Peptide-Carbon Quantum Dots conjugate, Derived from Human Retinoic Acid Receptor Responder Protein 2, against Antibiotic-Resistant Gram Positive and Gram Negative Pathogenic Bacteria

 , , , ,
, , , ,
Abstract
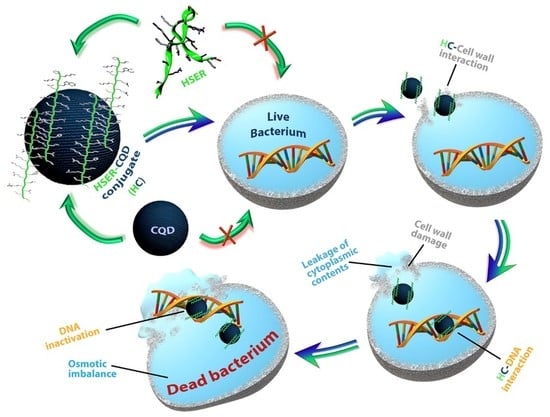
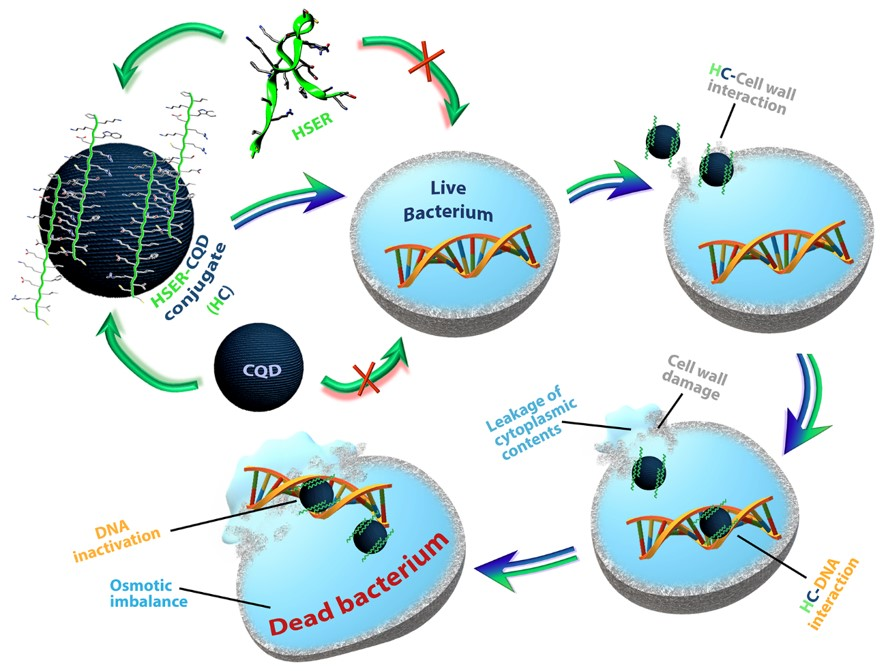
1. Introduction
2. Experimental
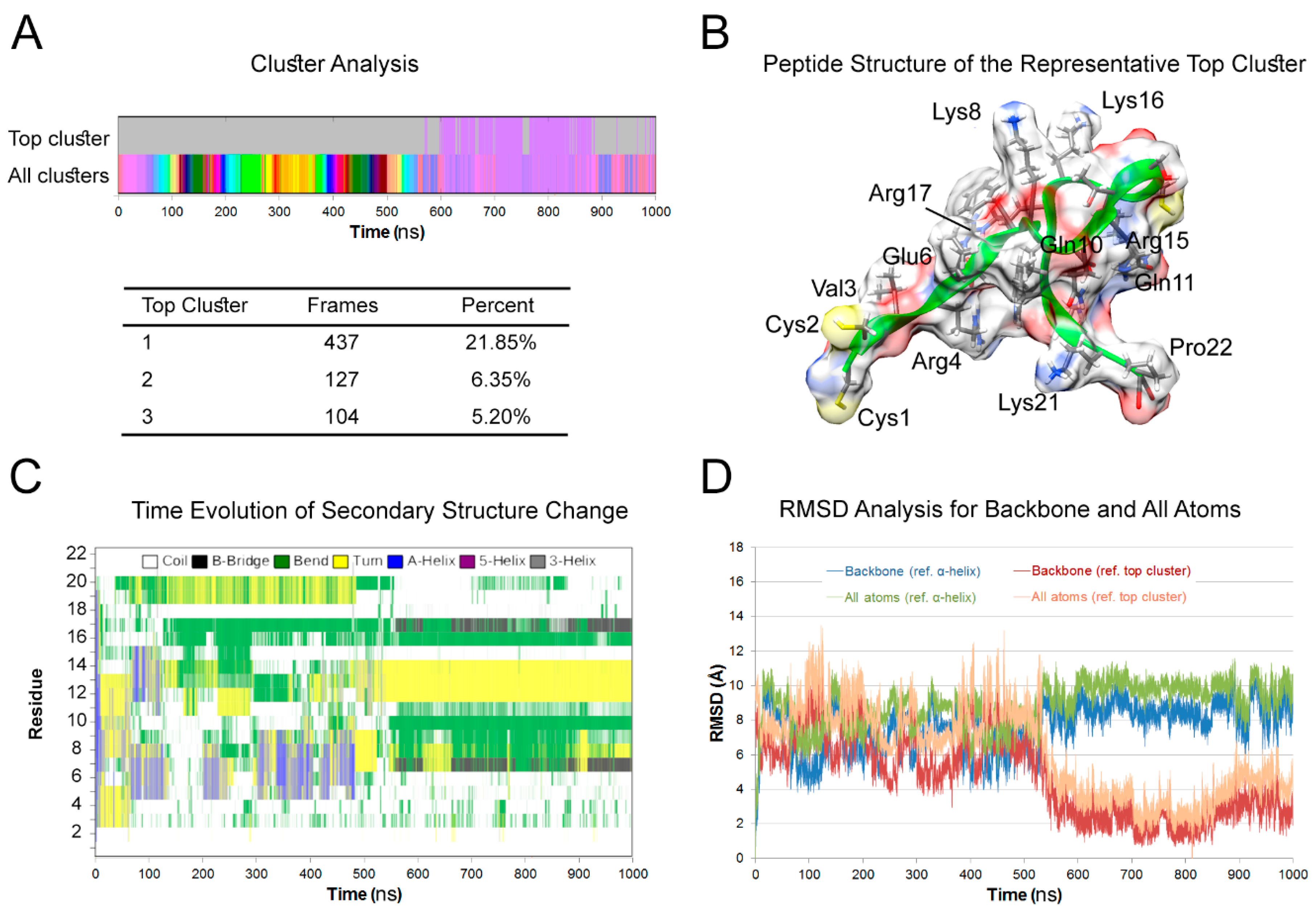
2.1. Molecular Dynamics (MD) Simulation
2.2. Chemicals and Synthesis of the HSER-CQDs Conjugate
2.3. Characterization of the HSER-CQDs Conjugate
2.4. Cultivation of Bacteria
2.5. Minimum Inhibitory Concentration (MIC) Determination
2.6. Growth Curves and Viability Percentage
2.7. Microscopy of Antibacterial Agents against Bacteria in Ambient Light and Live/Dead Cell Assay
2.8. Cell Membrane Break and Leakage Assay and Interaction with DNA
2.9. Influence and Toxicity of HSER–CQDs on Eukaryotic Cells (Cytotoxicity Assay and Hemolysis Assay)
2.10. Endogenous Reactive Oxygen Species (ROS) Production Assay
2.11. Microscopic Analysis of HSER-CQDs Interaction against Eukaryotic Cells
3. Results and Discussion
3.1. Peptide Structure
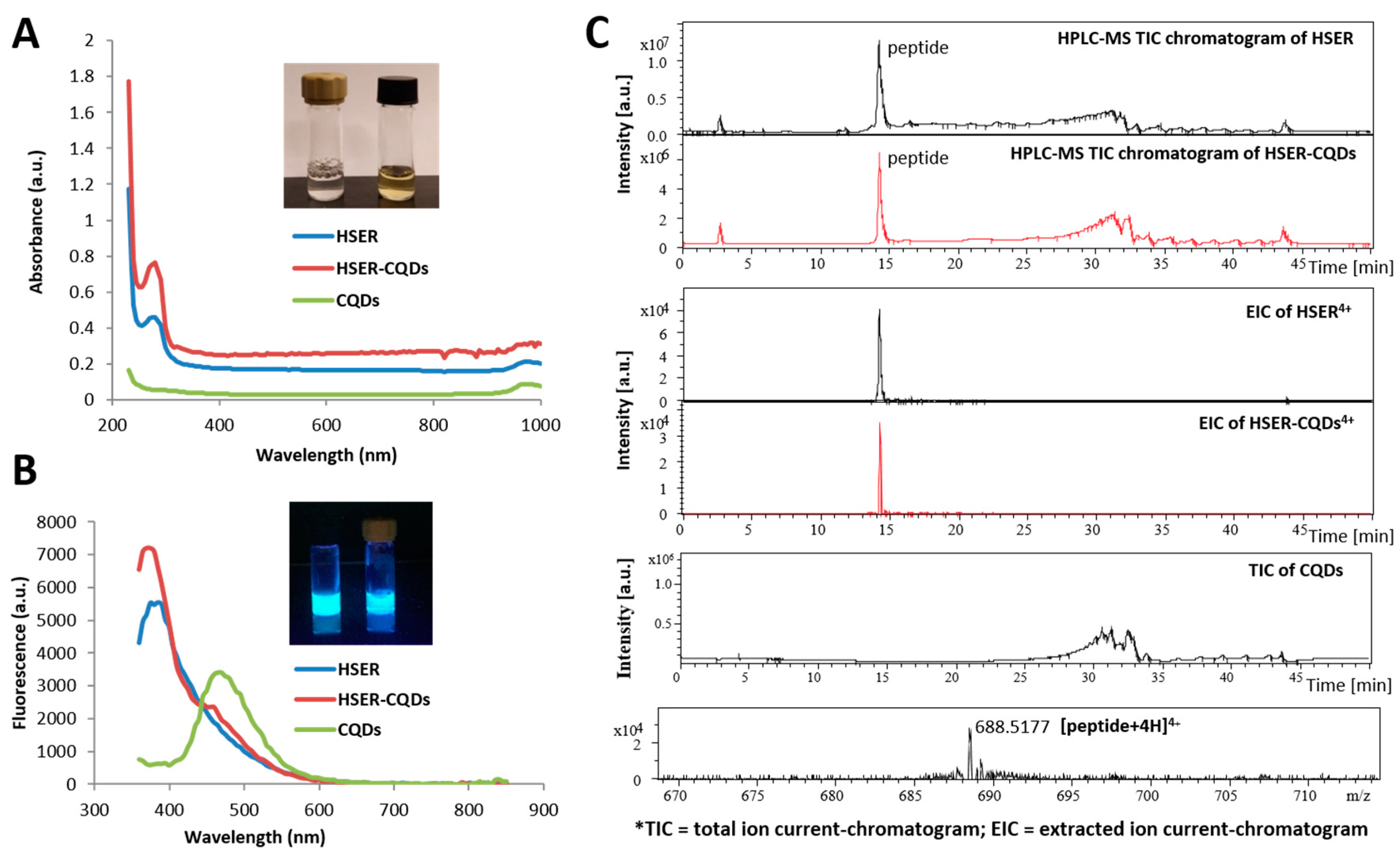
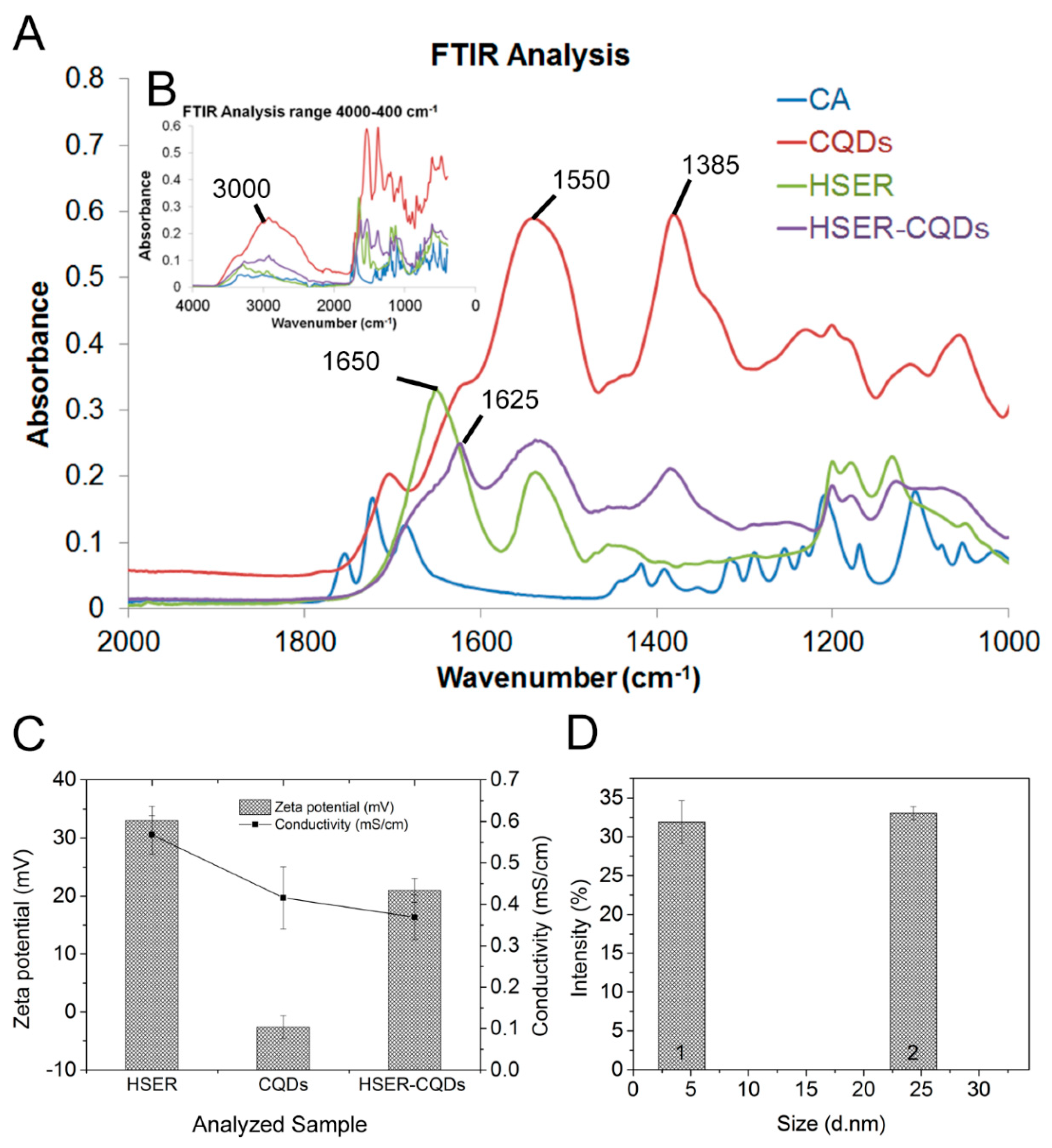
3.2. Synthesis and Characterization of the HSER-CQD Conjugate
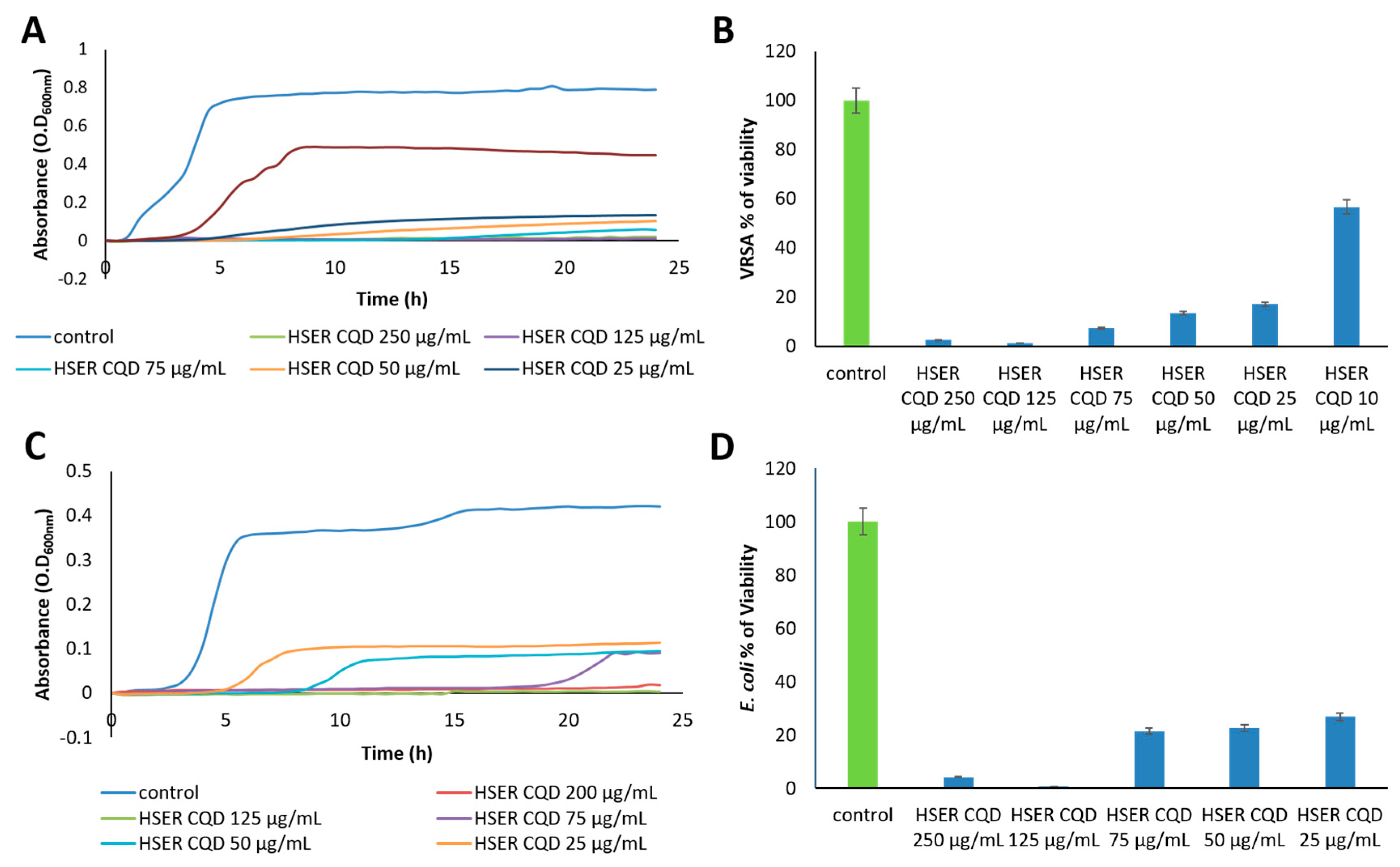
3.3. Effect of HSER-CQDs on Different Bacterial Strains
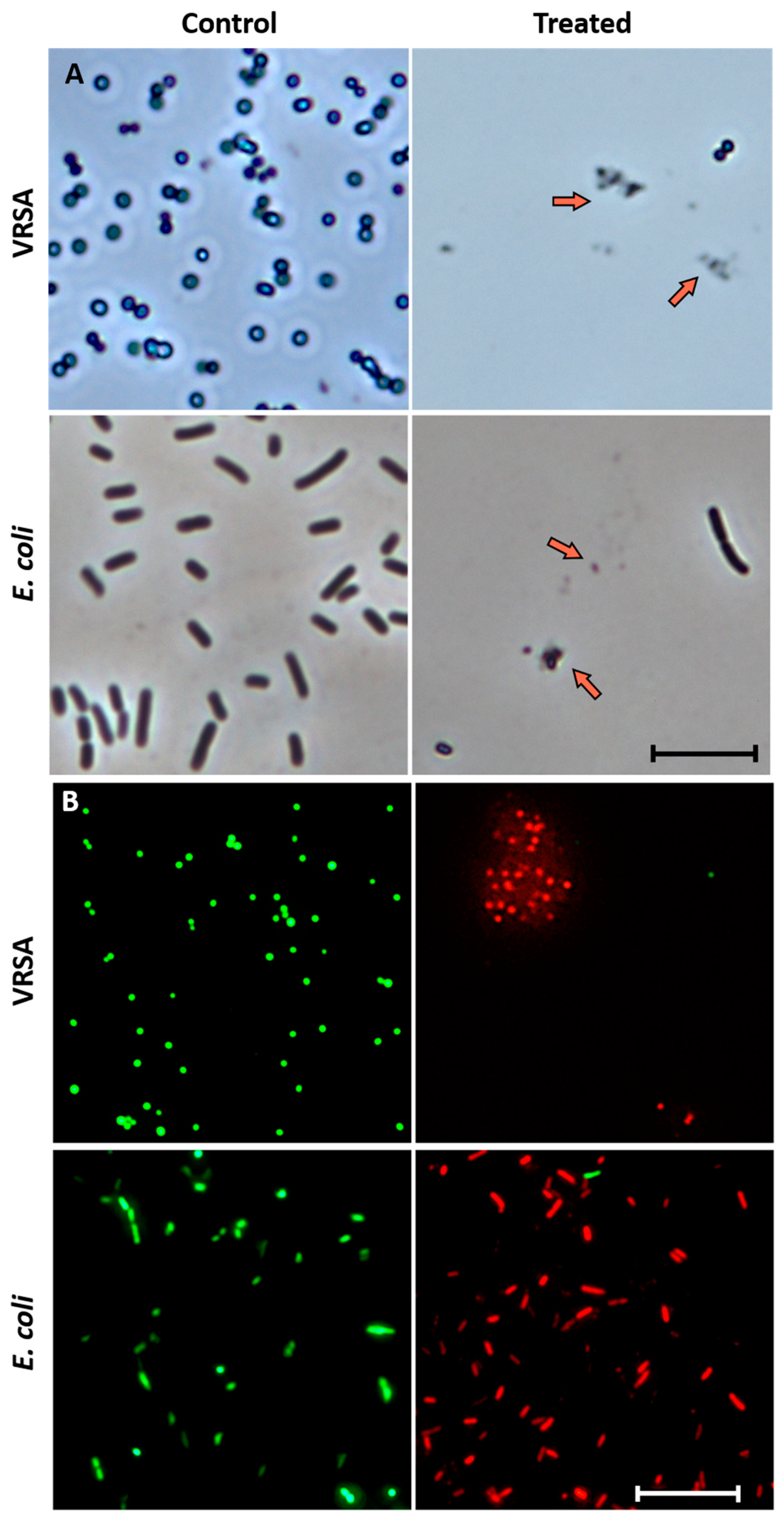
3.4. Microscopic Estimation of Live/Dead Cells and Damage of DNA (Bacterial Cells)
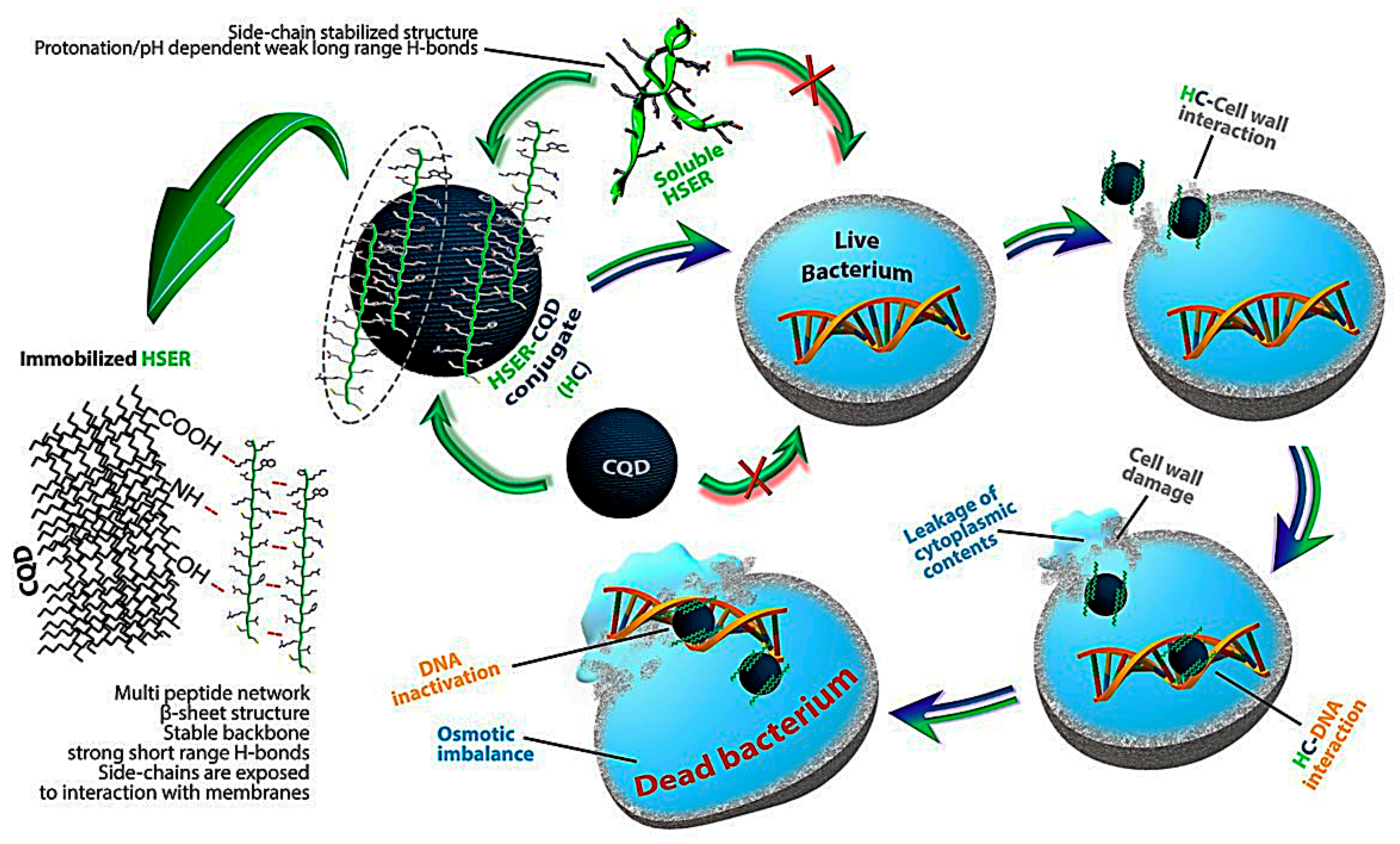
3.5. Mechanism of Action by Interaction with Bacterial Cells, DNA, Changes in Secondary Structure, and Membrane Insertion
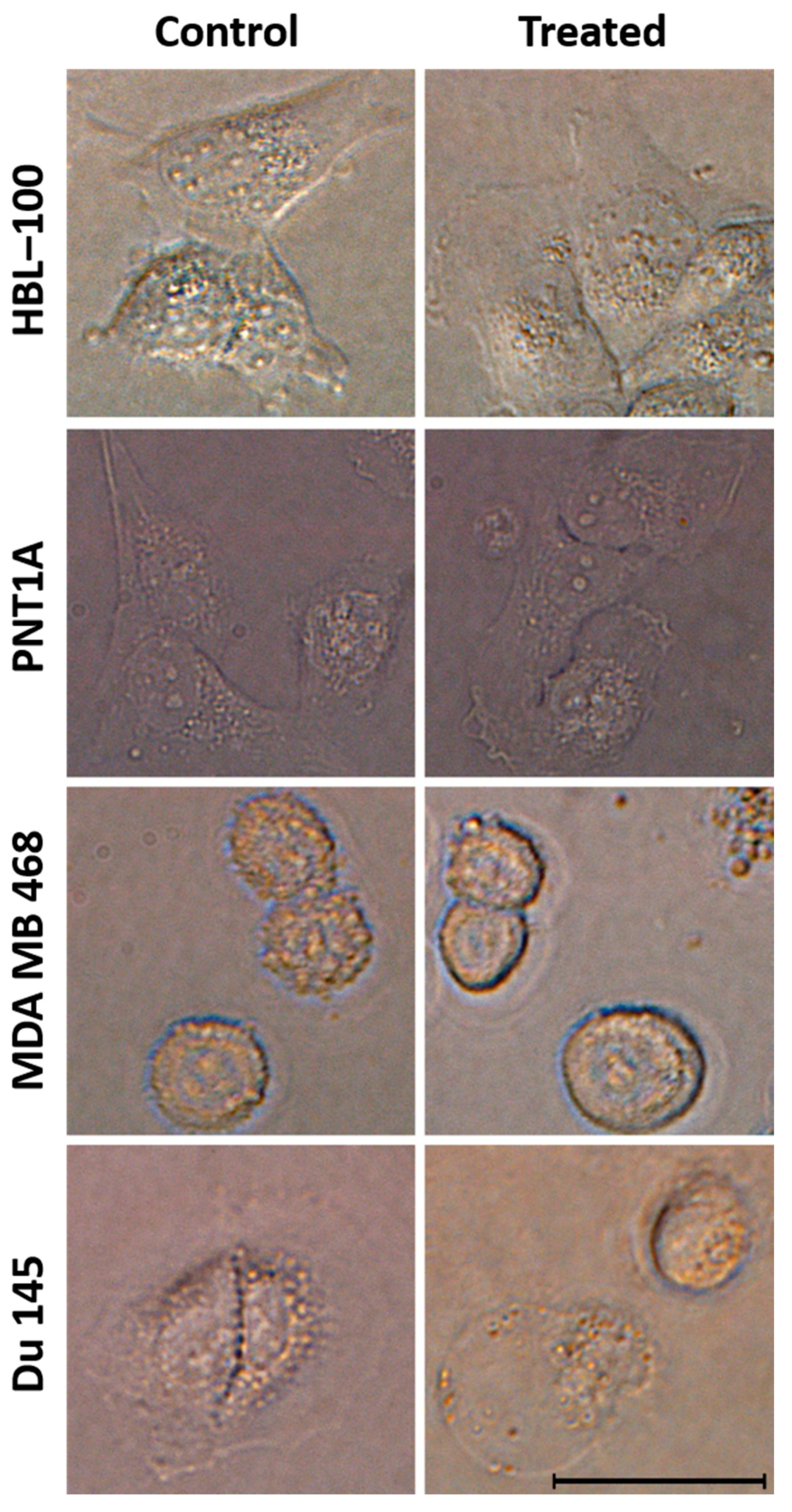
3.6. Influence and Cytotoxicity Test of HSER–CQD on Eukaryotic Cells
