Investigation of the Ionic Liquid Graphene Electric Double Layer in Supercapacitors Using Constant Potential Simulations


.jpg)
Abstract

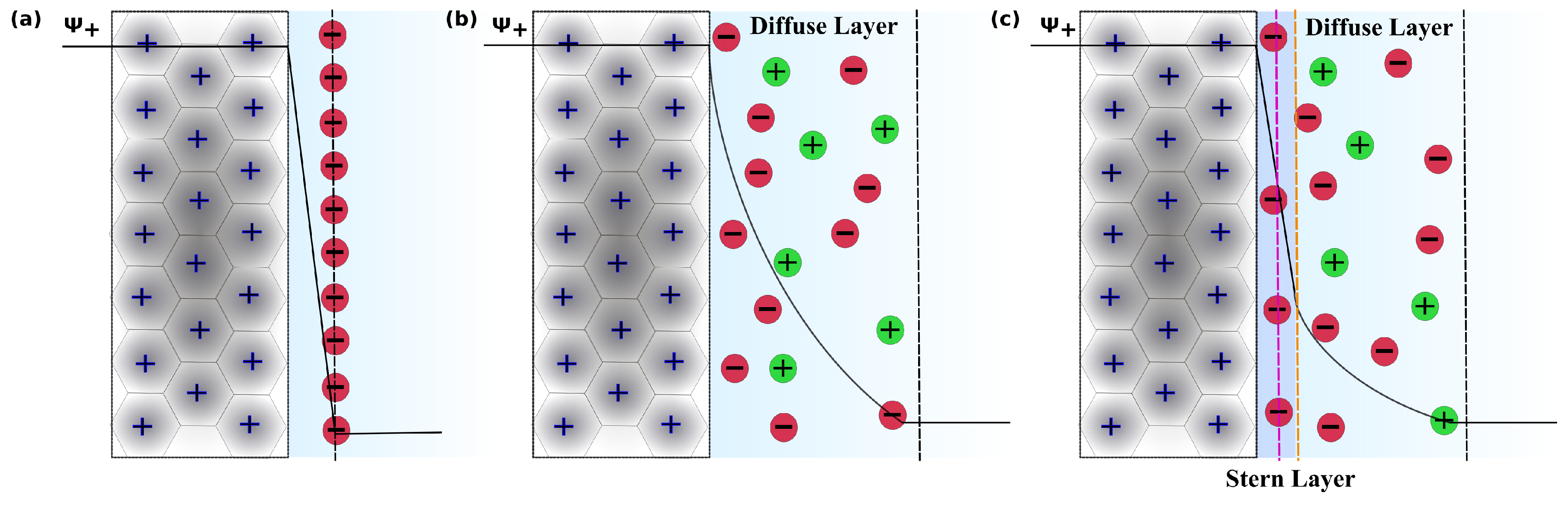
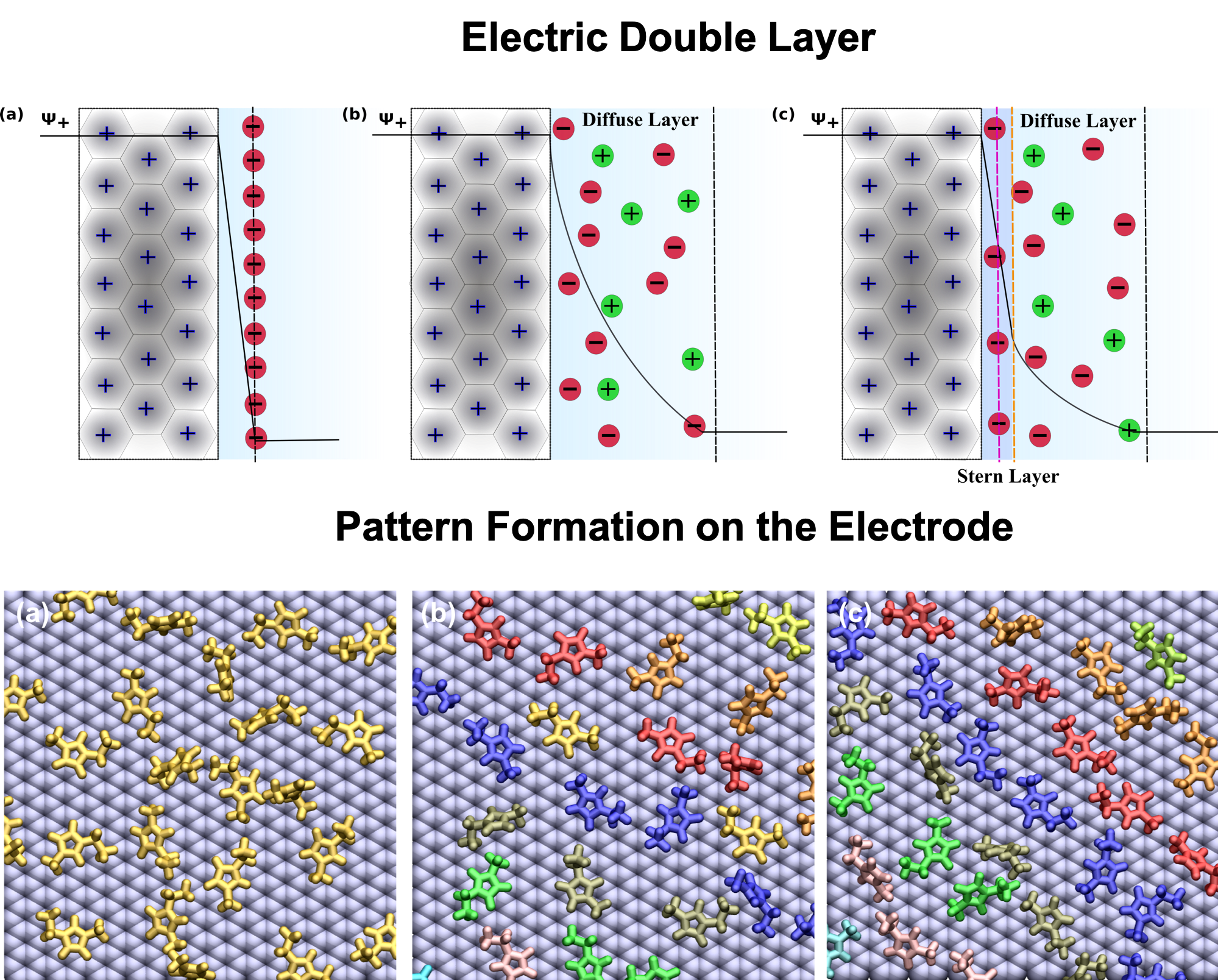{kind=link}
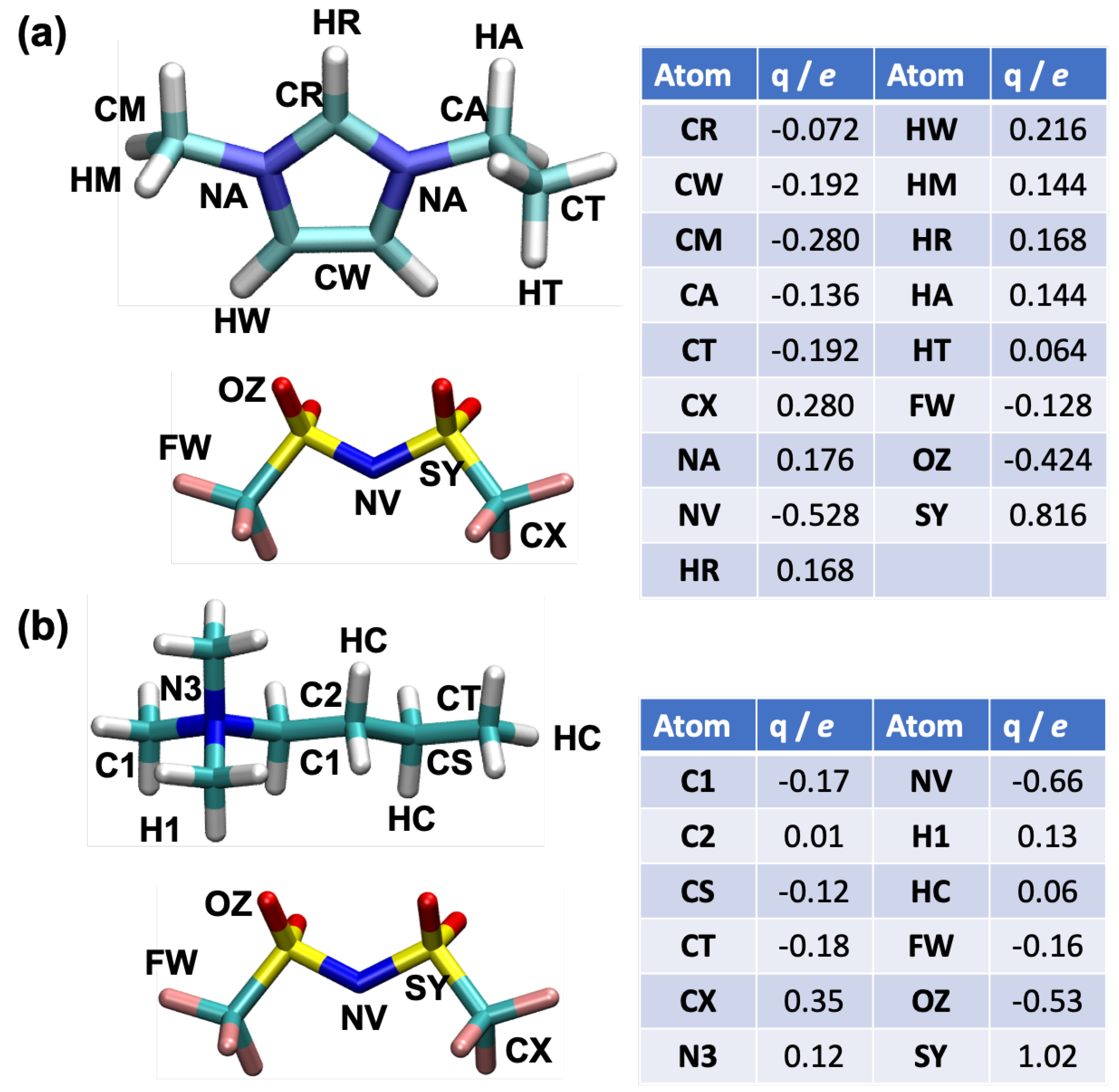
{kind=link}
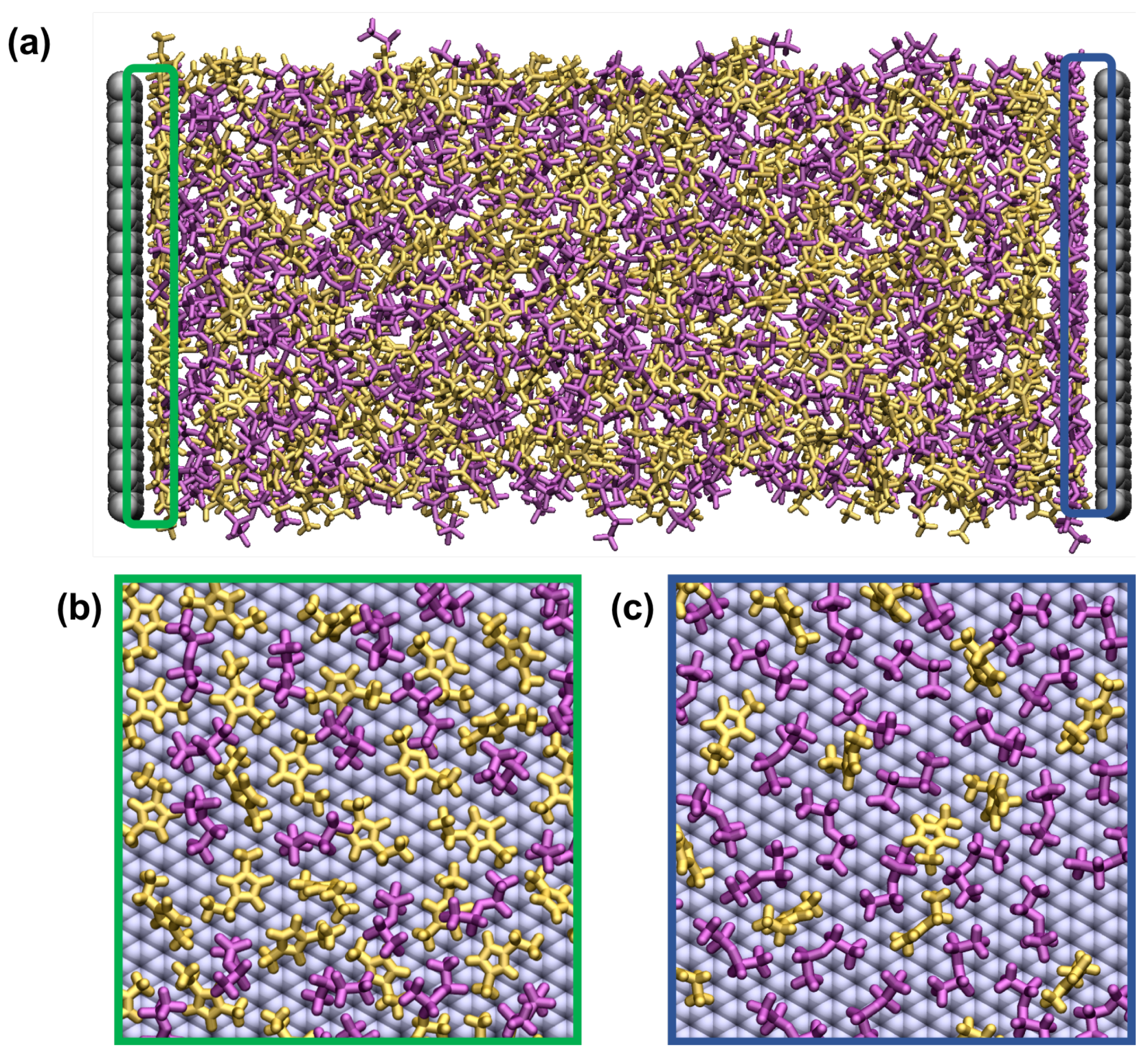
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Models and Simulation Methods
2.1. Sample Preparation Procedure
2.2. Constant Potential Simulations
3. Results and Discussion
3.1. Bulk Properties of Ionic Liquids
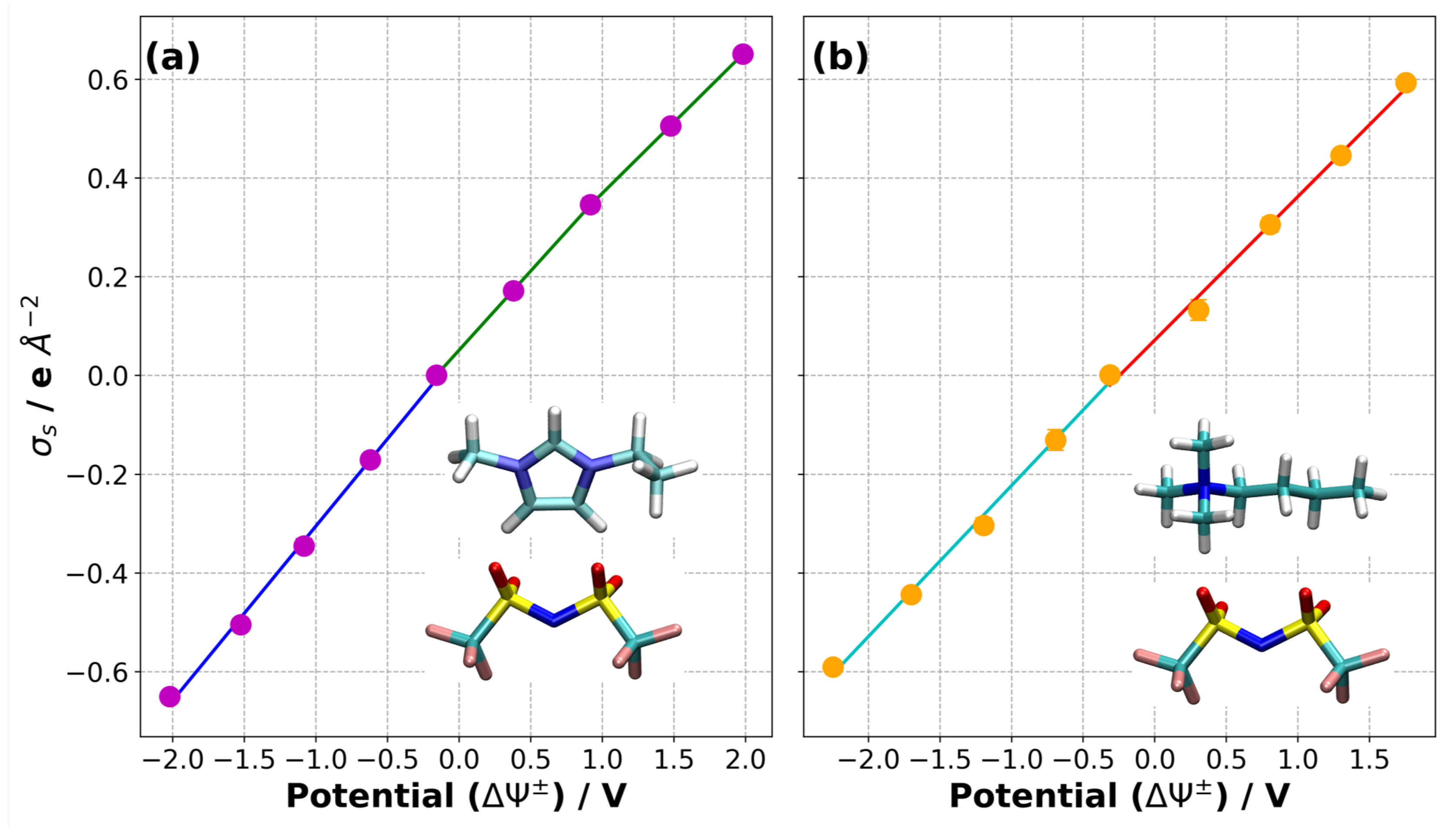
3.2. Equilibration of Supercapacitors
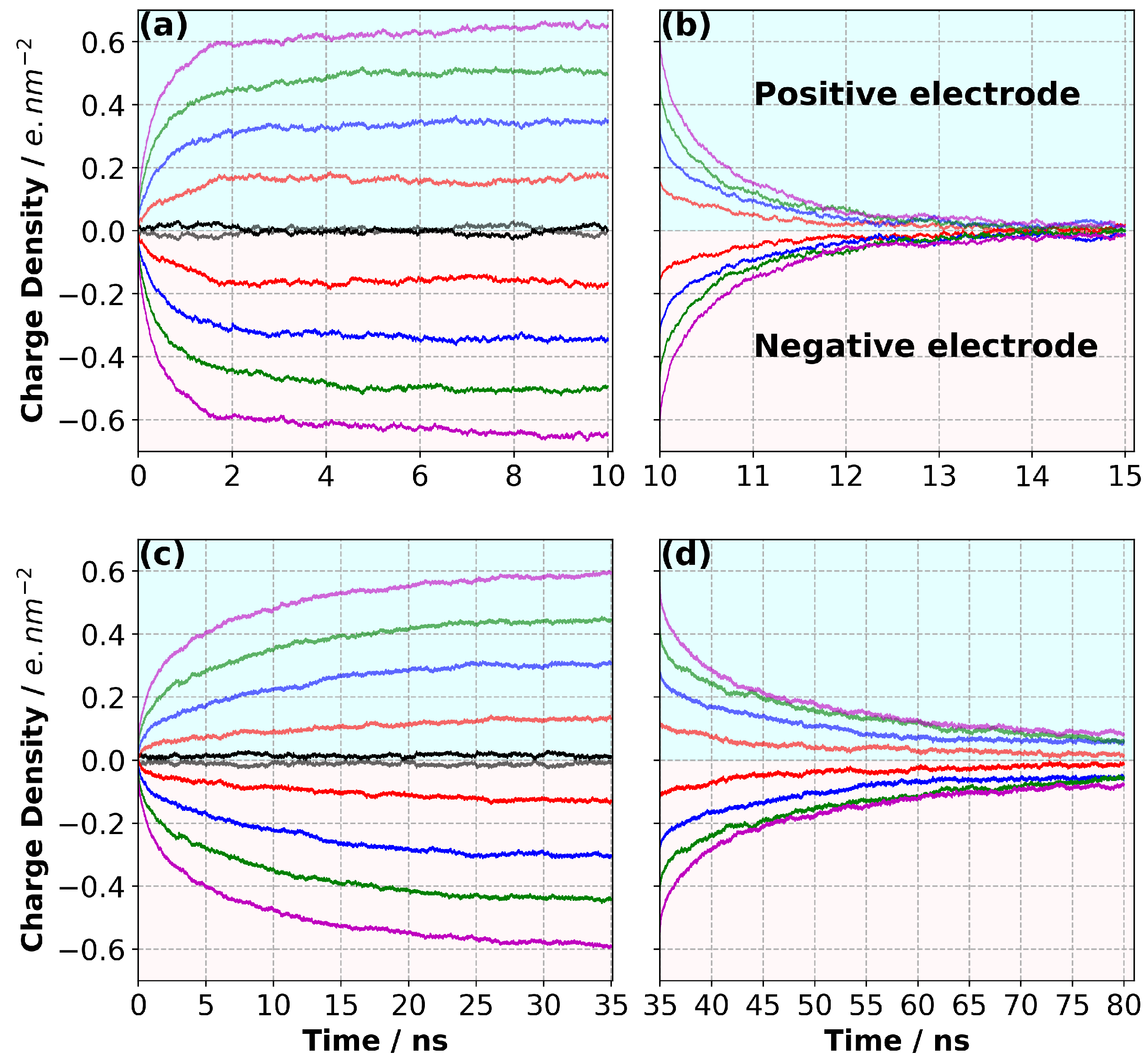
3.3. Charge/Discharge Dynamics of the Electrode
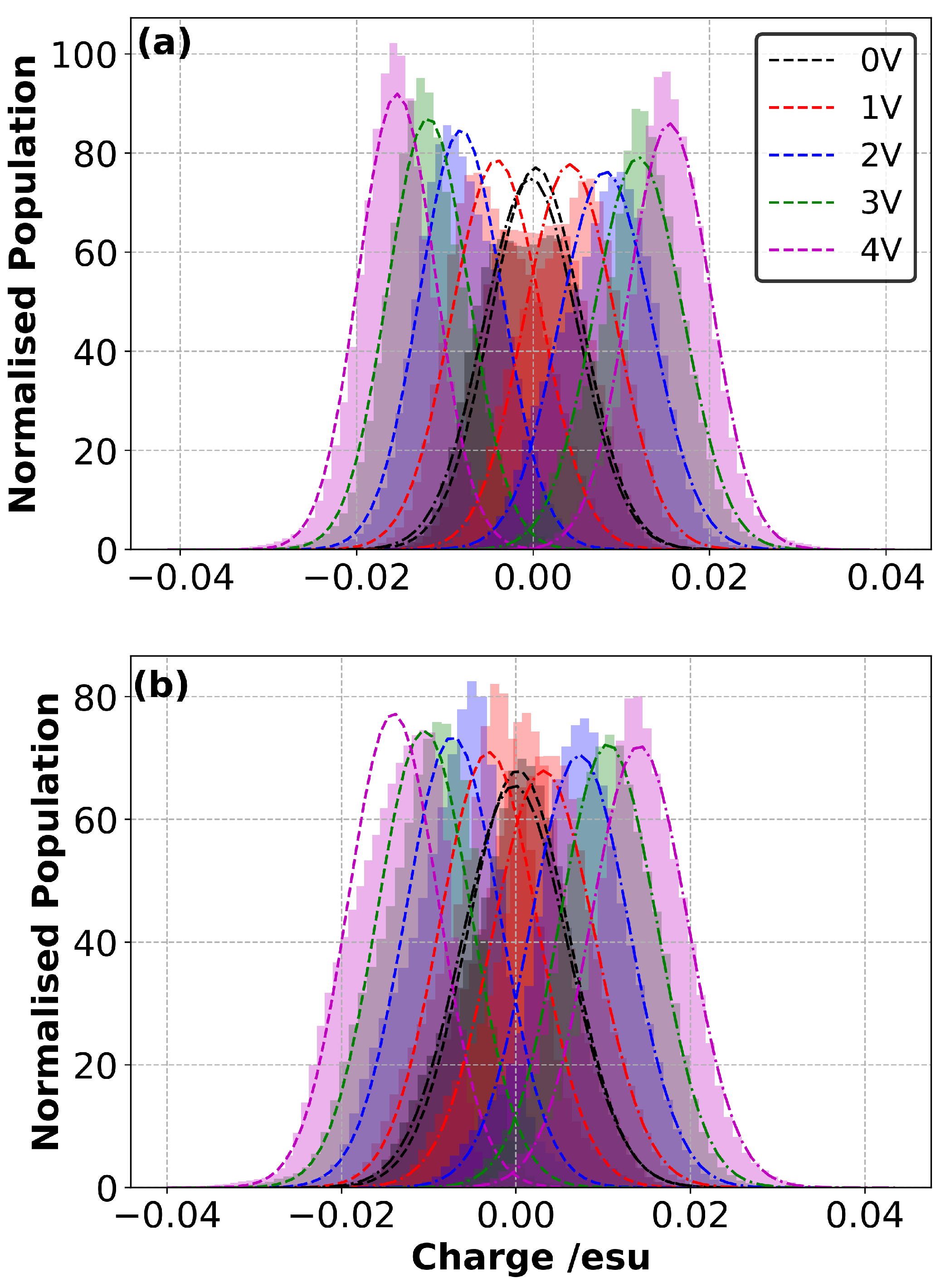
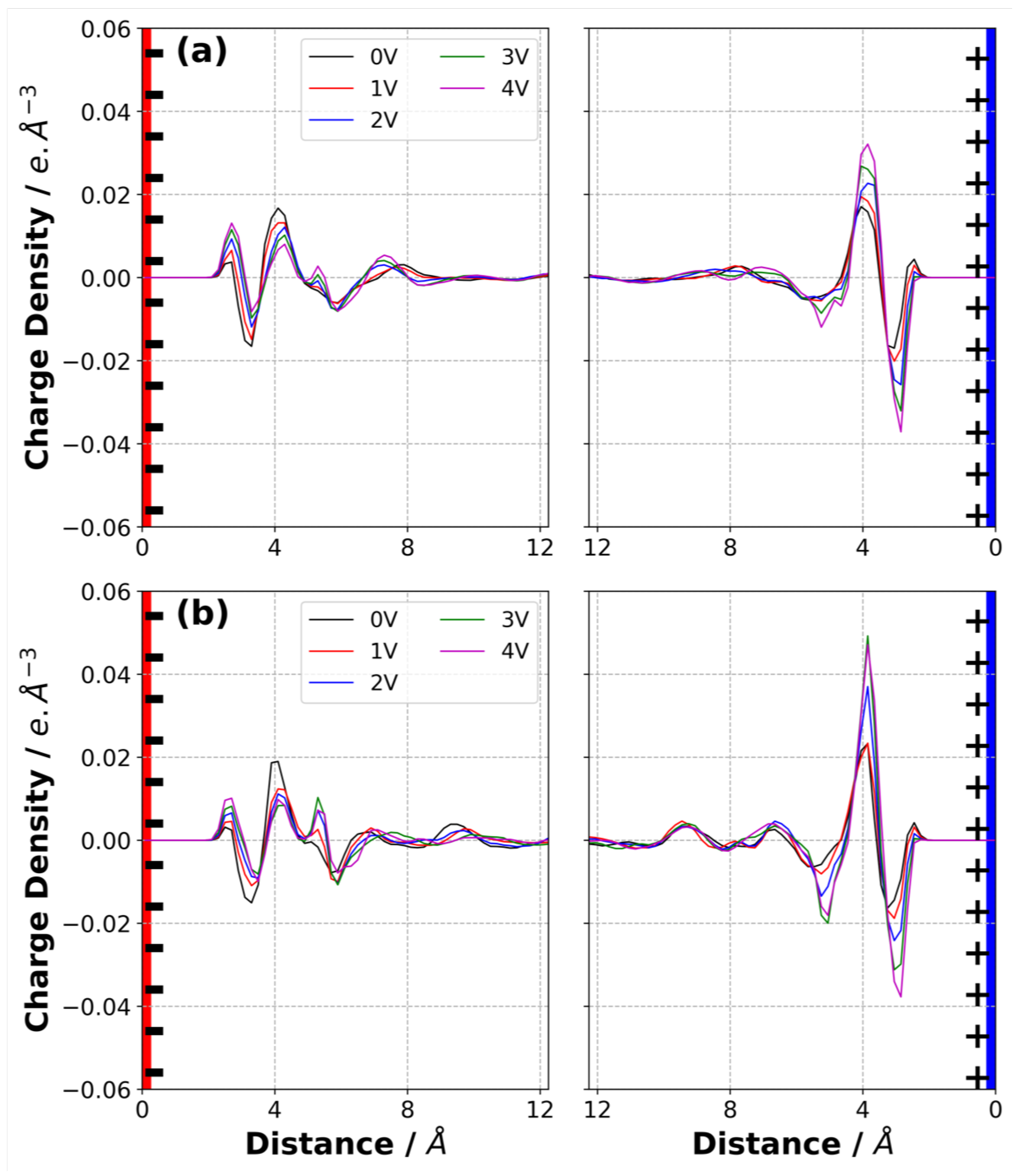
3.4. Charge Density Distribution on the Electrodes
3.5. Charge Evolution in the Electrolyte
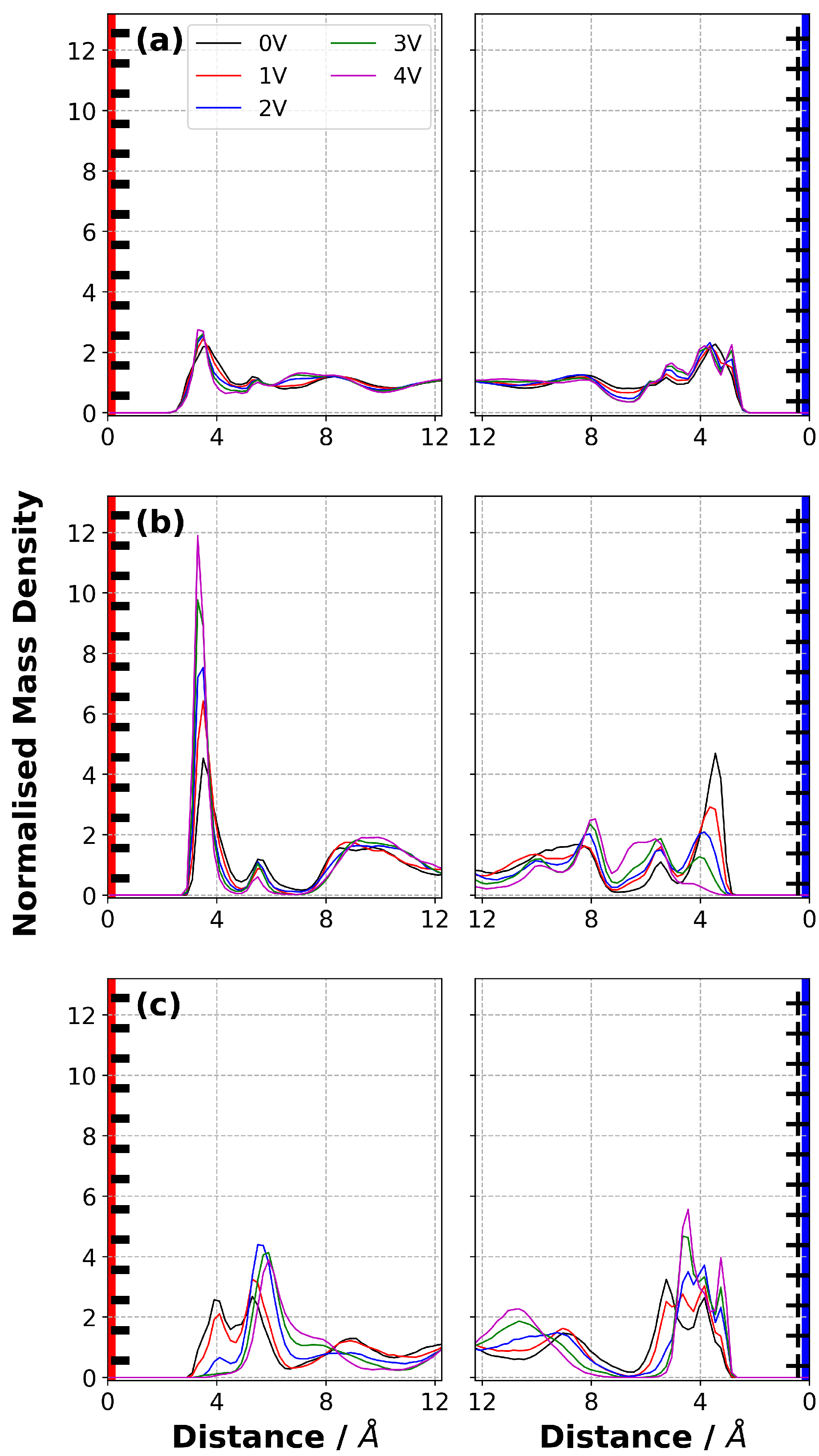
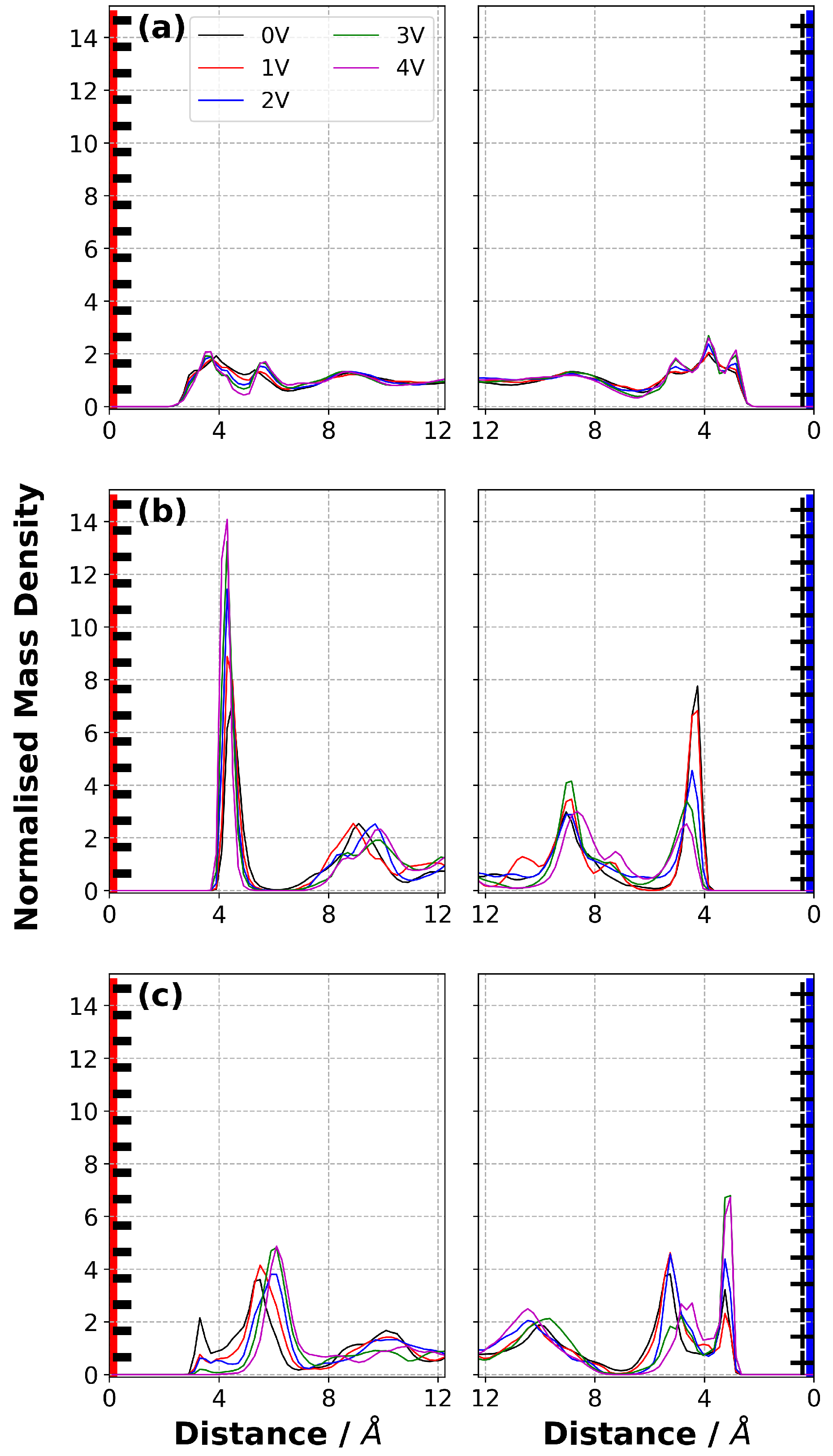
3.6. Normalised Mass Density
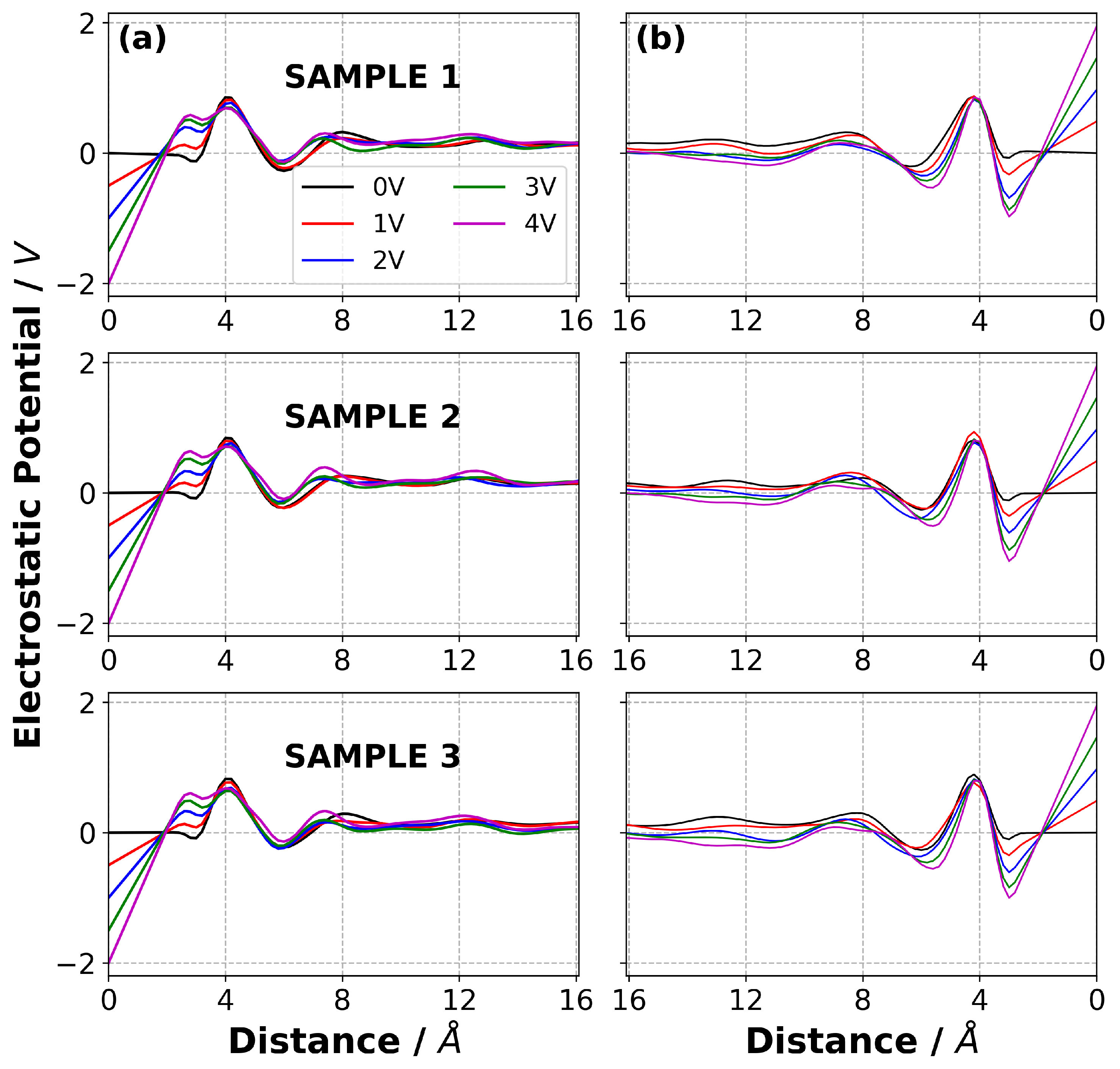
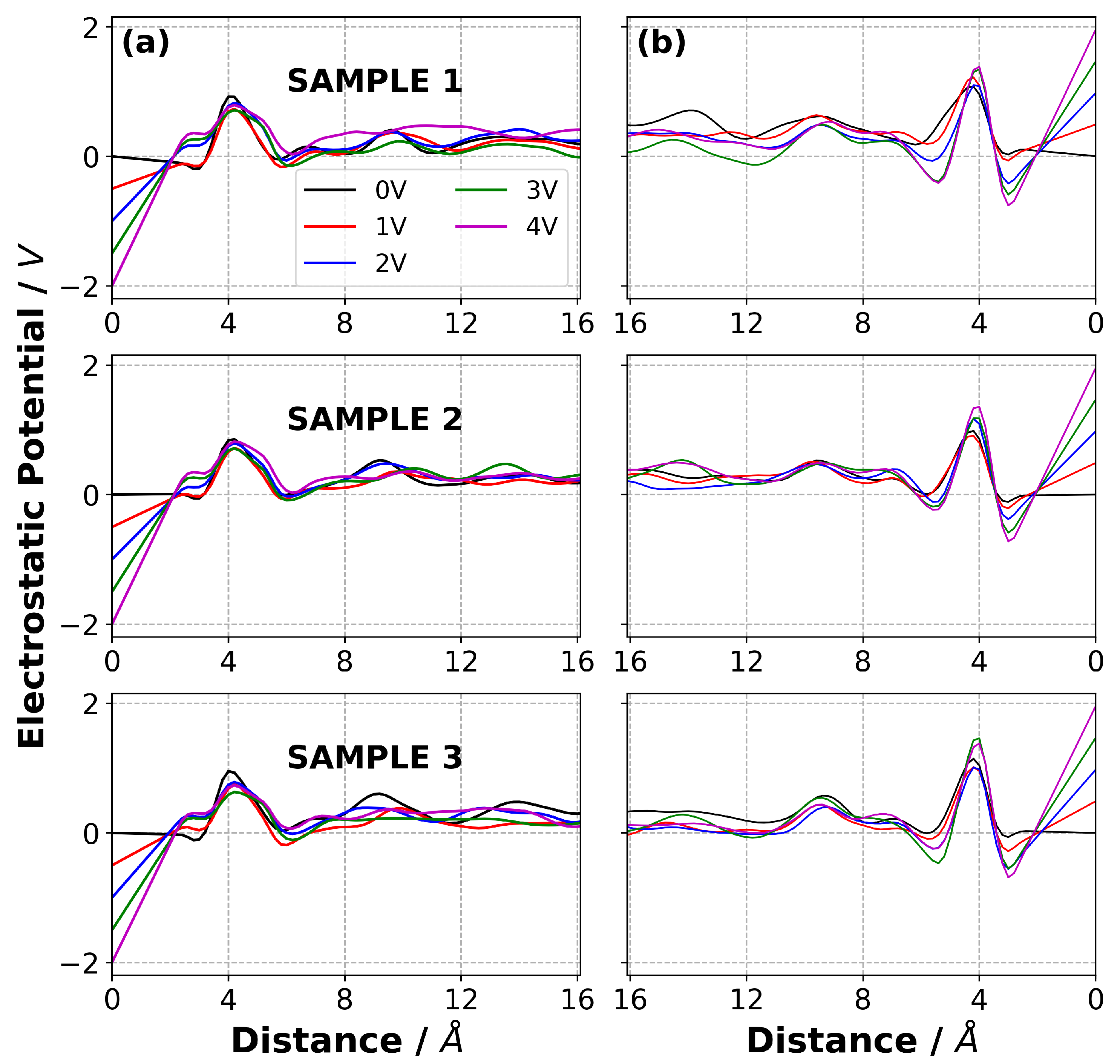
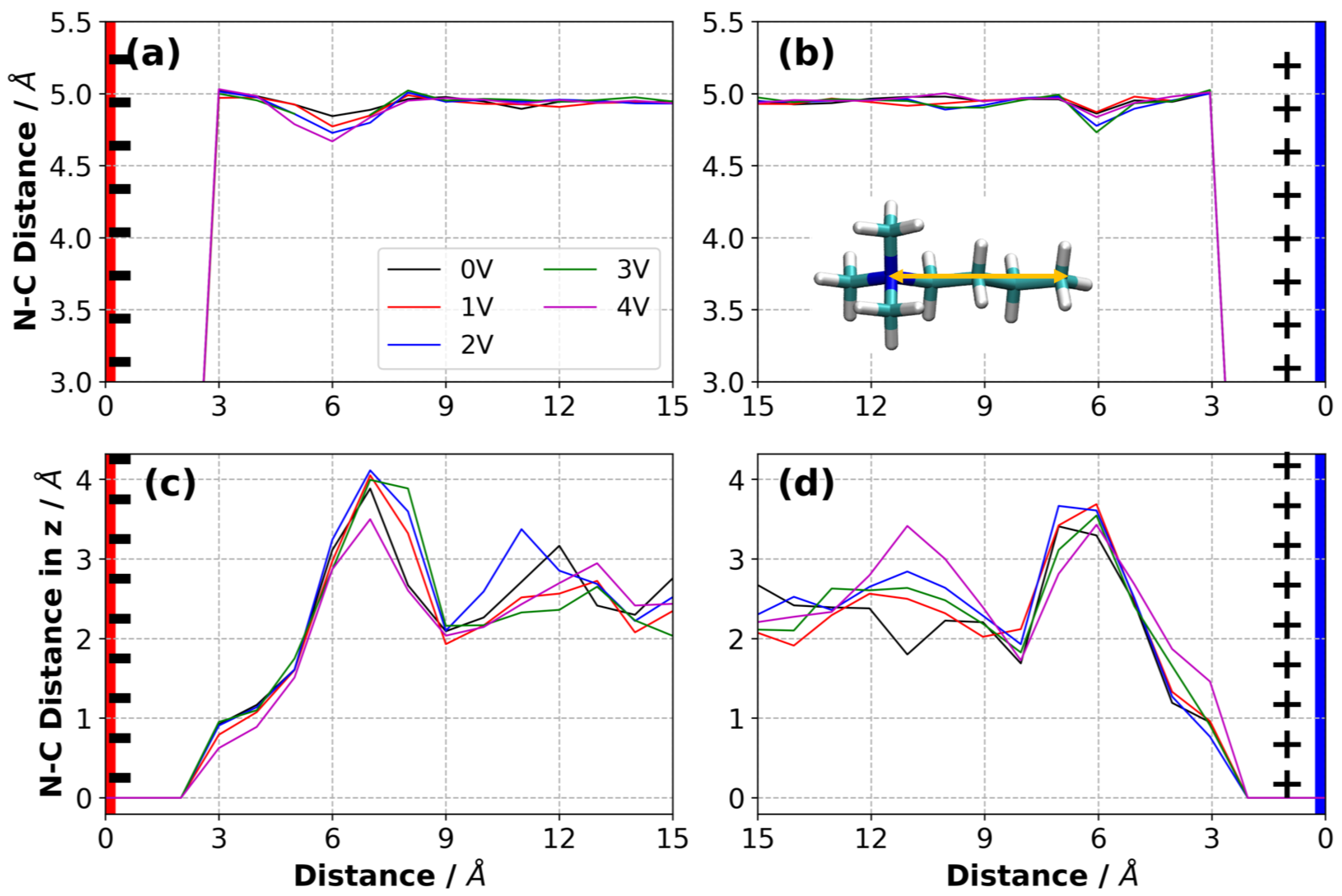
3.7. Charge Density Distribution in Electrolyte
3.8. Differences in Residency Time for Ions in the EDL
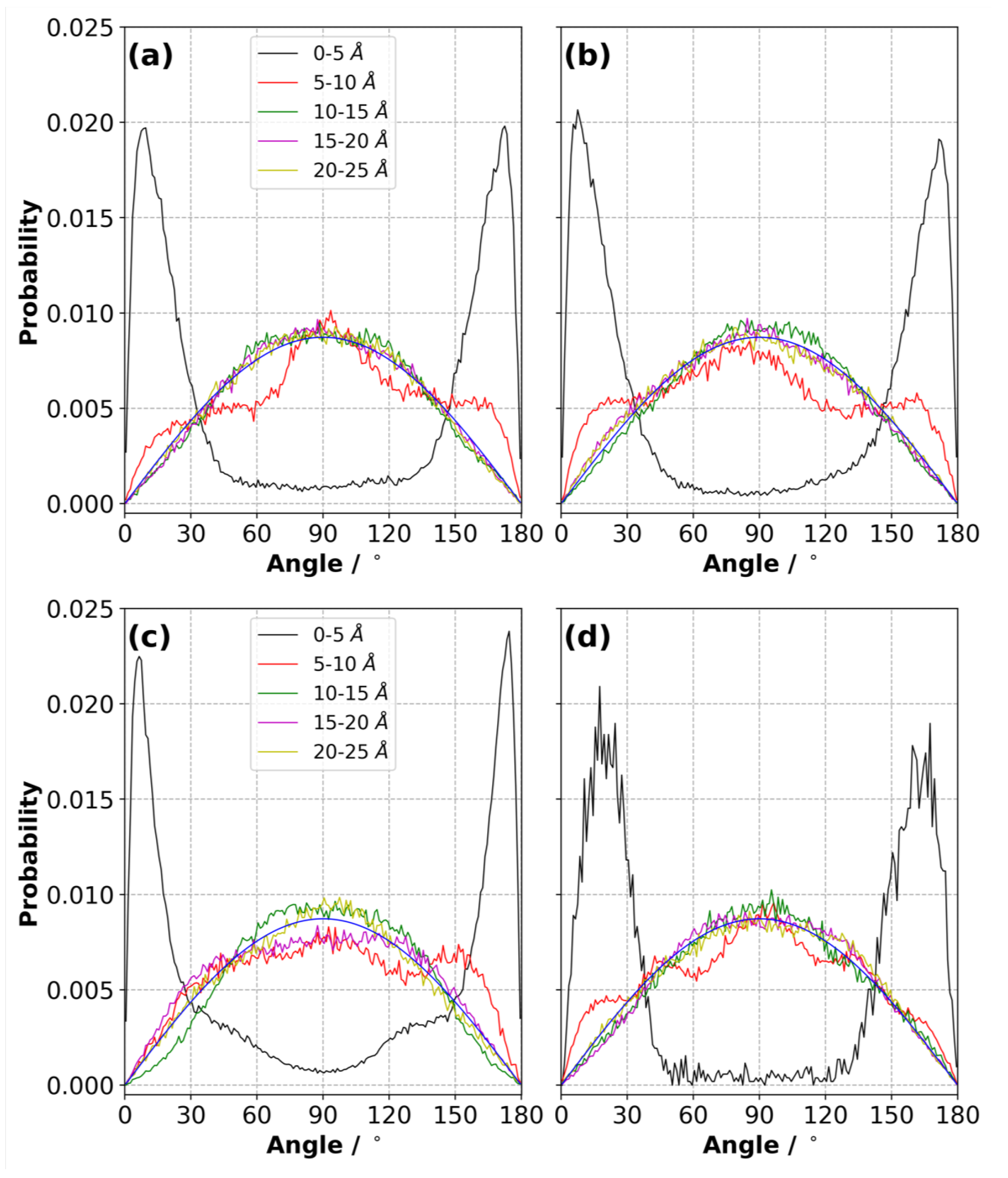
3.9. Orientation of Ions in the Electric Double Layer
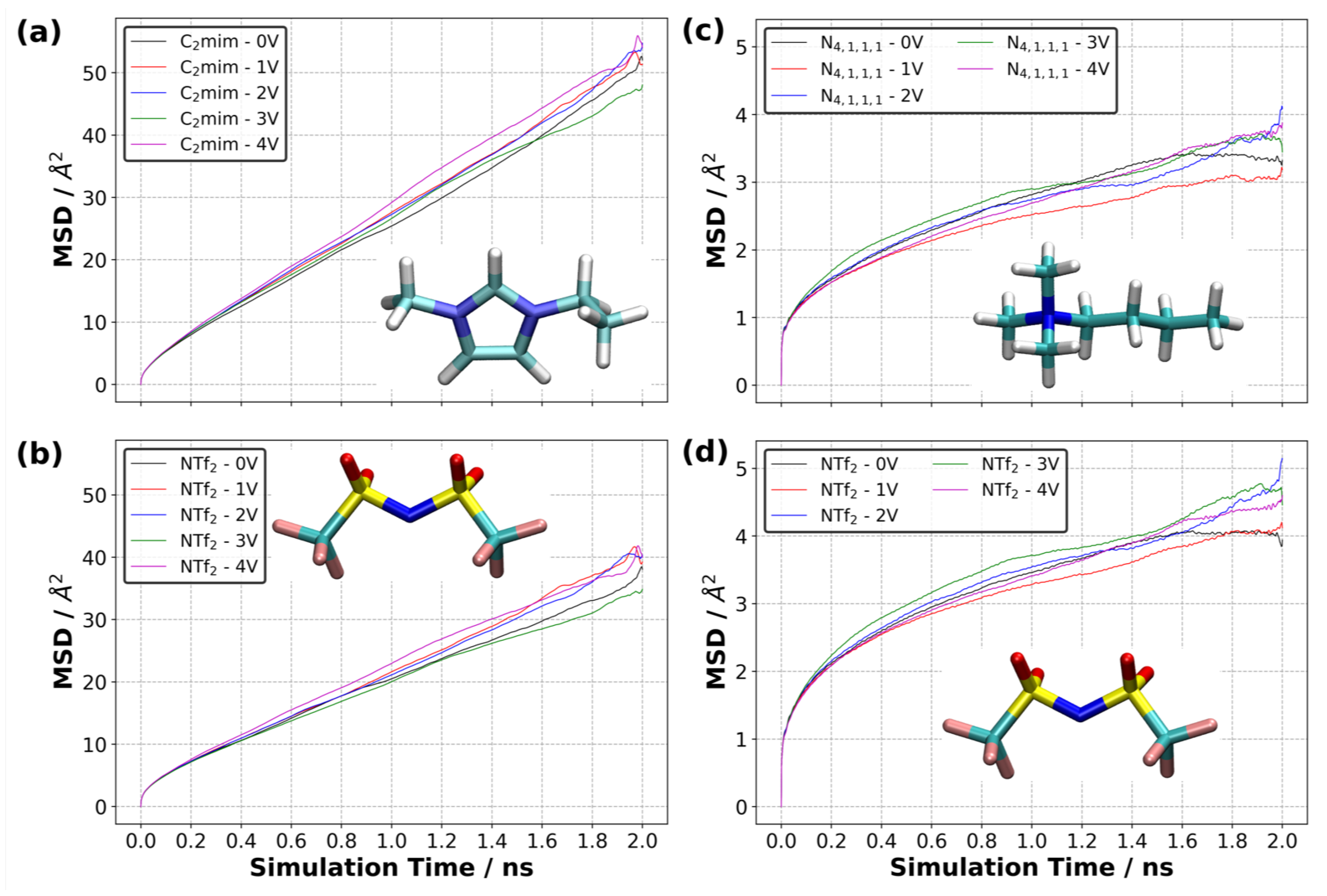
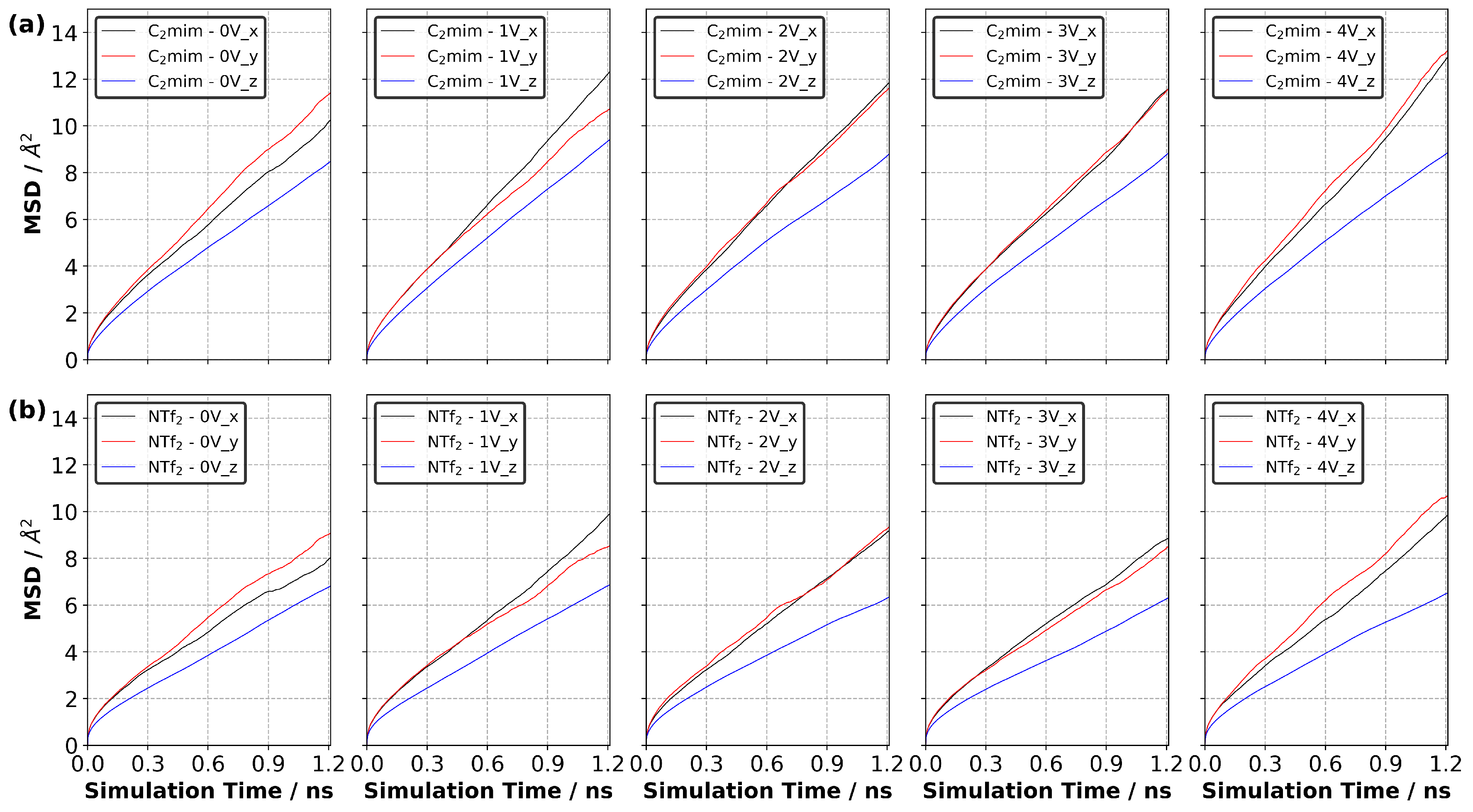
