
Cell Surface and Membrane Engineering: Emerging Technologies and Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basic Strategies: Cell-Based vs. Synthetic Systems
2.1. Biological Membrane Systems
2.2. Cell-Free, Artificial Membrane Systems
2.3. Characterization Technologies are Propelling Membrane Engineering Applications
3. Biomedical and Pharmaceutical Applications
3.1. Membrane-Based Receptors are Critical, but Difficult to Study, Drug Targets
3.2. Overcoming Challenges in the Over-Expression of Membrane Proteins
3.2.1. Cells Have a Limited Capacity to Produce Membrane Components, Including Embedded Proteins
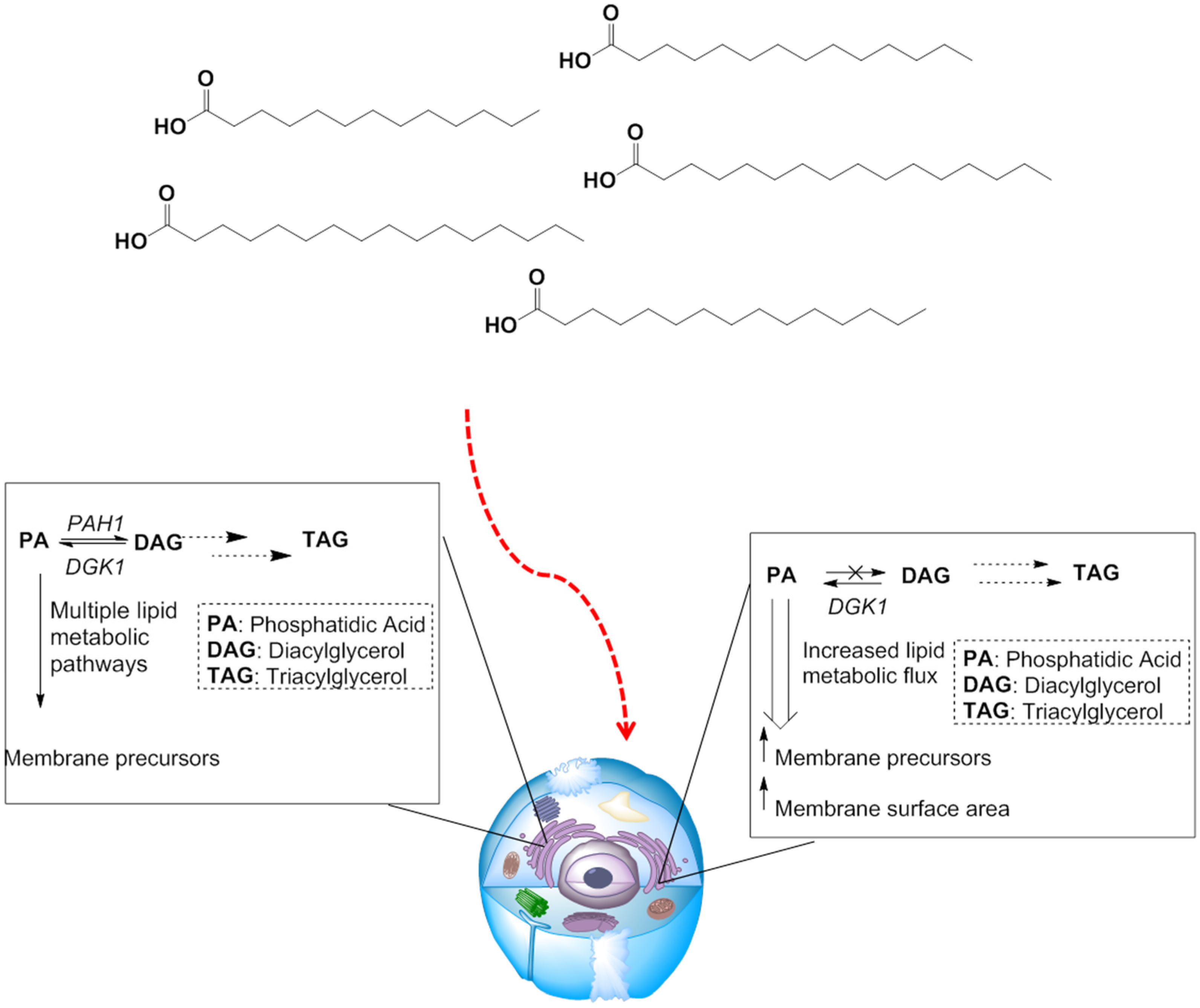
3.2.2. Manipulating Lipid Metabolism
3.3. Synthetic Membrane-Based Systems for Drug Development
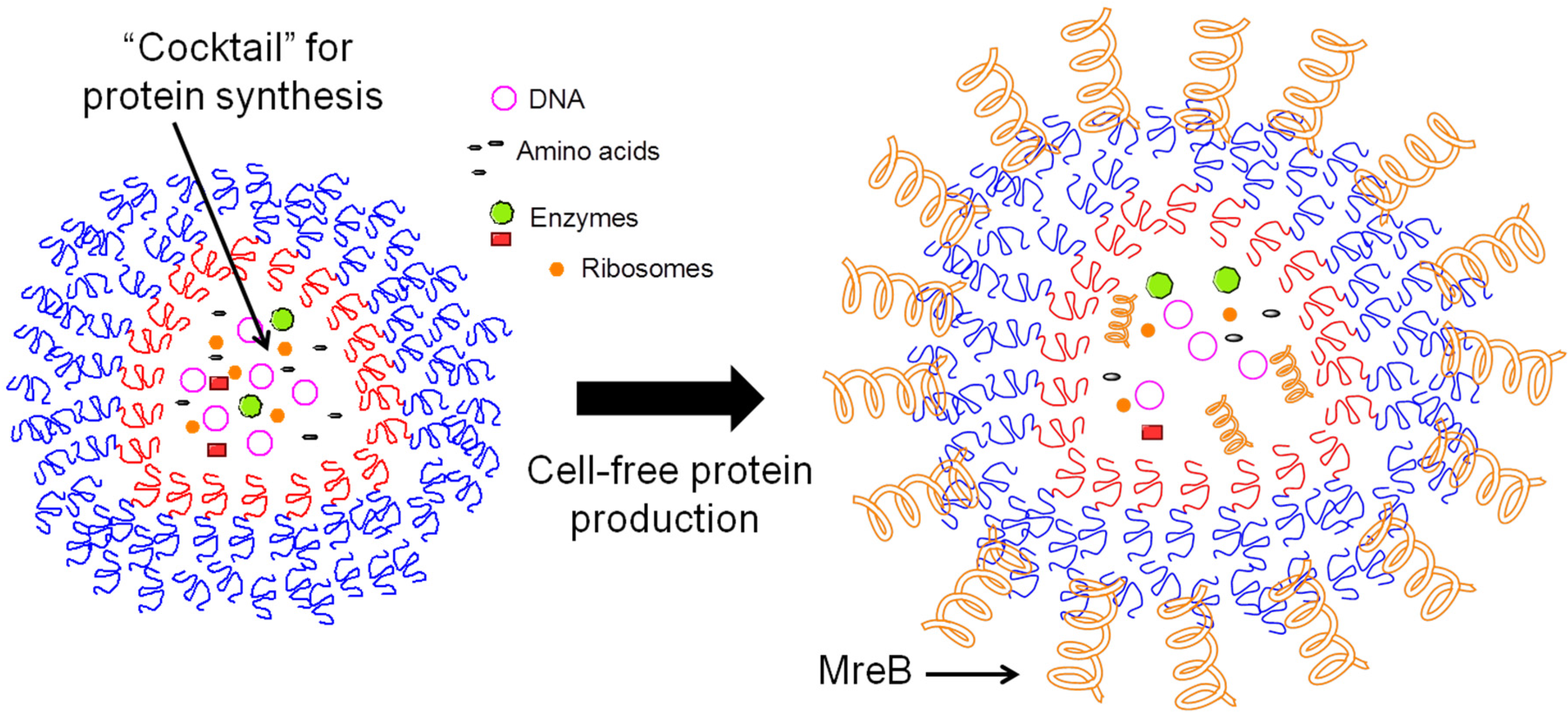
3.3.1. Polymersomes for Drug Development
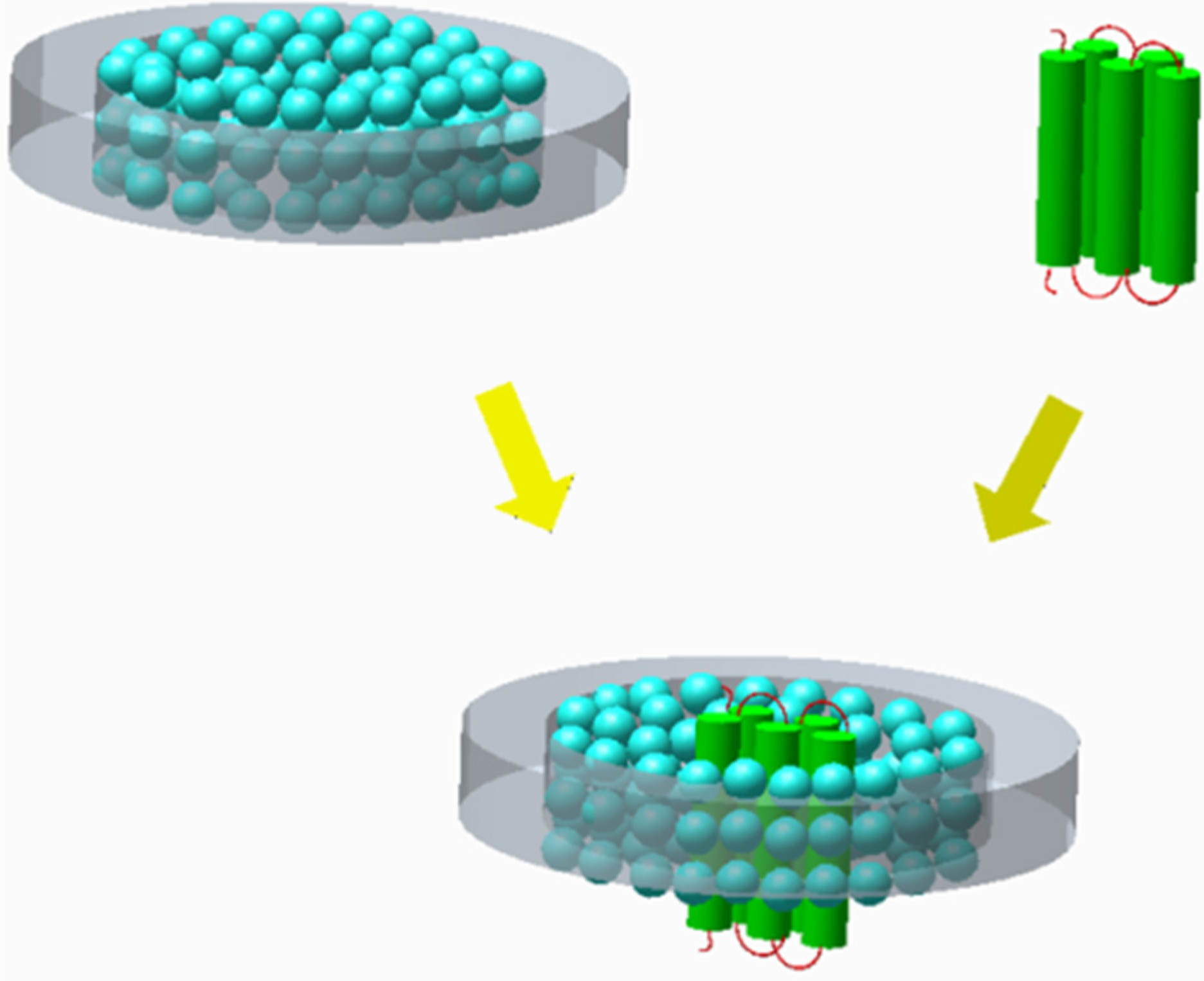
3.3.2. Nanolipoprotein Biolayer Discs for Membrane Proteins
4. Biofuels Production
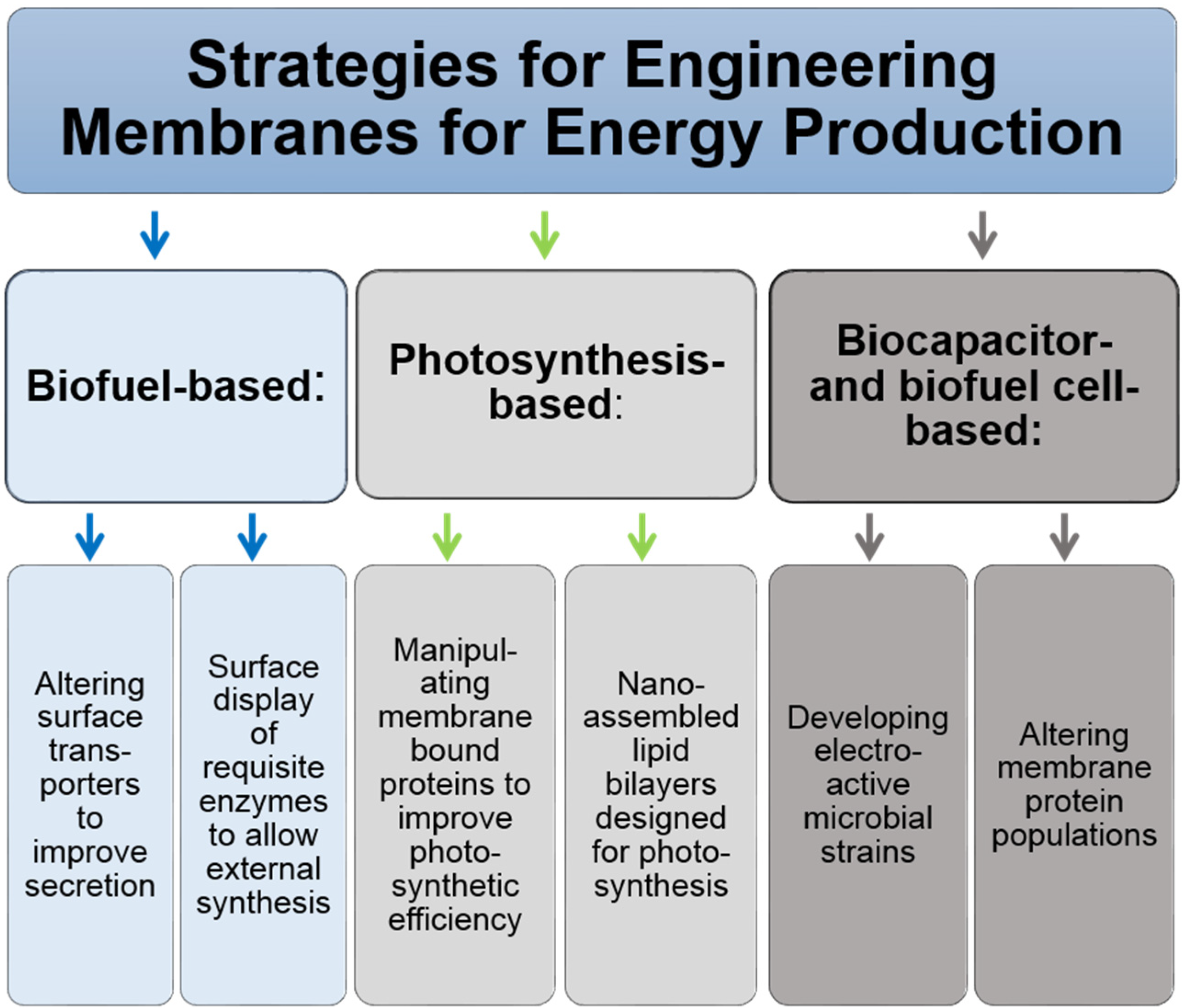
4.1. The Promise of Photosynthesis-Based Fuel Production
4.2. Manipulating Existing Photosynthetic Pathways to Avoid Inefficient Steps