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- McNeece, G.; Naughton, V.; Woodward, M.J.; Dooley, J.S.G.; Naughton, P.J. Array based detection of antibiotic resistance genes in Gram negative bacteria isolated from retail poultry meat in the UK and Ireland. Int. J. Food Microbiol. 2014, 179, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. MMBR 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Juknius, T.; Tamulevičius, T.; Gražulevičiūtė, I.; Klimienė, I.; Matusevičius, A.P.; Tamulevičius, S. In-situ measurements of bacteria resistance to antimicrobial agents employing leaky mode sub-wavelength diffraction grating. Sens. Actuators B Chem. 2014, 204, 799–806. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Alexander, E.L.; Gardete, S.; Bar, H.Y.; Wells, M.T.; Tomasz, A.; Rhee, K.Y. Intermediate-Type Vancomycin Resistance (VISA) in Genetically-Distinct Staphylococcus aureus Isolates Is Linked to Specific, Reversible Metabolic Alterations. PLoS ONE 2014, 9, e97137. [Google Scholar] [CrossRef]
- Doi, Y.; Paterson, D.L. Carbapenemase-producing Enterobacteriaceae. Semin. Respir. Crit. Care Med. 2015, 36, 74–84. [Google Scholar]
- World Health Organization. Guidelines for the Prevention and Control of Carbapenem-Resistant Enterobacteriaceae, Acinetobacter Baumannii and Pseudomonas Aeruginosa in Health Care Facilities; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Phe, K.; Lee, Y.; McDaneld, P.M.; Prasad, N.; Yin, T.; Figueroa, D.A.; Musick, W.L.; Cottreau, J.M.; Hu, M.; Tam, V.H. In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob. Agents Chemother. 2014, 58, 2740–2746. [Google Scholar] [CrossRef]
- Amladi, A.U.; Abirami, B.; Devi, S.M.; Sudarsanam, T.D.; Kandasamy, S.; Kekre, N.; Veeraraghavan, B.; Sahni, R.D. Susceptibility profile, resistance mechanisms & efficacy ratios of fosfomycin, nitrofurantoin & colistin for carbapenem-resistant Enterobacteriaceae causing urinary tract infections. Indian J. Med Res. 2019, 149, 185–191. [Google Scholar]
