2D Monomolecular Nanosheets Based on Thiacalixarene Derivatives: Synthesis, Solid State Self-Assembly and Crystal Polymorphism


 , , ,
, , , 
Abstract
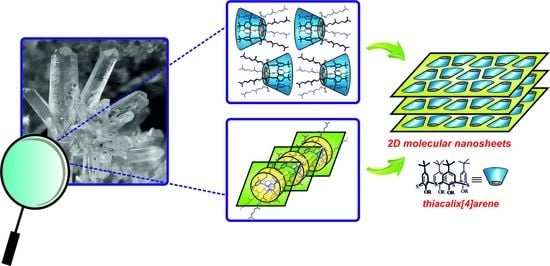
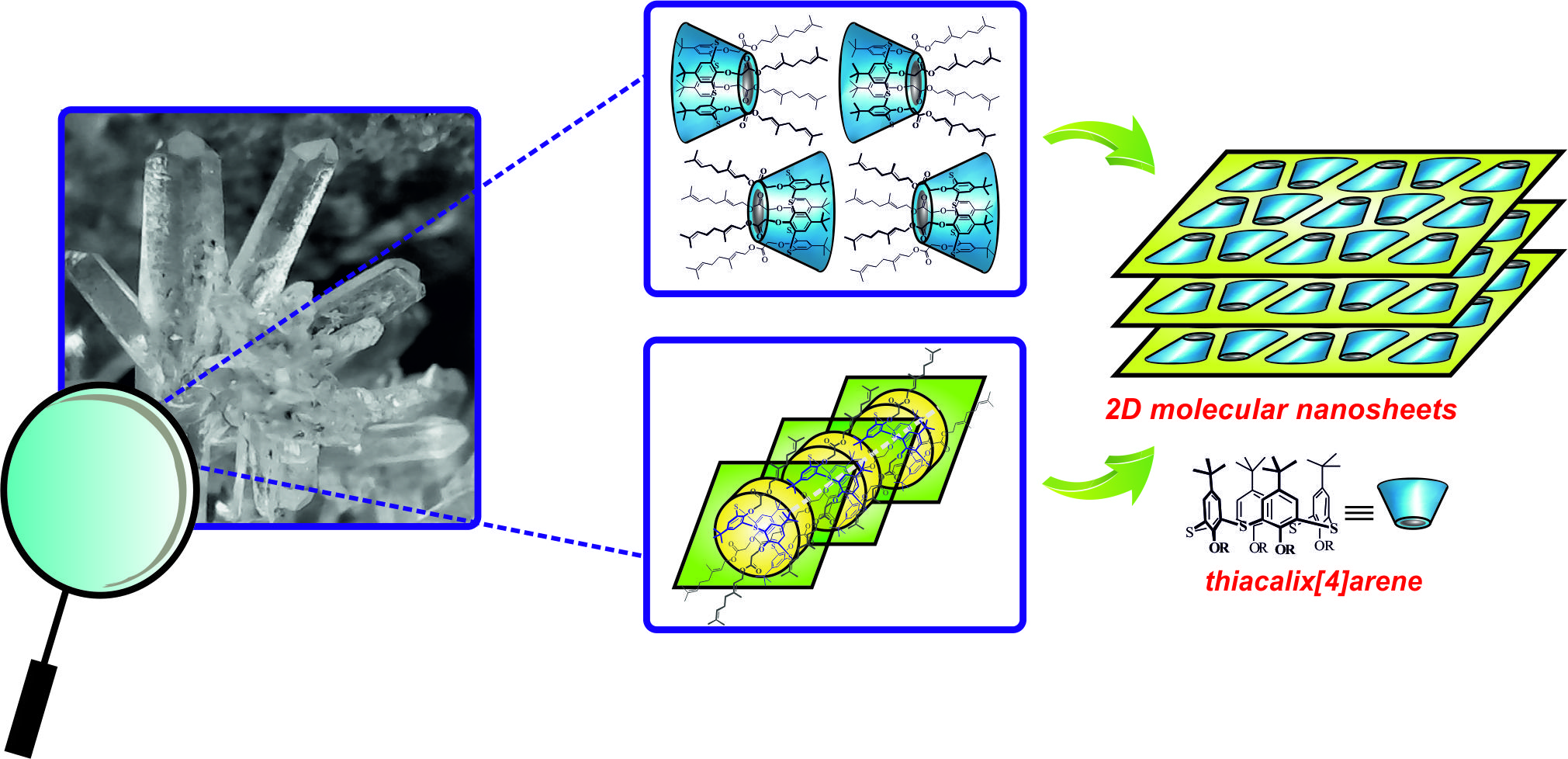{kind=link}
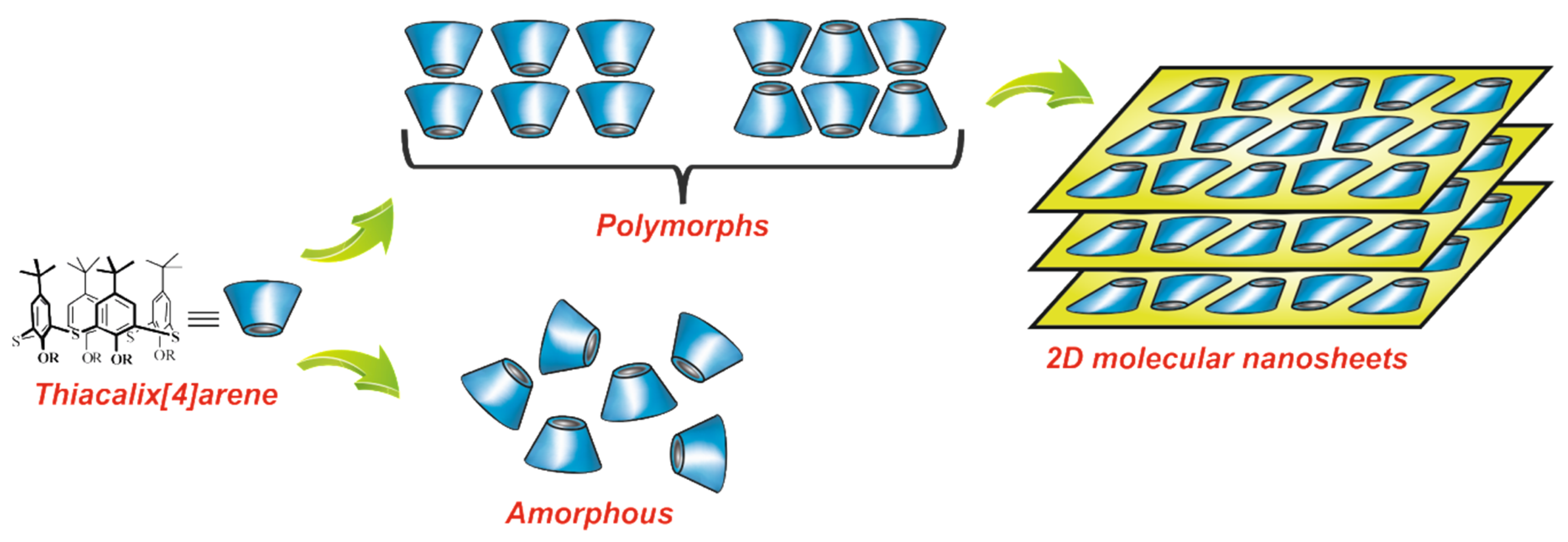{kind=link}
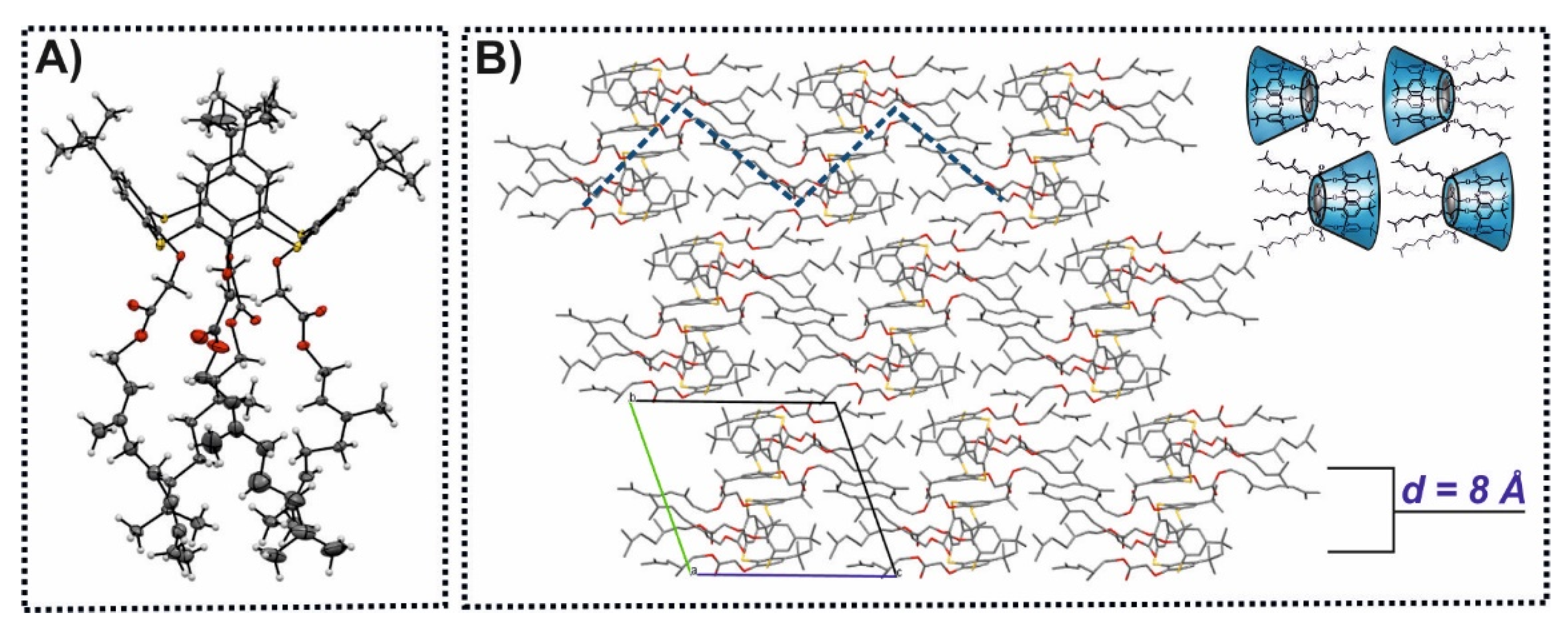{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis
2.2.1. Geranyl Bromoacetate (2)
2.2.2. 5,11,17,23-Tetra-Tert-Butyl-25,26,27,28-Tetra[(Geranyloxycarbonyl)-Methoxy]-2,8,14,20-Tetrathiacalix[4]arene (Cone-4)
2.2.3. 5,11,17,23-Tetra-Tert-Butyl-25,26,27,28-Tetra[(Geranyloxycarbonyl)-Methoxy]-2,8,14,20-Tetrathiacalix[4]arene (1,3-Alternate-5)
2.3. Differential Scanning Calorimetry (DSC)
2.4. Fast Scanning Calorimetry (FSC)
2.5. Powder X-ray Diffraction (PXRD) Experiment
2.6. X-ray Diffraction (XRD)
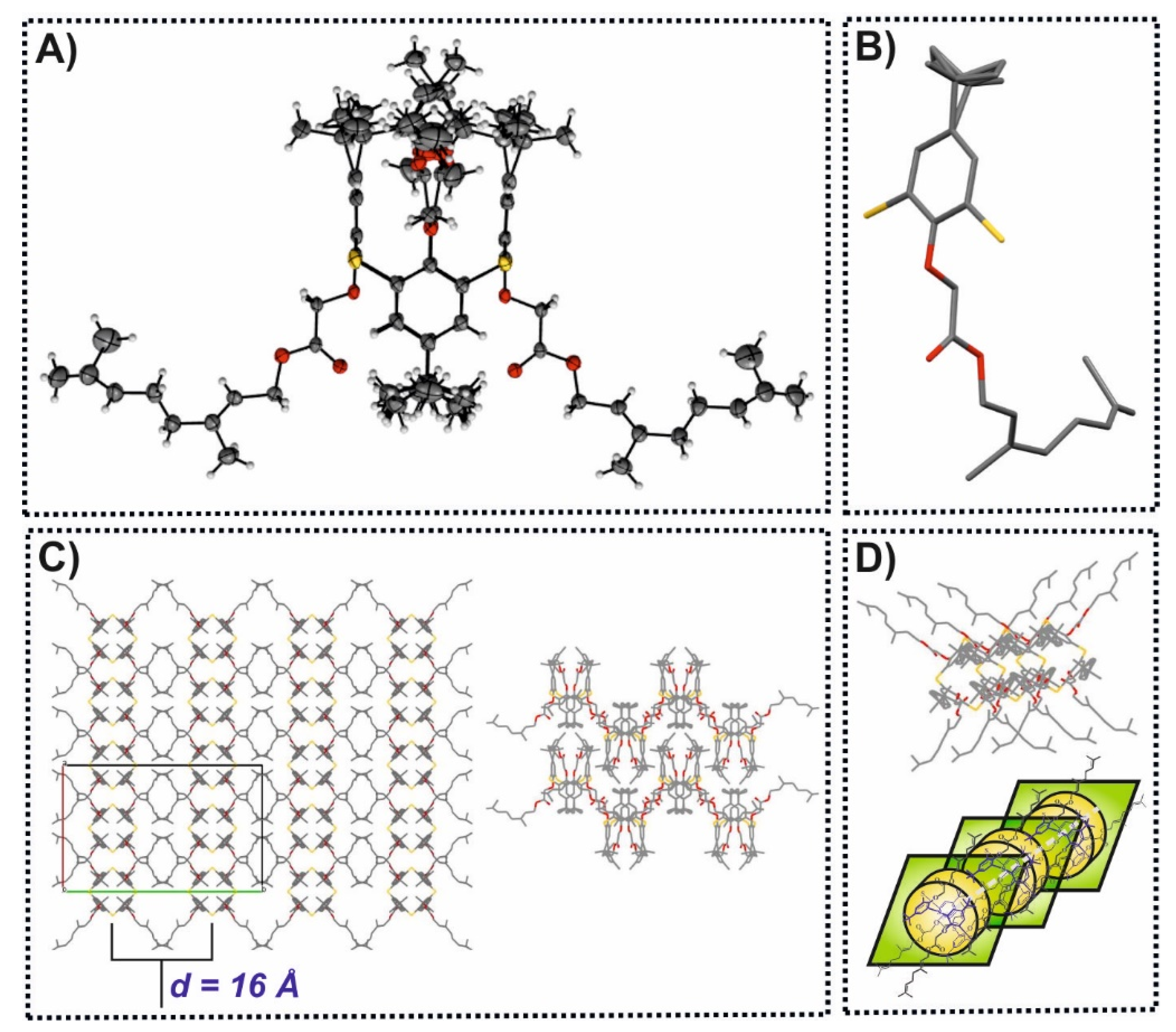
3. Results
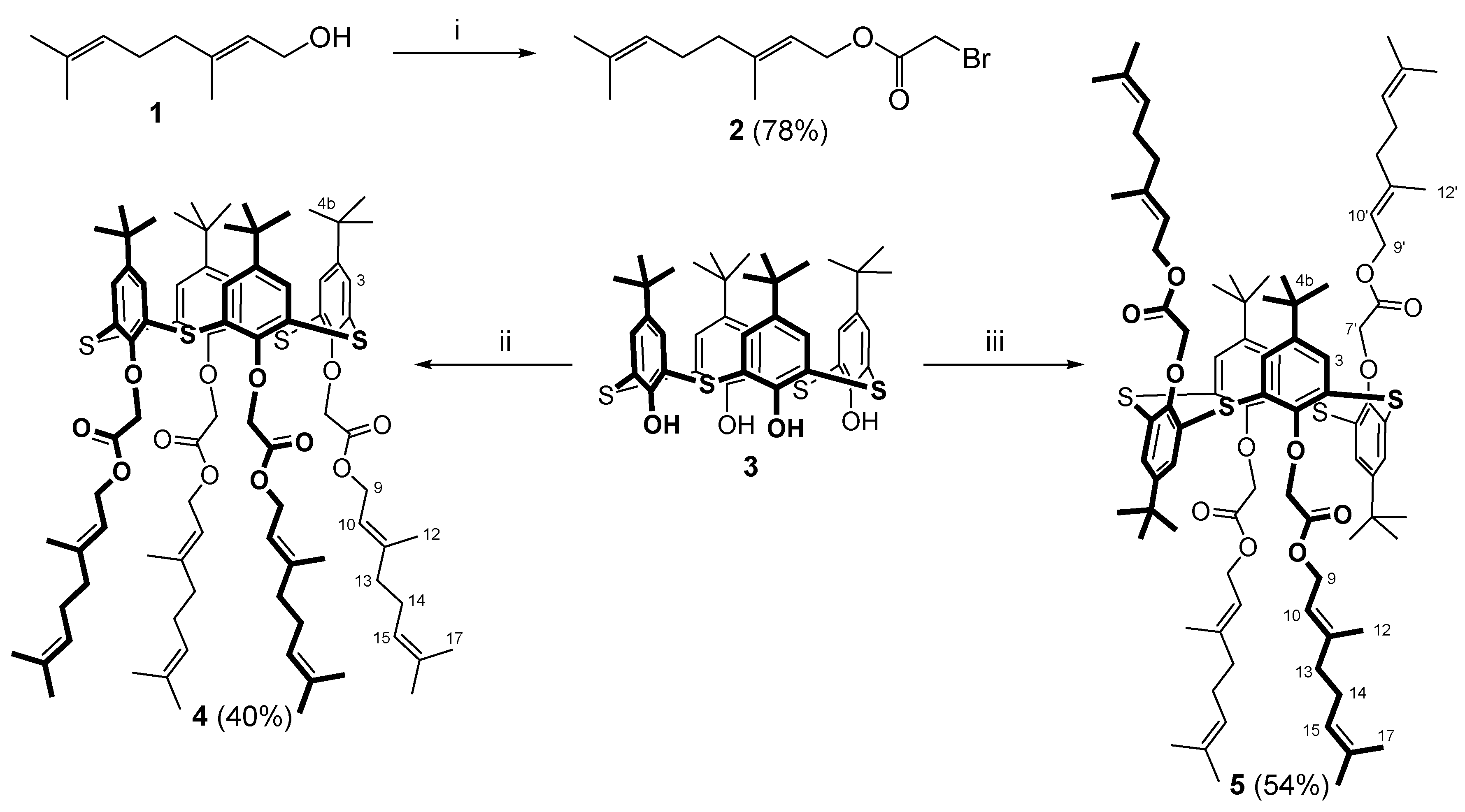
3.1. Synthesis of Thiacalix[4]arene Derivatives
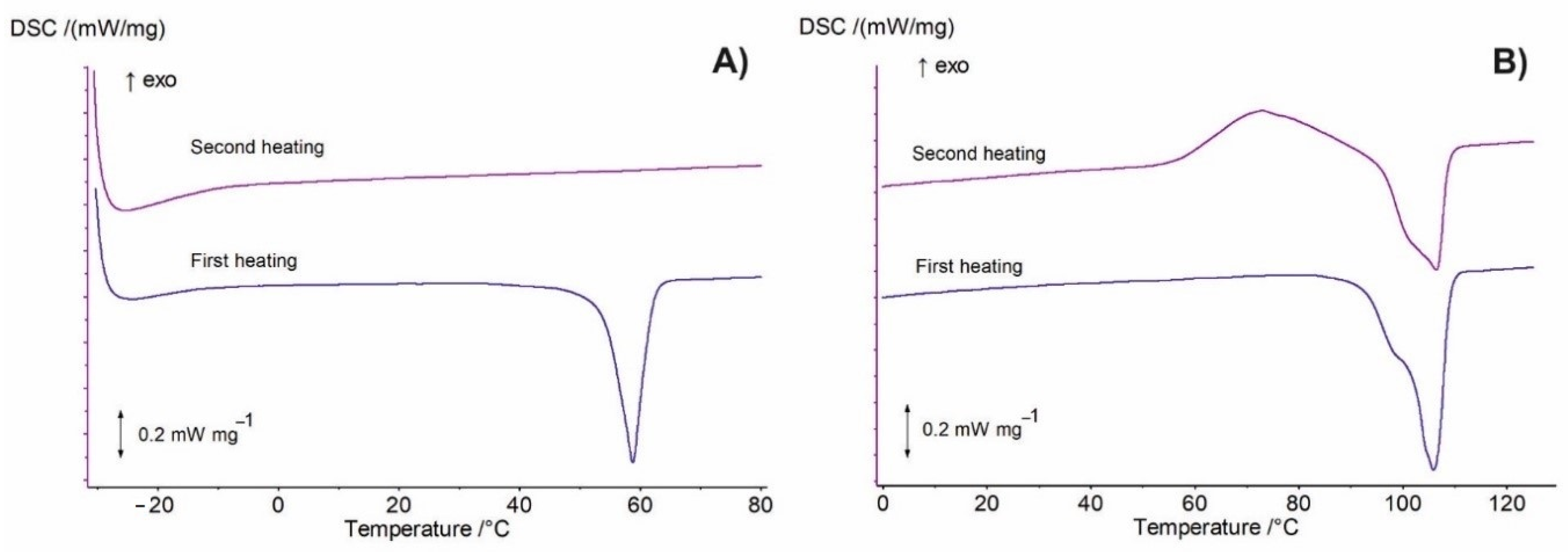
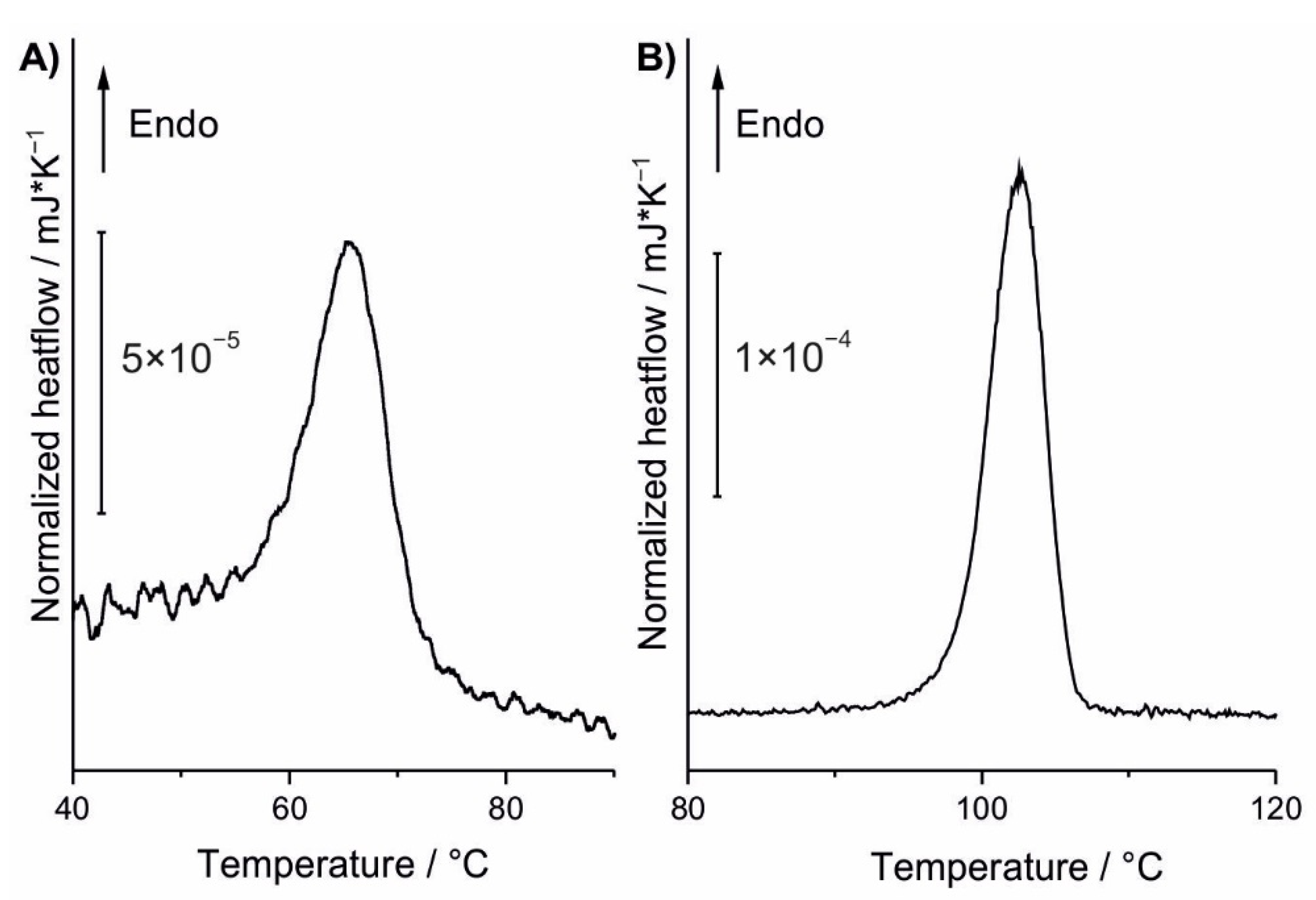
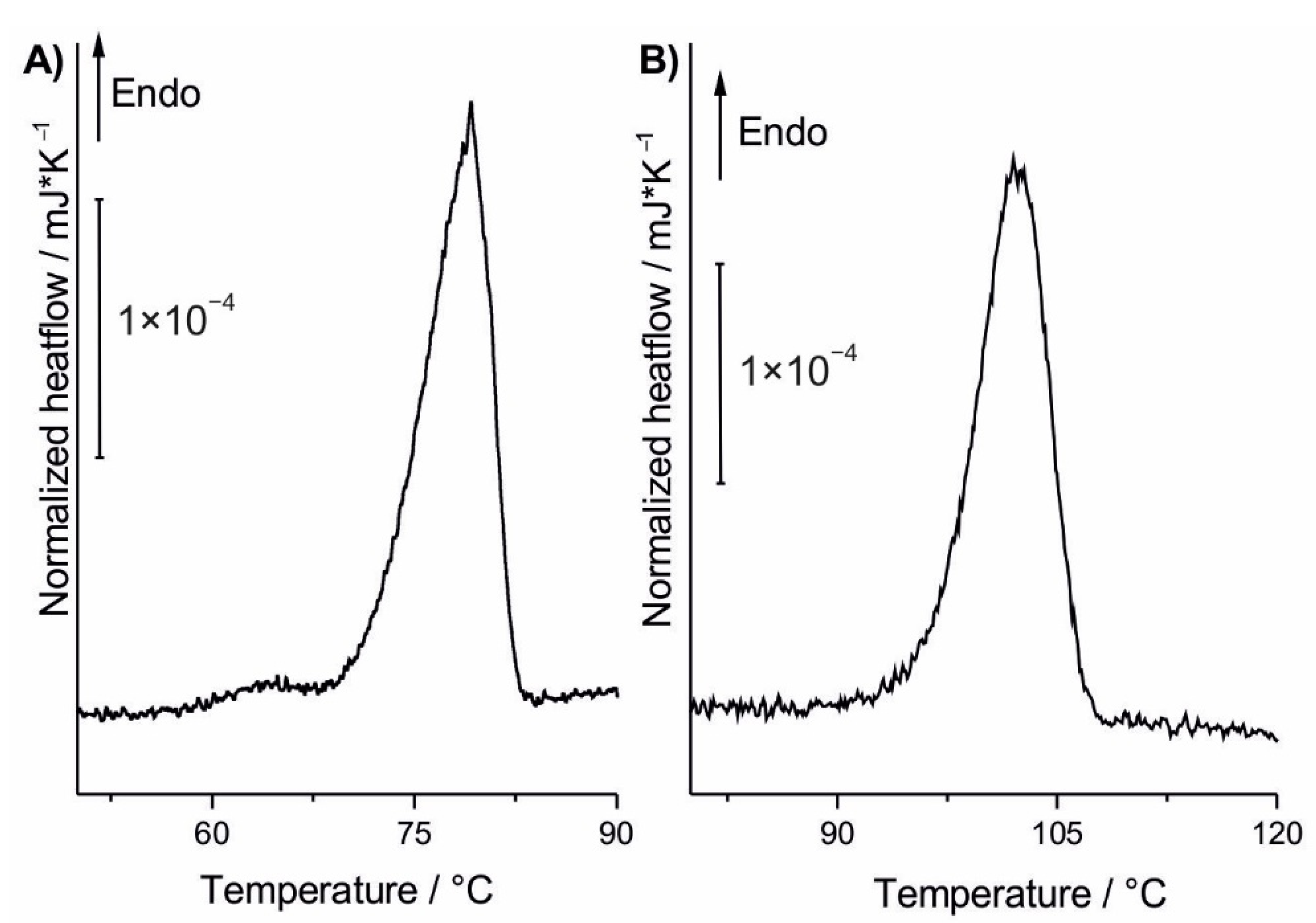
3.2. DSC Analysis
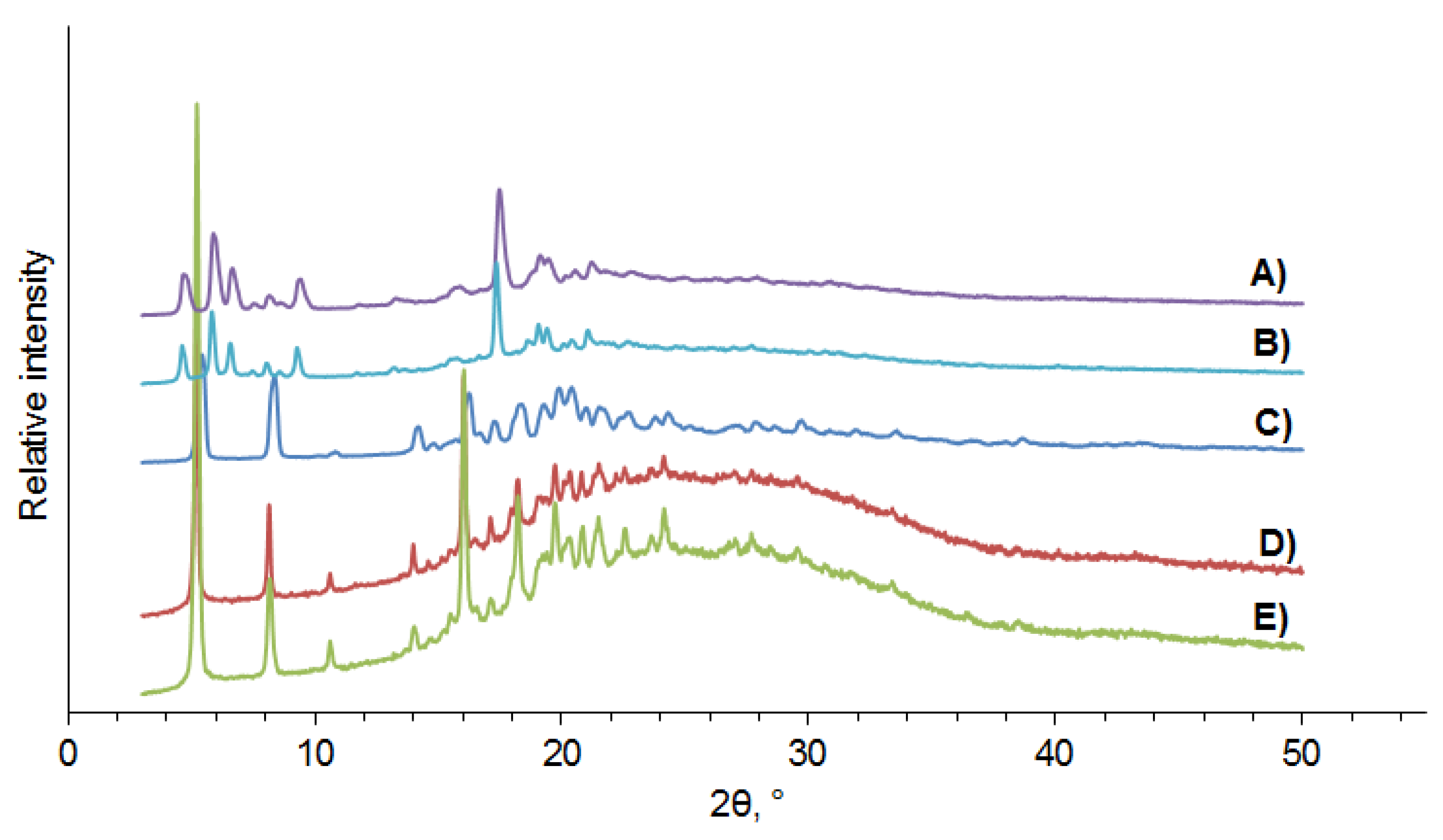
3.3. PXRD Data
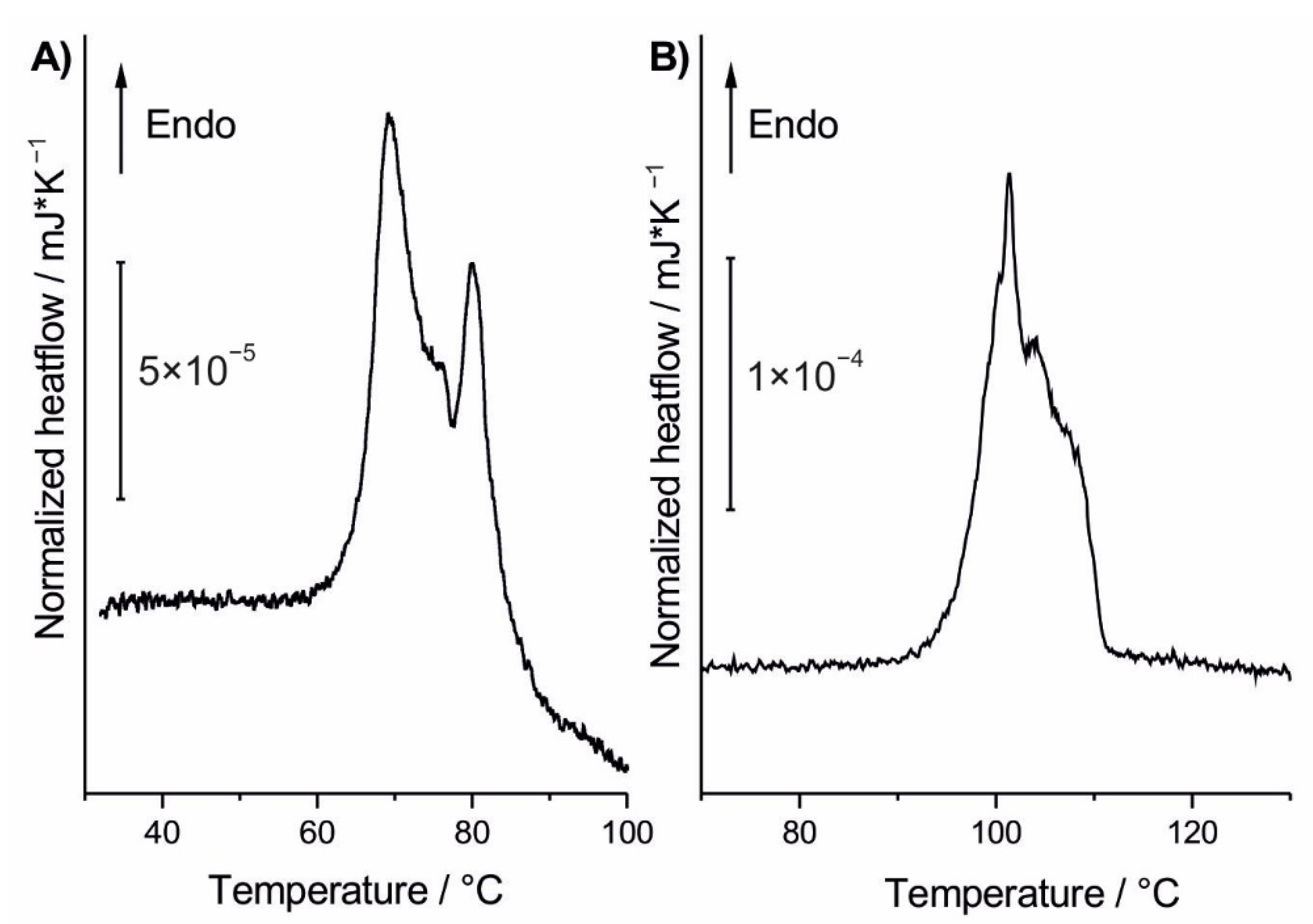
3.4. Fast Scanning Calorimetry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, Y.; Liang, J.; Wang, C.; Yin, S.; Fu, W.; Zhu, H.; Wan, C. Hybrid superlattices of two-dimensional materials and organics. Chem. Soc. Rev. 2020, 49, 6866–6883. