.; Henderson, J.T.; Rauth, A.M.; Wu, X.Y. Multitargeted Nanoparticles Deliver Synergistic Drugs across the Blood–Brain Barrier to Brain Metastases of Triple Negative Breast Cancer Cells and Tumor-Associated Macrophages. Adv. Healthc. Mater. 2019, 8, 1900543. [Google Scholar] [CrossRef]
- Yathindranath, V.; Rebbouh, L.; Moore, D.F.; Miller, D.W.; van Lierop, J.; Hegmann, T. A Versatile Method for the Reductive, One-Pot Synthesis of Bare, Hydrophilic and Hydrophobic Magnetite Nanoparticles. Adv. Funct. Mater. 2011, 21, 1457–1464. [Google Scholar] [CrossRef]
- Mu, Q.; Kievit, F.M.; Kant, R.J.; Lin, G.; Jeon, M.; Zhang, M. Anti-HER2/neu peptide-conjugated iron oxide nanoparticles for targeted delivery of paclitaxel to breast cancer cells. Nanoscale 2015, 7, 18010–18014. [Google Scholar] [CrossRef]
- Norouzi, M.; Boroujeni, S.M.; Omidvarkordshouli, N.; Soleimani, M. Advances in Skin Regeneration: Application of Electrospun Scaffolds. Adv. Healthc. Mater. 2015, 4, 1114–1133. [Google Scholar] [CrossRef]
- Tran, T.T.-D.; Van Vo, T.; Tran, P.H.-L. Design of iron oxide nanoparticles decorated oleic acid and bovine serum albumin for drug delivery. Chem. Eng. Res. Design 2015, 94, 112–118. [Google Scholar] [CrossRef]
- Hervault, A.; Dunn, A.E.; Lim, M.; Boyer, C.; Mott, D.; Maenosono, S.; Thanh, N.T. Doxorubicin loaded dual pH-and thermo-responsive magnetic nanocarrier for combined magnetic hyperthermia and targeted controlled drug delivery applications. Nanoscale 2016, 8, 12152–12161. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zeng, Y.; Qi, X.; Chen, Y.; Ge, Z.; Jiang, Z.; Zhang, X.; Dong, Y.; Chen, H.; Yu, Z. Targeted salinomycin delivery with EGFR and CD133 aptamers based dual-ligand lipid-polymer nanoparticles to both osteosarcoma cells and cancer stem cells. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2115–2127. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q.; Yu, Z. Poly (lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537. [Google Scholar]
- Sun, Z.; Worden, M.; Wroczynskyj, Y.; Yathindranath, V.; van Lierop, J.; Hegmann, T.; Miller, D.W. Magnetic field enhanced convective diffusion of iron oxide nanoparticles in an osmotically disrupted cell culture model of the blood-brain barrier. Int. J. Nanomed. 2014, 9, 3013–3026. [Google Scholar] [CrossRef]
- Iwadate, Y.; Namba, H.; Saegusa, T.; Sueyoshi, K. Intra-arterial mannitol infusion in the chemotherapy for malignant brain tumors. J. Neuro Oncol. 1993, 15, 185–193. [Google Scholar] [CrossRef]
- Choi, C.; Kim, H.M.; Shon, J.; Park, J.; Kim, H.-T.; Oh, S.-H.; Kim, N.K.; Kim, O.J. Additional increased effects of mannitol-temozolomide combined treatment on blood-brain barrier permeability. Biochem. Biophys. Res. Commun. 2018, 497, 769–775. [Google Scholar] [CrossRef]
- McCarty, D.; DiRosario, J.; Gulaid, K.; Muenzer, J.; Fu, H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009, 16, 1340. [Google Scholar] [CrossRef]
- Corot, C.; Robert, P.; Idée, J.-M.; Port, M. Recent advances in iron oxide nanocrystal technology for medical imaging. Adv. Drug Deliv. Rev. 2006, 58, 1471–1504. [Google Scholar] [CrossRef]
- Chen, J.; Glaus, C.; Laforest, R.; Zhang, Q.; Yang, M.; Gidding, M.; Welch, M.J.; Xia, Y. Gold nanocages as photothermal transducers for cancer treatment. Small 2010, 6, 811–817. [Google Scholar] [CrossRef]
- Melamed, J.R.; Riley, R.S.; Valcourt, D.M.; Day, E.S. Using gold nanoparticles to disrupt the tumor microenvironment: An emerging therapeutic strategy. ACS Nano 2016, 10, 10631–10635. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Wang, Y.-X.J.; Leung, K.C.-F.; Lee, S.-F.; Zhao, F.; Wang, D.-W.; Lai, J.M.; Wan, C.; Cheng, C.H.; Ahuja, A.T. Enhanced cellular uptake of aminosilane-coated superparamagnetic iron oxide nanoparticles in mammalian cell lines. Int. J. Nanomed. 2012, 7, 953. [Google Scholar]