- Rincón, S.; Panesso, D.; Díaz, L.; Carvajal, L.P.; Reyes, J.; Munita, J.M.; Arias, C.A. Resistencia a antibióticos de última línea en cocos Gram positivos: La era posterior a la vancomicina. Biomed. Rev. Inst. Nac. Salud 2014, 34 (Suppl. 1), 191–208. [Google Scholar] [CrossRef]
- Zhu, W.L.; Lan, H.; Park, I.-S.; Kim, J.I.; Jin, H.Z.; Hahm, K.-S.; Shin, S.Y. Design and mechanism of action of a novel bacteria-selective antimicrobial peptide from the cell-penetrating peptide Pep-1. Biochem. Biophys. Res. Commun. 2006, 349, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Groh, T.; Hrabeta, J.; Khalil, M.A.; Doktorova, H.; Eckschlager, T.; Stiborova, M. The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. Int. J. Oncol. 2015, 47, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Q.; Zeng, X.; Ye, Q.; Huang, S.; Yu, H.; Yang, T.; Qiao, S. Use of the Antimicrobial Peptide Sublancin with Combined Antibacterial and Immunomodulatory Activities To Protect against Methicillin-Resistant Staphylococcus aureus Infection in Mice. J. Agric. Food Chem. 2017, 65, 8595–8605. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, X.; Yang, Q.; Qiao, S. Antimicrobial Peptides as Potential Alternatives to Antibiotics in Food Animal Industry. Int. J. Mol. Sci. 2016, 17, 603. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, J.; Falciani, C.; Roscia, G.; Pollini, S.; Bindi, S.; Scali, S.; Arrieta, U.C.; Gómez-Vallejo, V.; Quercini, L.; Ibba, E.; et al. In vitro and in vivo efficacy, toxicity, bio-distribution and resistance selection of a novel antibacterial drug candidate. Sci. Rep. 2016, 6, 26077. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, P.; Mazumdar, A.; Sur, V.P.; Kociova, S.; Dolezelikova, K.; Jimenez, A.M.J.; Koudelkova, Z.; Mishra, P.K.; Smerkova, K.; Heger, Z.; et al. Nanoparticle-drug conjugates treating bacterial infections. J. Control. Release 2019, 307, 166–185. [Google Scholar] [CrossRef]