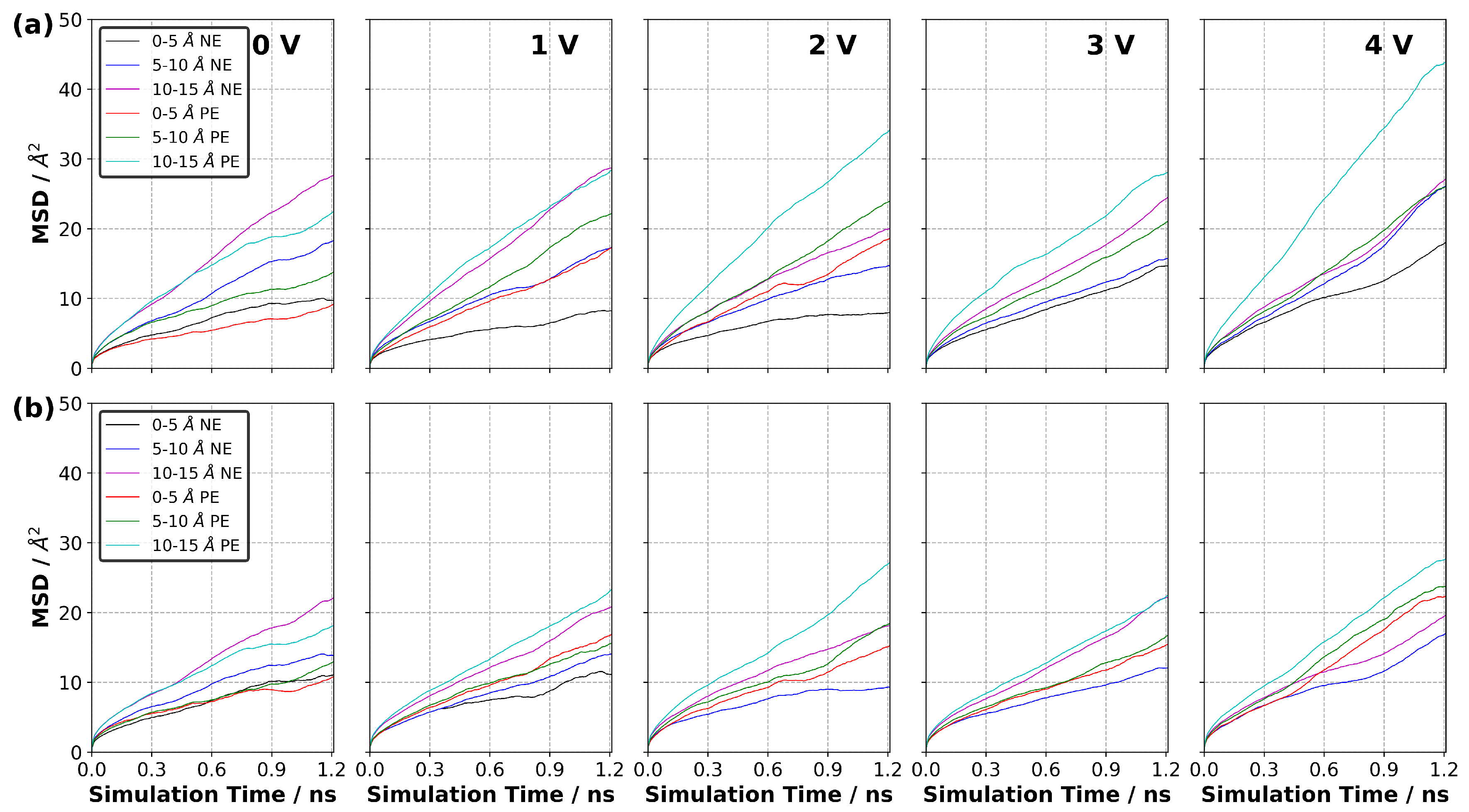
3.10. Diffusion of Ions in EDL
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EDLSC | Electric double-layer supercapacitor |
| EDL | Electric double layer |
| IL | Ionic liquid |
| MD | Molecular dynamics |
| CPM | Constant potential method |
| FCM | Fixed charge method |
| SA | Simulated annealing |
| MSD | Mean square displacement |
References
- Helmholtz, H. Ueber einige Gesetze der Vertheilung elektrischer Ströme in körperlichen Leitern mit Anwendung auf die thierisch-elektrischen Versuche. Ann. Phys. 1853, 89, 211–233. [Google Scholar] [CrossRef]
- Gouy, M. Sur la constitution de la charge electriquea la surface’d’ue electrolyte. J. Phys. Theor. Appl. 1910, 9, 457–468. [Google Scholar] [CrossRef]
- Stern, O. Zur Theorie der Elektrolytischen Doppelschich. Elektrochem. Angew. Phys. Chem. 1924, 149, 508–516. [Google Scholar]
- Yan, J.W.; Tian, Z.Q.; Mao, B.W. Molecular-level understanding of electric double layer in ionic liquids. Curr. Opin. Electrochem. 2017, 4, 105–111. [Google Scholar] [CrossRef]
- Atkin, R.; Warr, G.G. Structure in Confined Room-Temperature Ionic Liquids. J. Phys. Chem. C 2007, 111, 5162–5168. [Google Scholar] [CrossRef]
- Black, J.M.; Zhu, M.; Zhang, P.; Unocic, R.R.; Guo, D.; Okatan, M.B.; Dai, S.; Cummings, P.T.; Kalinin, S.V.; Feng, G.; et al. Fundamental Aspects of Electric Double Layer Force-Distance Measurements at Liquid-Solid Interfaces Using Atomic Force Microscopy. Sci. Rep. 2016, 6, 32389. [Google Scholar] [CrossRef]