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Ma, R.; Sakai, N.; Nurdiwijayanto, L.; Sasaki, T. Unilamellar metallic MoS2/graphene superlattice for efficient sodium storage and hydrogen evolution. ACS Energy Lett. 2018, 3, 997–1005. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, P.; Zhou, J.; Qi, S.; Yamauchi, Y.; Shi, R.; Fang, R.; Ishida, Y.; Wang, S.; Tomsia, A.P.; et al. Layered nanocomposites by shear-flow-induced alignment of nanosheets. Nature 2020, 580, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kempt, R.; Kuc, A.; Han, J.H.; Cheon, J.; Heine, T. 2D Crystals in Three Dimensions: Electronic Decoupling of Single-Layered Platelets in Colloidal Nanoparticles. Small 2018, 14, 1803910. [Google Scholar] [CrossRef] [PubMed]
- Miró, P.; Audiffred, M.; Heine, T. An atlas of two-dimensional materials. Chem. Soc. Rev. 2014, 43, 6537–6554. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Erdemir, D.; Myerson, A.S. Crystal polymorphism in chemical process development. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 259–280. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals, 2nd ed.; Oxford University Press: Oxford, UK, 2020; Volume 30, p. 608. [Google Scholar]
- Lee, E.H. A practical guide to pharmaceutical polymorph screening & selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef]
- Aitipamula, S. Polymorphism in molecular crystals and cocrystals. In Advances in Organic Crystal Chemistry, 1st ed.; Miyata, M., Tamura, R., Eds.; Springer: Tokyo, Japan, 2015; pp. 265–298. [Google Scholar]