- Schenk, M.; Aykut, B.; Teske, C.; Giese, N.A.; Weitz, J.; Welsch, T. Salinomycin inhibits growth of pancreatic cancer and cancer cell migration by disruption of actin stress fiber integrity. Cancer Lett. 2015, 358, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, A.; Delgado, M.; Antoszczak, M.; Huczyński, A.; Chambers, T.C. Salinomycin derivatives exhibit activity against primary acute lymphoblastic leukemia (ALL) cells in vitro. Biomed. Pharmacother. 2018, 99, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-Y.; Yu, S.-N.; Lee, S.-Y.; Chun, S.-S.; Choi, Y.-L.; Park, Y.-M.; Song, C.S.; Chatterjee, B.; Ahn, S.-C. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem. Biophys. Res. Commun. 2011, 413, 80–86. [Google Scholar] [CrossRef]
- Tung, C.-L.; Chen, J.-C.; Wu, C.-H.; Peng, Y.-S.; Chen, W.-C.; Zheng, H.-Y.; Jian, Y.-J.; Wei, C.-L.; Cheng, Y.-T.; Lin, Y.-W. Salinomycin acts through reducing AKT-dependent thymidylate synthase expression to enhance erlotinib-induced cytotoxicity in human lung cancer cells. Exp. Cell Res. 2017, 357, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Teksen, F. Apoptotic effects of salinomycin on human ovarian cancer cell line (OVCAR-3). Tumor Biol. 2016, 37, 3897–3903. [Google Scholar] [CrossRef]
- Zhao, S.-J.; Wang, X.-J.; Wu, Q.-J.; Liu, C.; Li, D.-W.; Fu, X.-T.; Zhang, H.-F.; Shao, L.-R.; Sun, J.-Y.; Sun, B.-L. Induction of G1 Cell Cycle Arrest in Human Glioma Cells by Salinomycin Through Triggering ROS-Mediated DNA Damage In Vitro and In Vivo. Neurochem. Res. 2017, 42, 997–1005. [Google Scholar] [CrossRef]
- Xipell, E.; Gonzalez-Huarriz, M.; Martinez de Irujo, J.J.; Garcia-Garzon, A.; Lang, F.F.; Jiang, H.; Fueyo, J.; Gomez-Manzano, C.; Alonso, M.M. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget 2016, 7, 30626–30641. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Q.; Ning, B.; Luo, L.; Fang, Y. β-Asarone promotes Temozolomide’s entry into glioma cells and decreases the expression of P-glycoprotein and MDR1. Biomed. Pharmacother. 2017, 90, 368–374. [Google Scholar] [CrossRef]
- Geppert, M.; Hohnholt, M.C.; Nürnberger, S.; Dringen, R. Ferritin up-regulation and transient ROS production in cultured brain astrocytes after loading with iron oxide nanoparticles. Acta Biomater. 2012, 8, 3832–3839. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Endres, M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hui, P.; Meng, W.; Wang, N.; Xiang, S. Ku70 inhibits gemcitabine-induced DNA damage and pancreatic cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2017, 484, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Subramanian, C.; Andrews, P.C.; Kwok, R.P. Cytosolic Ku70 regulates Bax-mediated cell death. Tumor Biol. 2016, 37, 13903–13914. [Google Scholar] [CrossRef]
- Järvinen, T.A.; Tanner, M.; Rantanen, V.; Bärlund, M.; Borg, Å.; Grénman, S.; Isola, J. Amplification and deletion of topoisomerase IIα associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am. J. Pathol. 2000, 156, 839–847. [Google Scholar] [CrossRef]
- Romero, A.; Martín, M.; Cheang, M.C.; García-Asenjo, J.A.L.; Oliva, B.; He, X.; de la Hoya, M.; Sáenz, J.Á.G.; Fernández, M.A.; Rubio, E.D. Assessment of topoisomerase II α status in breast cancer by quantitative PCR, gene expression microarrays, immunohistochemistry, and fluorescence in situ hybridization. Am. J. Pathol. 2011, 178, 1453–1460. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843. [Google Scholar] [CrossRef]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.L.; Shiras, A. Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell. Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013, 4, e500. [Google Scholar] [CrossRef]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef]
- Yin, D.; He, X.; Zhang, E.; Kong, R.; De, W.; Zhang, Z. Long noncoding RNA GAS5 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer. Med. Oncol. 2014, 31, 253. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Shi, X.; Zhu, Q.; Li, Q.; Liu, Y.; Yao, Y.; Song, Y. The growth arrest-specific transcript 5 (GAS5): A pivotal tumor suppressor long noncoding RNA in human cancers. Tumor Biol. 2016, 37, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, P.; Liu, J.; Zheng, J.; Liu, Y.; Chen, J.; Xue, Y. Gas5 exerts tumor-suppressive functions in human glioma cells by targeting miR-222. Mol. Ther. 2015, 23, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.; Fersht, A. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226. [Google Scholar] [CrossRef]