- Mezger, M.; Schröder, H.; Reichert, H.; Schramm, S.; Okasinski, J.S.; Schöder, S.; Honkimäki, V.; Deutsch, M.; Ocko, B.M.; Ralston, J.; et al. Molecular Layering of Fluorinated Ionic Liquids at a Charged Sapphire (0001) Surface. Science 2008, 322, 424–428. [Google Scholar] [CrossRef]
- Begić, S.; Li, H.; Atkin, R.; Hollenkamp, A.F.; Howlett, P.C. A Comparative AFM Study of the Interfacial Nanostructure in Imidazolium or Pyrrolidinium Ionic Liquid Electrolytes for Zinc Electrochemical Systems. Phys. Chem. Chem. Phys. 2016, 18, 29337–29347. [Google Scholar] [CrossRef]
- Jurado, L.A.; Espinosa-Marzal, R.M. Insight into the Electrical Double Layer of an Ionic Liquid on Graphene. Sci. Rep. 2017, 7, 4225. [Google Scholar] [CrossRef]
- Ruzanov, A.; Lembinen, M.; Jakovits, P.; Srirama, S.N.; Voroshylova, I.V.; Cordeiro, M.N.D.S.; Pereira, C.M.; Rossmeisl, J.; Ivaništšev, V.B. On the Thickness of the Double Layer in Ionic Liquids. Phys. Chem. Chem. Phys. 2018, 20, 10275–10285. [Google Scholar] [CrossRef]
- Ers, H.; Lembinen, M.; Mišin, M.; Seitsonen, A.P.; Fedorov, M.V.; Ivaništšev, V.B. Graphene–Ionic Liquid Interfacial Potential Drop from Density Functional Theory-Based Molecular Dynamics Simulations. J. Phys. Chem. C 2020, 124, 19548–19555. [Google Scholar] [CrossRef]