- Moradlou, O.; Rabiei, Z.; Delavari, N.J.J.o.P.; Chemistry, P.A. Antibacterial effects of carbon quantum dots@ hematite nanostructures deposited on titanium against Gram-positive and Gram-negative bacteria. J. Photochem. Photobiol. A Chem. 2019, 379, 144–149. [Google Scholar] [CrossRef]
- Kováčová, M.; Špitalská, E.; Markovic, Z.; Špitálský, Z. Carbon Quantum Dots As Antibacterial Photosensitizers and Their Polymer Nanocomposite Applications. Part. Part. Syst. Charact. 2020, 37, 1900348. [Google Scholar] [CrossRef]
- Dong, X.; Liang, W.; Meziani, M.J.; Sun, Y.-P.; Yang, L.J.T. Carbon Dots as Potent Antimicrobial Agents. Theranostics 2020, 10, 671. [Google Scholar] [CrossRef]
- Lin, F.; Bao, Y.W.; Wu, F.G. Carbon dots for sensing and killing microorganisms. C J. Carbon Res. 2019, 5, 33. [Google Scholar] [CrossRef]
- Zabel, B.A.; Kwitniewski, M.; Banas, M.; Zabieglo, K.; Murzyn, K.; Cichy, J. Chemerin regulation and role in host defense. Am. J. Clin. Exp. Immunol. 2014, 3, 1–19. [Google Scholar] [PubMed]
- Milosavljevic, V.; Nguyen, H.V.; Michalek, P.; Moulick, A.; Kopel, P.; Kizek, R.; Adam, V. Synthesis of carbon quantum dots for DNA labeling and its electrochemical, fluorescent and electrophoretic characterization. Chem. Pap. 2015, 69, 192–201. [Google Scholar] [CrossRef]
- Wang, F.; Pang, S.; Wang, L.; Li, Q.; Kreiter, M.; Liu, C.-y. One-SteSynthesis of Highly Luminescent Carbon Dots in Noncoordinating Solvents. Chem. Mater. 2010, 22, 4528–4530. [Google Scholar] [CrossRef]