4.3. Engineering Membrane Transporters for Improved Secretion and Export of Synthesized Biofuels
4.4. Circumventing Biomass Transport for Improved Efficiency of Biofuel Production
4.5. Biocapacitors and Biofuel Cells through Membrane Engineering
4.6. Developing Artificial Nano Assemblies to Convert CO2 and Water into Fuel
5. Biosensors
5.1. Surface Engineering Cells for Bioelectric and Photo Sensing
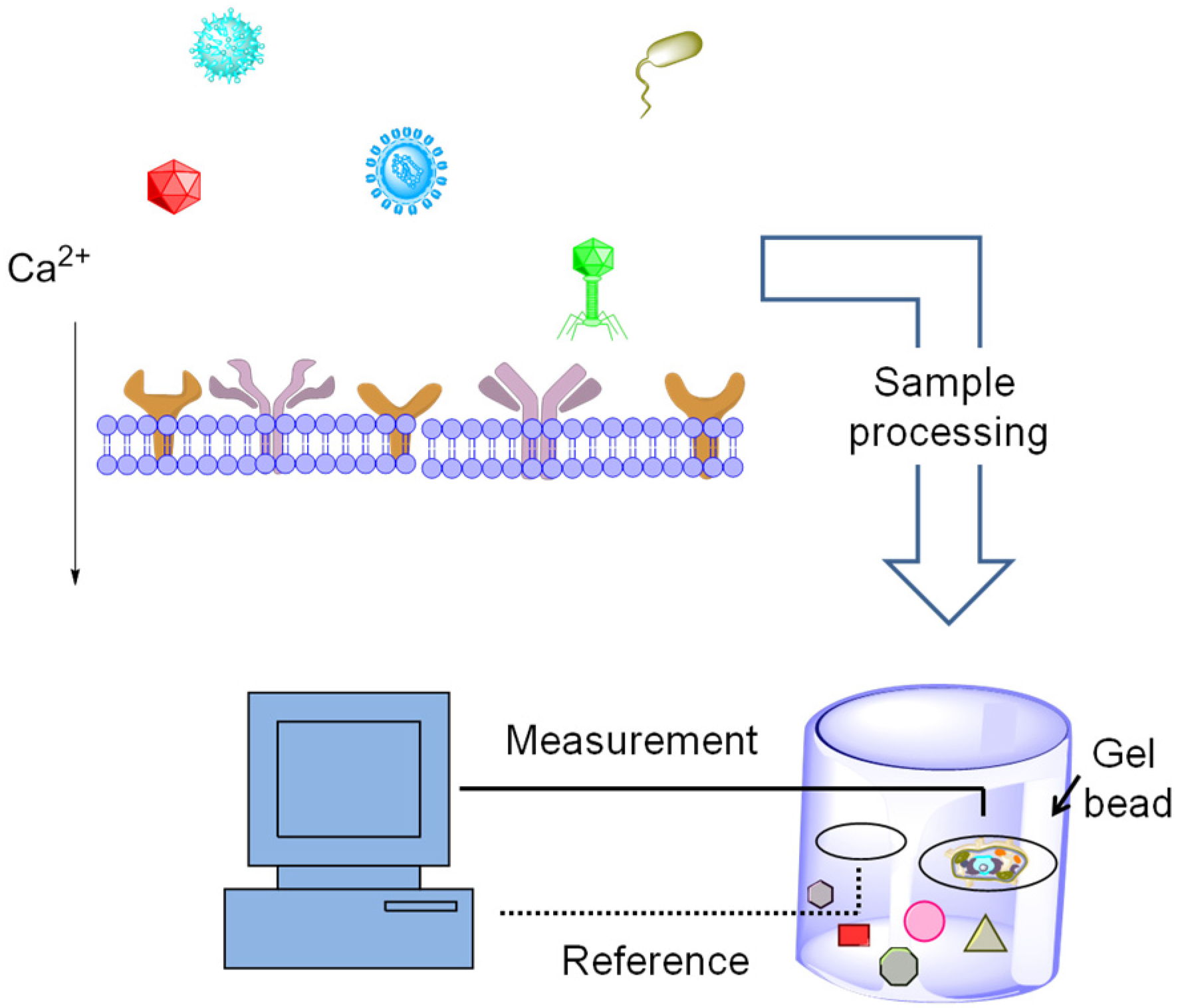
5.2. Exploiting Cellular Networks for Biosensing
5.3. Surface Engineered Microbes as an in Vitro Tool for Environmental Monitoring
5.4. Towards Cell-Free, High-Throughput Membrane-Based Biosensors
6. Engineered Membranes for Use in and the Development of Bio 3D Printing Applications
7. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Life Science Tools and Reagents: Global Markets. BCC Research LLC.: Wellesley, MA, USA, April 2011.
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Ann. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Delmar, J.A.; Bolla, J.R.; Su, C.-C.; Yu, E.W. Crystallization of membrane proteins by vapor diffusion. Methods Enzymol. 2015, 557, 363–392. [Google Scholar] [PubMed]