- Siepmann, J.I.; Sprik, M. Influence of surface topology and electrostatic potential on water/electrode systems. J. Chem. Phys. 1995, 102, 511–524. [Google Scholar] [CrossRef]
- Reed, S.K.; Lanning, O.J.; Madden, P.A. Electrochemical interface between an ionic liquid and a model metallic electrode. J. Chem. Phys. 2007, 126, 084704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Y.; Olmsted, D.L.; Asta, M.; Laird, B.B. Evaluation of the constant potential method in simulating electric double-layer capacitors. J. Chem. Phys. 2014, 141, 184102. [Google Scholar] [CrossRef]
- Noh, C.; Jung, Y. Understanding the charging dynamics of an ionic liquid electric double layer capacitor via molecular dynamics simulations. Phys. Chem. Chem. Phys. 2019, 21, 6790–6800. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Smolyanitsky, A. Simulation Study of the Capacitance and Charging Mechanisms of Ionic Liquid Mixtures near Carbon Electrodes. J. Phys. Chem. C 2019, 123, 1610–1618. [Google Scholar] [CrossRef]
- Jiang, S.; Hu, Y.; Wang, Y.; Wang, X. Viscosity of Typical Room-Temperature Ionic Liquids: A Critical Review. J. Phys. Chem. Ref. Data 2019, 48, 033101. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Canongia Lopes, J.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids Composed of Triflate or Bistriflylimide Anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory. Comput. 