- Möhwald, H. Phospholipid Monolayers. In Handbook of Biological Physics—Structure and Dynamics of Membranes, 1st ed.; Lipowsky, R., Sackman, E., Eds.; Elsevier: Amsterdam, Netherlands, 1995; Volume 1A, pp. 161–211. [Google Scholar]
- Santos, H.A.; García-Morales, V.; Pereira, C.M. Electrochemical properties of phospholipid monolayers at liquid–liquid interfaces. Chem. Phys. Chem. 2010, 11, 28–41. [Google Scholar] [CrossRef]
- Pichot, R.; Watson, R.L.; Norton, I.T. Phospholipids at the interface: Current trends and challenges. Int. J. Mol. Sci. 2013, 14, 11767–11794. [Google Scholar] [CrossRef]
- Ariga, K.; Yuki, H.; Kikuchi, J.I.; Dannemuller, O.; Albrecht-Gary, A.M.; Nakatani, Y.; Ourisson, G. Monolayer studies of single-chain polyprenyl phosphates. Langmuir 2005, 21, 4578–4583. [Google Scholar] [CrossRef]
- Harper, J.K.; Grant, D.M. Solid-state 13C chemical shift tensors in terpenes. 3. Structural characterization of polymorphous verbenol. J. Am. Chem. Soc. 2000, 122, 3708–3714. [Google Scholar] [CrossRef]
- Corvis, Y.; Négrier, P.; Massip, S.; Leger, J.M.; Espeau, P. Insights into the crystal structure, polymorphism and thermal behavior of menthol optical isomers and racemates. CrystEngComm 2012, 14, 7055–7064. [Google Scholar] [CrossRef]
- Pozzi, G.; Birault, V.; Werner, B.; Dannenmuller, O.; Nakatani, Y.; Ourisson, G.; Terakawa, S. Single-Chain Polyprenyl Phosphates Form “Primitive” Membranes. Angew. Chem. Int. Ed. 1996, 35, 177–180. [Google Scholar] [CrossRef]