- Moulick, A.; Heger, Z.; Milosavljevic, V.; Richtera, L.; Barroso-Flores, J.; Merlos Rodrigo, M.A.; Buchtelova, H.; Podgajny, R.; Hynek, D.; Kopel, P.; et al. Real-Time Visualization of Cell Membrane Damage Using Gadolinium–Schiff Base Complex-Doped Quantum Dots. ACS Appl. Mater. Interfaces 2018, 10, 35859–35868. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, P.; Splichal, Z.; Jimenez, A.M.J.; Haddad, Y.; Mazumdar, A.; Sur, V.P.; Milosavljevic, V.; Kopel, P.; Buchtelova, H.; Guran, R.; et al. Novel vancomycin-peptide conjugate as potent antibacterial agent against vancomycin-resistant Staphylococcus aureus. Infect. Drug Resist. 2018, 11, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, P.; Vesely, R.; Cihalova, K.; Hegerova, D.; Ananbeh, H.A.A.A.; Richtera, L.; Smerkova, K.; Brtnicky, M.; Kynicky, J.; Moulick, A.; et al. Effect of arsenic (III and V) on oxidative stress parameters in resistant and susceptible Staphylococcus aureus. Environ. Res. 2018, 166, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Heger, Z.; Merlos Rodrigo, M.A.; Michalek, P.; Polanska, H.; Masarik, M.; Vit, V.; Plevova, M.; Pacik, D.; Eckschlager, T.; Stiborova, M.; et al. Sarcosine Up-Regulates Expression of Genes Involved in Cell Cycle Progression of Metastatic Models of Prostate Cancer. PLoS ONE 2016, 11, e0165830. [Google Scholar] [CrossRef]
- Agbor, T.A.; Demma, Z.; Mrsny, R.J.; Castillo, A.; Boll, E.J.; McCormick, B.A.J.C.m. The oxido–reductase enzyme glutathione peroxidase 4 (GPX4) governs S almonella T yphimurium–induced neutrophil transepithelial migration. Cell. Microbiol. 2014, 16, 1339–1353. [Google Scholar] [CrossRef]
- Li, Y.; Lv, M.; Su, C.; Long, S.; Zhang, W.; Conway, K.L.; Li, W.; Xavier, R.J.; Shi, H.N.J.F.i.i. p40phox-Deficient Mice Exhibit Impaired Bacterial Clearance and Enhanced Pro-inflammatory Responses during Salmonella enterica serovar Typhimurium Infection. Front. Immunol. 2017, 8, 1270. [Google Scholar] [CrossRef]