2017, 13, 6131–6145. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Hanwell, M.; Curtis, D.; Lonie, D.; Vandermeersch, T.; Zurek, E.; Hutchison, G. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Hockney, R.; Eastwood, J. Computer Simulation Using Particles; Adam Hilger: New York, NY, USA, 1989. [Google Scholar]
- Yeh, I.C.; Berkowitz, M.L. Ewald summation for systems with slab geometry. J. Chem. Phys. 1999, 111, 3155–3162. [Google Scholar] [CrossRef]
- Eyckens, D.J.; Demir, B.; Walsh, T.R.; Welton, T.; Henderson, L.C. Determination of Kamlet–Taft parameters for selected solvate ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 13153–13157. [Google Scholar] [CrossRef] [PubMed]
- Eyckens, D.J.; Servinis, L.; Scheffler, C.; Wolfel, E.; Demir, B.; Walsh, T.R.; Henderson, L.C. Synergistic interfacial effects of ionic liquids as sizing agents and surface modified carbon fibers. J. Mater. Chem. A 2018, 6, 4504–4514. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Hoover, W.G. Constant-pressure equations of motion. Phys. Rev. A 1986, 34, 2499–2500. [Google Scholar] [CrossRef]
- Fröba, A.P.; Kremer, H.; Leipertz, A. Density, Refractive Index, Interfacial Tension, and Viscosity of Ionic Liquids [EMIM][EtSO4], [EMIM][NTf2], [EMIM][N(CN)2], and [OMA][NTf2] in Dependence on Temperature at Atmospheric Pressure. J. Phys. Chem. B 2008, 112, 12420–12430. [Google Scholar] [CrossRef]