- Hubbell, J.A.; Langer, R. Translating materials design to the clinic. Nat. Mater. 2013, 12, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Dosoky, N.S.; Williams, J.D. Engineering lipid bilayer membranes for protein studies. Int. J. Mol. Sci. 2013, 14, 21561–21597. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.D.; Franco, O.L.; Nascimento, J.M.; de Melo, C.P.; Andrade, C.A. Mechanistic aspects of peptide-membrane interactions determined by optical, dielectric and piezoelectric techniques: An overview. Curr. Protein Pept. Sci. 2013, 14, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.R.; Avelino, K.Y.; Ribeiro, K.L.; Franco, O.L.; Oliveira, M.D.; Andrade, C.A. Optical and dielectric sensors based on antimicrobial peptides for microorganism diagnosis. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Turzhitsky, V.; Qiu, L.; Itzkan, I.; Novikov, A.A.; Kotelev, M.S.; Getmanskiy, M.; Vinokurov, V.A.; Muradov, A.V.; Perelman, L.T. Spectroscopy of scattered light for the characterization of micro and nanoscale objects in biology and medicine. Appl. Spectrosc. 2014, 68, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Drug candidates derailed in case of mistaken identity. Nature 2012, 482, 519. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ellis, L.M. Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Tan, H.T.; Chung, M.C.M. Membrane proteins and membrane proteomics. Proteomics 2008, 8, 3924–2932. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.J.; Milligan, G. Allostery at G protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol. Rev. 2010, 62, 701–725. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R. The cost and value of three-dimensional protein structure. Drug Disc. World 2003, 4, 35–48. [Google Scholar]
- Stevens, R. Long live structural biology. Nat. Struct. Mol. Biol. 2004, 11, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Schein, C. Production of soluble recombinant proteins in bacteria. Nat. Biotechnol. 1989, 7, 1141–1147. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, W.; Hong, M.; Pan, Z.; Sinko, P.J.; Ma, J.; You, G. The role of N-linked glycosylation in protein folding, membrane targeting, and substrate binding of human organic anion transporter hOAT4. Mol. Pharmacol. 