- Streiff, S.; Ribeiro, N.; Wu, Z.; Gumienna-Kontecka, E.; Elhabiri, M.; Albrecht-Gary, A.M.; Ourisson, G.; Nakatani, Y. “Primitive” membrane from polyprenyl phosphates and polyprenyl alcohols. Chem. Biol. 2007, 14, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Aslanov, L.A.; Fetisov, G.V.; Paseshnichenko, K.A.; Dunaev, S.F. Liquid phase methods for design and engineering of two-dimensional nanocrystals. Coord. Chem. Rev. 2017, 352, 220–248. [Google Scholar] [CrossRef]
- Stoikov, I.I.; Yantemirova, A.A.; Nosov, R.V.; Rizvanov, I.K.; Julmetov, A.R.; Klochkov, V.V.; Antipin, I.S.; Konovalov, A.I.; Zharov, I. p-tert-Butyl thiacalix[4]arenes functionalized at the lower rim by amide, hydroxyl and ester groups as anion receptors. Org. Biomol. Chem. 2011, 9, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Stoikov, I.I.; Yantemirova, A.A.; Nosov, R.V.; Julmetov, A.R.; Klochkov, V.V.; Antipin, I.S.; Konovalov, A.I. Chemo-and stereocontrolled alkylation of 1, 2-disubstituted at the lower rim 1,2-alternate p-tert-butylthiacalix[4]arene. Mendeleev Commun. 2011, 21, 41–43. [Google Scholar] [CrossRef]
- Stoikov, I.I.; Mostovaya, O.A.; Yantemirova, A.A.; Antipin, I.S.; Konovalov, A.I. Phosphorylation of p-tert-butyl (thia)calixarenes by ethylene chlorophosphite. Mendeleev Commun. 2012, 22, 21–22. [Google Scholar] [CrossRef]