- Tavares, A.F.N.; Teixeira, M.; Romão, C.C.; Seixas, J.D.; Nobre, L.S.; Saraiva, L.M. Reactive Oxygen Species Mediate Bactericidal Killing Elicited by Carbon Monoxide-releasing Molecules. J. Biol. Chem. 2011, 286, 26708–26717. [Google Scholar] [CrossRef]
- Antosiewicz, M.J.; Shugar, D. UV–Vis spectroscopy of tyrosine side-groups in studies of protein structure. Part 2: Selected applications. Biophys. Rev. 2016, 8, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Emam, A.N.; Loutfy, S.A.; Mostafa, A.A.; Awad, H.; Mohamed, M.B. Cyto-toxicity, biocompatibility and cellular response of carbon dots–plasmonic based nano-hybrids for bioimaging. RSC Adv. 2017, 7, 23502–23514. [Google Scholar] [CrossRef]
- Xu, M.; He, G.; Li, Z.; He, F.; Gao, F.; Su, Y.; Zhang, L.; Yang, Z.; Zhang, Y. A green heterogeneous synthesis of N-doped carbon dots and their photoluminescence applications in solid and aqueous states. Nanoscale 2014, 6, 10307–10315. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lu, Y.; Yuan, H.; Ren, Z.; Xu, C.; Chen, J. Microplasma-assisted rapid synthesis of luminescent nitrogen-doped carbon dots and their application in pH sensing and uranium detection. Nanoscale 2015, 7, 20743–20748. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, Y.; Zhang, R.; He, S.; Ju, J.; Liu, M.; Li, L.; Chen, W. One-pot synthesis of carbon nanodots for fluorescence turn-on detection of Ag+ based on the Ag+-induced enhancement of fluorescence. J. Mater. Chem. C 2015, 3, 2302–2309. [Google Scholar] [CrossRef]
- Pan, K.; Chen, H.; Baek, S.J.; Zhong, Q. Self-assembled curcumin-soluble soybean polysaccharide nanoparticles, physicochemical properties and in vitro anti-proliferation activity against cancer cells. Food Chem. 2018, 246, 82–89. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of infrared spectra, a practical approach. Encycl. Anal. Chem. Appl. Theory Instrum. 2006. [Google Scholar] [CrossRef]