- Jacquemin, J.; Husson, P.; Padua, A.A.H.; Majer, V. Density and viscosity of several pure and water-saturated ionic liquids. Green Chem. 2006, 8, 172–180. [Google Scholar] [CrossRef]
- Radchenko, A.V.; Chabane, H.; Demir, B.; Searles, D.J.; Duchet-Rumeau, J.; Gerard, J.F.; Baudoux, J.; Livi, S. New Epoxy Thermosets Derived from Bisimidazolium Ionic Liquid Monomer: An Experimental and Modelling Investigation. ACS Sustain. Chem. Eng. 2020, 8, 12208–12221. [Google Scholar] [CrossRef]
- Demir, B.; Chan, K.Y.; Searles, D.J. Structural Electrolytes Based on Epoxy Resins and Ionic Liquids: A Molecular-Level Investigation. Macromolecules 2020, 53, 7635–7649. [Google Scholar] [CrossRef]
- Gorlov, M.; Kloo, L. Ionic liquid electrolytes for dye-sensitized solar cells. Dalton Trans. 2008, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Bazant, M.Z.; Storey, B.D.; Kornyshev, A.A. Double Layer in Ionic Liquids: Overscreening versus Crowding. Phys. Rev. Lett. 2011, 106, 046102. [Google Scholar] [CrossRef]
- Zhou, H.; Rouha, M.; Feng, G.; Lee, S.S.; Docherty, H.; Fenter, P.; Cummings, P.T.; Fulvio, P.F.; Dai, S.; McDonough, J.; et al. Nanoscale Perturbations of Room Temperature Ionic Liquid Structure at Charged and Uncharged Interfaces. ACS Nano 2012, 6, 9818–9827. [Google Scholar] [CrossRef]