- Vavilova, A.A.; Stoikov, I.I. p-tert-Butylthiacalix[4]arenes functionalized by N-(4’-nitrophenyl) acetamide and N,N-diethylacetamide fragments: Synthesis and binding of anionic guests. Beilstein J. Org. Chem. 2017, 13, 1940–1949. [Google Scholar] [CrossRef]
- Mostovaya, O.A.; Padnya, P.L.; Vavilova, A.A.; Shurpik, D.N.; Khairutdinov, B.I.; Mukhametzyanov, T.A.; Khannanov, A.A.; Kutyreva, M.P.; Stoikov, I.I. Tetracarboxylic acids on a thiacalixarene scaffold: Synthesis and binding of dopamine hydrochloride. New J. Chem. 2018, 42, 177–183. [Google Scholar] [CrossRef]
- Vavilova, A.A.; Gorbachuk, V.V.; Shurpik, D.N.; Gerasimov, A.V.; Yakimova, L.S.; Padnya, P.L.; Stoikov, I.I. Synthesis, self-assembly and the effect of the macrocyclic platform on thermal properties of lactic acid oligomer modified by p-tert-butylthiacalix[4]arene. J. Mol. Liq. 2019, 281, 243–251. [Google Scholar] [CrossRef]
- Yakimova, L.S.; Nugmanova, A.R.; Mostovaya, O.A.; Vavilova, A.A.; Shurpik, D.N.; Mukhametzyanov, T.A.; Stoikov, I.I. Nanostructured Polyelectrolyte Complexes Based on Water-Soluble Thiacalix[4]Arene and Pillar[5]Arene: Self-Assembly in Micelleplexes and Polyplexes at Packaging DNA. Nanomaterials 2020, 10, 777. [Google Scholar] [CrossRef] [PubMed]
- Yuskova, E.A.; Ignacio-de Leon, P.A.A.; Khabibullin, A.; Stoikov, I.I.; Zharov, I. Silica nanoparticles surface-modified with thiacalixarenes selectively adsorb oligonucleotides and proteins. J. Nanopart. Res. 2013, 15, 2012. [Google Scholar] [CrossRef]