- Jackson, M.; Mantsch, H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta (BBA)-Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Barth, A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Stiefel, P.; Schmidt-Emrich, S.; Maniura-Weber, K.; Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol. 2015, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell–penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, H.; He, Q. Taming cell penetrating peptides: Never too old to teach old dogs new tricks. Mol. Pharm. 2015, 12, 3105–3118. [Google Scholar] [CrossRef]
- Soares, J.W.; Kirby, R.; Doherty, L.A.; Meehan, A.; Arcidiacono, S. Immobilization and orientation–dependent activity of a naturally occurring antimicrobial peptide. J. Pept. Sci. 2015, 21, 669–679. [Google Scholar] [CrossRef]
- Dorosz, J.; Gofman, Y.; Kolusheva, S.; Otzen, D.; Ben-Tal, N.; Nielsen, N.C.; Jelinek, R. Membrane interactions of novicidin, a novel antimicrobial peptide, phosphatidylglycerol promotes bilayer insertion. J. Phys. Chem. B 2010, 114, 11053–11060. [Google Scholar] [CrossRef]
- Balakrishnan, S.V.; Vad, B.S.; Otzen, D.E. Novicidin’s membrane permeabilizing activity is driven by membrane partitioning but not by helicity: A biophysical study of the impact of lipid charge and cholesterol. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 996–1002. [Google Scholar] [CrossRef]
- Kim, S.R.; Park, M.J.; Lee, M.K.; Sung, S.H.; Park, E.J.; Kim, J.; Kim, S.Y.; Oh, T.H.; Markelonis, G.J.; Kim, Y.C. Flavonoids of Inula britannica protect cultured cortical cells from necrotic cell death induced by glutamate. Free Radic. Biol. Med. 2002, 32, 596–604. [Google Scholar] [CrossRef]