- Montes-Campos, H.; Otero-Mato, J.M.; Méndez-Morales, T.; Cabeza, O.; Gallego, L.J.; Ciach, A.; Varela, L.M. Two-dimensional pattern formation in ionic liquids confined between graphene walls. Phys. Chem. Chem. Phys. 2017, 19, 24505–24512. [Google Scholar] [CrossRef]
- Su, Y.Z.; Fu, Y.C.; Yan, J.W.; Chen, Z.B.; Mao, B.W. Double Layer of Au(100)/Ionic Liquid Interface and Its Stability in Imidazolium-Based Ionic Liquids. Angew. Chem. Int. Edit. 2009, 48, 5148–5151. [Google Scholar] [CrossRef]
- Rotenberg, B.; Salanne, M. Structural Transitions at Ionic Liquid Interfaces. J. Phys. Chem. Lett. 2015, 6, 4978–4985. [Google Scholar] [CrossRef]
- Jo, S.; Park, S.W.; Shim, Y.; Jung, Y. Effects of Alkyl Chain Length on Interfacial Structure and Differential Capacitance in Graphene Supercapacitors: A Molecular Dynamics Simulation Study. Electrochim. Acta 2017, 247, 634–645. [Google Scholar] [CrossRef]
- D’Agostino, C.; Mantle, M.D.; Mullan, C.L.; Hardacre, C.; Gladden, L.F. Diffusion, Ion Pairing and Aggregation in 1-Ethyl-3-Methylimidazolium-Based Ionic Liquids Studied by 1H and 19F PFG NMR: Effect of Temperature, Anion and Glucose Dissolution. ChemPhysChem 2018, 19, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Simonnin, P.; Noetinger, B.; Nieto-Draghi, C.; Marry, V.; Rotenberg, B. Diffusion under Confinement: Hydrodynamic Finite-Size Effects in Simulation. J. Chem. Theory Comput. 