- Mostovaya, O.A.; Gorbachuk, V.V.; Padnya, P.L.; Vavilova, A.A.; Evtugyn, G.A.; Stoikov, I.I. Modification of Oligo- and Polylactides With Macrocyclic Fragments: Synthesis and Properties. Front. Chem. 2019, 7, 554. [Google Scholar] [CrossRef]
- Rodik, R.V.; Boyko, V.I.; Kalchenko, V.I. Calixarenes in bio-medical researches. Curr. Med. Chem. 2009, 16, 1630–1655. [Google Scholar] [CrossRef]
- Schühle, D.T.; Peters, J.A.; Schatz, J. Metal binding calixarenes with potential biomimetic and biomedical applications. Coord. Chem. Rev. 2011, 255, 2727–2745. [Google Scholar] [CrossRef]
- Gataullina, K.V.; Ziganshin, M.A.; Stoikov, I.I.; Klimovitskii, A.E.; Gubaidullin, A.T.; Suwińska, K.; Gorbatchuk, V.V. Smart polymorphism of thiacalix[4]arene with long-chain amide containing substituents. Cryst. Growth Des. 2017, 17, 3512–3527. [Google Scholar] [CrossRef]
- Frolov, I.N.; Okhotnikova, E.S.; Ziganshin, M.A.; Firsin, A.A. Interpretation of Double-Peak Endotherm on DSC Heating Curves of Bitumen. Energ. Fuel. 2020, 34, 3960–3968. [Google Scholar] [CrossRef]
- Mathot, V.; Pyda, M.; Pijpers, T.; Poel, G.V.; Van de Kerkhof, E.; Van Herwaarden, S.; Van Herwaarden, F.; Leenaers, A. The Flash DSC 1, a power compensation twin-type, chip-based fast scanning calorimeter (FSC): First findings on polymers. Thermochim. Acta 2011, 522, 36–45. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT: Integrating space group determination and structure solution. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Guzei, I.A. An idealized molecular geometry library for refinement of poorly behaved molecular fragments with constraints. J. Appl. Crystallogr. 2014, 47, 806–809. [Google Scholar] [CrossRef]