- Shoemaker, M.; Cohen, I.; Campbell, M. Reduction of MTT by aqueous herbal extracts in the absence of cells. J. Ethnopharmacol. 2004, 93, 381–384. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | HSER-CQDs (μg/mL) | HSER (μg/mL) | CQDs (μg/mL) |
|---|---|---|---|
| VRSA | 25 | >125 | >125 |
| E. coli | 50 | >125 | >125 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazumdar, A.; Haddad, Y.; Milosavljevic, V.; Michalkova, H.; Guran, R.; Bhowmick, S.; Moulick, A. Peptide-Carbon Quantum Dots conjugate, Derived from Human Retinoic Acid Receptor Responder Protein 2, against Antibiotic-Resistant Gram Positive and Gram Negative Pathogenic Bacteria. Nanomaterials 2020, 10, 325. https://doi.org/10.3390/nano10020325
Mazumdar A, Haddad Y, Milosavljevic V, Michalkova H, Guran R, Bhowmick S, Moulick A. Peptide-Carbon Quantum Dots conjugate, Derived from Human Retinoic Acid Receptor Responder Protein 2, against Antibiotic-Resistant Gram Positive and Gram Negative Pathogenic Bacteria. Nanomaterials. 2020; 10(2):325. https://doi.org/10.3390/nano10020325
Chicago/Turabian StyleMazumdar, Aninda, Yazan Haddad, Vedran Milosavljevic, Hana Michalkova, Roman Guran, Sukanya Bhowmick, and Amitava Moulick. 2020. "Peptide-Carbon Quantum Dots conjugate, Derived from Human Retinoic Acid Receptor Responder Protein 2, against Antibiotic-Resistant Gram Positive and Gram Negative Pathogenic Bacteria" Nanomaterials 10, no. 2: 325. https://doi.org/10.3390/nano10020325
APA StyleMazumdar, A., Haddad, Y., Milosavljevic, V., Michalkova, H., Guran, R., Bhowmick, S., & Moulick, A. (2020). Peptide-Carbon Quantum Dots conjugate, Derived from Human Retinoic Acid Receptor Responder Protein 2, against Antibiotic-Resistant Gram Positive and Gram Negative Pathogenic Bacteria. Nanomaterials, 10(2), 325. https://doi.org/10.3390/nano10020325