2017, 13, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; McDaniel, J.G. Interference of electrical double layers: Confinement effects on structure, dynamics, and screening of ionic liquids. J. Chem. Phys. 2020, 152, 074709. [Google Scholar] [CrossRef]
- He, Y.; Qiao, R.; Vatamanu, J.; Borodin, O.; Bedrov, D.; Huang, J.; Sumpter, B.G. Importance of Ion Packing on the Dynamics of Ionic Liquids during Micropore Charging. J. Phys. Chem. Lett. 2016, 7, 36–42. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demir, B.; Searles, D.J. Investigation of the Ionic Liquid Graphene Electric Double Layer in Supercapacitors Using Constant Potential Simulations. Nanomaterials 2020, 10, 2181. https://doi.org/10.3390/nano10112181
Demir B, Searles DJ. Investigation of the Ionic Liquid Graphene Electric Double Layer in Supercapacitors Using Constant Potential Simulations. Nanomaterials. 2020; 10(11):2181. https://doi.org/10.3390/nano10112181
Chicago/Turabian StyleDemir, Baris, and Debra J. Searles. 2020. "Investigation of the Ionic Liquid Graphene Electric Double Layer in Supercapacitors Using Constant Potential Simulations" Nanomaterials 10, no. 11: 2181. https://doi.org/10.3390/nano10112181
APA StyleDemir, B., & Searles, D. J. (2020). Investigation of the Ionic Liquid Graphene Electric Double Layer in Supercapacitors Using Constant Potential Simulations. Nanomaterials, 10(11), 2181. https://doi.org/10.3390/nano10112181