- Lhotak, P. Chemistry of thiacalixarenes. Eur. J. Org. Chem. 2004, 8, 1675–1692. [Google Scholar] [CrossRef]
- Morohashi, N.; Narumi, F.; Iki, N.; Hattori, T.; Miyano, S. Thiacalixarenes. Chem. Rev. 2006, 106, 5291–5316. [Google Scholar] [CrossRef] [PubMed]
- Iki, N.; Narumi, F.; Fujimoto, T.; Morohashi, N.; Miyano, S. Selective synthesis of three conformational isomers of tetrakis[(ethoxycarbonyl)methoxy]thiacalix[4]arene and their complexation properties towards alkali metal ions. J. Chem. Soc. Perkin Trans. 1998, 12, 2745–2750. [Google Scholar] [CrossRef]
- Gataullina, K.V.; Buzyurov, A.V.; Ziganshin, M.A.; Padnya, P.L.; Stoikov, I.I.; Schick, C.; Gorbatchuk, V.V. Using fast scanning calorimetry to detect guest-induced polymorphism by irreversible phase transitions in the nanogram scale. CrystEngComm 2019, 21, 1034–1041. [Google Scholar] [CrossRef]
- Abdelaziz, A.; Zaitsau, D.H.; Mukhametzyanov, T.A.; Solomonov, B.N.; Cebe, P.; Verevkin, S.P.; Schick, C. Melting temperature and heat of fusion of cytosine revealed from fast scanning calorimetry. Thermochim. Acta 2017, 657, 47–55. [Google Scholar] [CrossRef]