- Qin, L.-S.; Jia, P.-F.; Zhang, Z.-Q.; Zhang, S.-M. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. J. Exp. Clin. Cancer Res. 2015, 34, 57. [Google Scholar] [CrossRef]
- Magrath, J.W.; Kim, Y. Salinomyci’s potential to eliminate glioblastoma stem cells and treat glioblastoma multiforme. Int. J. Oncol. 2017, 51, 753–759. [Google Scholar] [CrossRef][Green Version]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 497–505. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.; Ju, X.; Vetuschi, A.; Sferra, R.; Casimiro, M.C.; Pompili, S.; Festuccia, C.; Colapietro, A.; Gaudio, E. Cyclin D1 silencing suppresses tumorigenicity, impairs DNA double strand break repair and thus radiosensitizes androgen-independent prostate cancer cells to DNA damage. Oncotarget 2016, 7, 5383. [Google Scholar] [CrossRef]
- Bachmann, K.; Neumann, A.; Hinsch, A.; Nentwich, M.F.; El Gammal, A.T.; Vashist, Y.; Perez, D.; Bockhorn, M.; Izbicki, J.R.; Mann, O. Cyclin D1 is a strong prognostic factor for survival in pancreatic cancer: Analysis of CD G870A polymorphism, FISH and immunohistochemistry. J. Surg. Oncol. 2015, 111, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Arato-Ohshima, T.; Sawa, H. Over-expression of cyclin D1 induces glioma invasion by increasing matrix metalloproteinase activity and cell motility. Int. J. Cancer 1999, 83, 387–392. [Google Scholar] [CrossRef]
- Koo, K.; Kim, H.; Bae, Y.; Kim, K.; Park, B.; Lee, C.; Kim, Y. Salinomycin induces cell death via inactivation of Stat3 and downregulation of Skp2. Cell Death Dis. 2013, 4, e693. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, Y. Salinomycin Suppresses LRP6 Expression and Inhibits Both Wnt/β-catenin and mTORC1 Signaling in Breast and Prostate Cancer Cells. J. Cell. Biochem. 2014, 115, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; David, A.E.; Wang, J.; Galbán, C.J.; Yang, V.C. Magnetic brain tumor targeting and biodistribution of long-circulating PEG-modified, cross-linked starch-coated iron oxide nanoparticles. Biomaterials 2011, 32, 6291–6301. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward | Reverse |
|---|---|---|
| TOP2 | ATTCCCAAACTCGATGATGC | CCCCATATTTGTCTCTCCCA |
| Ku70 | CTGTCCAAGTTGGTCGCTTC | CTGCCCCTTAAACTGGTCAA |
| p53 | TCTGAGTCAGGCCCTTCTGT | GTTCCGAGAGCTGAATGAGG |
| Caspase 9 | CACGGCAGAAGTTCACATTG | AACAGGCAAGCAGCAAAGTT |
| Caspase 3 | CTCTGGTTTTCGGTGGGTGT | CGCTTCCATGTATGATCTTTGGTT |
| Cyclin D | GTCCCACTCCTACGATACGC | CAGGGCCGTTGGGTAGAAAA |
| Wnt1 | CAACAGCAGTGGCCGATGGTGG | CGGCCTGCCTCGTTGTTGTGAAG |
| Rbl2 | GGTTCCCACTGAGTGATTACTGT | AGAAGCCTCCTATGCTCACG |
| GAS5 | TGGTTCTGCTCCTGGTAACG | AGGATAACAGGTCTGCCTGC |
| MIR 155 | AATCGTGATAGGGGTTTTTGCC | ATGTAGGAGTCAGTTGGAGGC |
| β-actin | AATGCCAGGGTACATGGTGG | AGGAAGGAAGGCTGGAAGAGTG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Miller, D.W. Salinomycin-Loaded Iron Oxide Nanoparticles for Glioblastoma Therapy. Nanomaterials 2020, 10, 477. https://doi.org/10.3390/nano10030477
Norouzi M, Yathindranath V, Thliveris JA, Miller DW. Salinomycin-Loaded Iron Oxide Nanoparticles for Glioblastoma Therapy. Nanomaterials. 2020; 10(3):477. https://doi.org/10.3390/nano10030477
Chicago/Turabian StyleNorouzi, Mohammad, Vinith Yathindranath, James A. Thliveris, and Donald W. Miller. 2020. "Salinomycin-Loaded Iron Oxide Nanoparticles for Glioblastoma Therapy" Nanomaterials 10, no. 3: 477. https://doi.org/10.3390/nano10030477
APA StyleNorouzi, M., Yathindranath, V., Thliveris, J. A., & Miller, D. W. (2020). Salinomycin-Loaded Iron Oxide Nanoparticles for Glioblastoma Therapy. Nanomaterials, 10(3), 477. https://doi.org/10.3390/nano10030477