- Perrenot, B.; Widmann, G. Polymorphism by differential scanning calorimetry. Thermochim. Acta 1994, 234, 31–39. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vavilova, A.A.; Padnya, P.L.; Mukhametzyanov, T.A.; Buzyurov, A.V.; Usachev, K.S.; Islamov, D.R.; Ziganshin, M.A.; Boldyrev, A.E.; Stoikov, I.I. 2D Monomolecular Nanosheets Based on Thiacalixarene Derivatives: Synthesis, Solid State Self-Assembly and Crystal Polymorphism. Nanomaterials 2020, 10, 2505. https://doi.org/10.3390/nano10122505
Vavilova AA, Padnya PL, Mukhametzyanov TA, Buzyurov AV, Usachev KS, Islamov DR, Ziganshin MA, Boldyrev AE, Stoikov II. 2D Monomolecular Nanosheets Based on Thiacalixarene Derivatives: Synthesis, Solid State Self-Assembly and Crystal Polymorphism. Nanomaterials. 2020; 10(12):2505. https://doi.org/10.3390/nano10122505
Chicago/Turabian StyleVavilova, Alena A., Pavel L. Padnya, Timur A. Mukhametzyanov, Aleksey V. Buzyurov, Konstantin S. Usachev, Daut R. Islamov, Marat A. Ziganshin, Artur E. Boldyrev, and Ivan I. Stoikov. 2020. "2D Monomolecular Nanosheets Based on Thiacalixarene Derivatives: Synthesis, Solid State Self-Assembly and Crystal Polymorphism" Nanomaterials 10, no. 12: 2505. https://doi.org/10.3390/nano10122505
APA StyleVavilova, A. A., Padnya, P. L., Mukhametzyanov, T. A., Buzyurov, A. V., Usachev, K. S., Islamov, D. R., Ziganshin, M. A., Boldyrev, A. E., & Stoikov, I. I. (2020). 2D Monomolecular Nanosheets Based on Thiacalixarene Derivatives: Synthesis, Solid State Self-Assembly and Crystal Polymorphism. Nanomaterials, 10(12), 2505. https://doi.org/10.3390/nano10122505