2005, 67, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Popov, M.; Tam, L.; Li, J.; Reithmeier, R. Mapping the ends of transmembrane segments in a polytopic membrane protein—Scanning N-glycosylation mutagenesis of extracytosolic loops in the anion exchanger, band 3. J. Biol. Chem. 1997, 272, 18325–18332. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Bobrowicz, P.; Bobrowicz, B.; Davidson, R.; Li, H.; Mitchell, T.; Nett, J.H.; Rausch, S.; Stadheim, T.A.; Wischnewski, H.; Wildt, S.; Gerngross, T.U. Production of complex human glycoproteins in yeast. Science 2003, 301, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Oberg, F.; Hedfalk, K. Recombinant production of the human aquaporins in the yeast Pichia pastoris. Mol. Membr. Biol. 2013, 30, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Takagi, Y.; Schultz, P.; Berger, I. Baculovirus expression: tackling the complexity challenge. Curr. Opin. Struct. Biol. 2013, 23, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, M.M.; Pijlman, G.P.; Vlak, J.M. Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J. Gen. Virol. 2015, 96, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Palmberger, D.; Klausberger, M.; Berger, I.; Grabherr, R. MultiBac turns sweet. Bioengineered 2013, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Marchal, I.; Jarvis, D.L.; Cacan, R.; Verbert, A. Glycoproteins from insect cells: sialylated or not? Biol. Chem. 2001, 382, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, D.; Zhang, M.; Hurtado-Ziola, N.; Varki, A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol. Genet. Eng. Rev. 2012, 28, 147–175. [Google Scholar] [CrossRef] [PubMed]
- Tomiya, N.; Narang, S.; Lee, Y.C.; Betenbaugh, M.J. Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconj. J. 2004, 21, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Granell, A.E.; Palter, K.B.; Akan, I.; Aich, U.; Yarema, K.J.; Betenbaugh, M.J.; Thornhill, W.B.; Recio-Pinto, E. DmSAS is required for sialic acid biosynthesis in cultured Drosophila third instar larvae CNS neurons. ACS Chem. Biol. 2011, 6, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Patnaik, S.K. Chinese hamster ovary (CHO) glycosylation mutants for glycan engineering. In Handbook of Carbohydrate Engineering; Yarema, K.J., Ed.; Francis & Taylor/CRC Press: Boca Raton, FL, USA, 2005; pp. 371–385. [Google Scholar]
- Zhang, P.; Chan, K.F.; Haryadi, R.; Bardor, M.; Song, Z. CHO glycosylation mutants as potential host cells to produce therapeutic proteins with enhanced efficacy. Adv. Biochem. Eng. Biotechnol. 2013, 131, 63–87. [Google Scholar] [PubMed]
- Gubellini, F.; Verdon, G.; Karpowich, N.K.; Luff, J.D.; Boel, G.; Gauthier, N.; Handelman, S.K.; Ades, S.E.; Hunt, J.F. Physiological response to membrane protein overexpression in E. coli. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Davletov, B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006, 440, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; O’Hara, L.; Carman, G.M.; Siniossoglou, S. An unconventional diacylglycerol kinase that regulates phospholipid synthesis and nuclear membrane growth. J. Biol. Chem. 2008, 283, 20433–20442. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, H.H.; Zorec, R. Adipocyte cell size enlargement involves plasma membrane area increase. Arch. Physiol. Biochem. 2012, 118, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Barbosa, A.D.; Sembongi, H.; Mari, M.; Han, G.; Reggiori, F.; Carman, G.M.; Siniossoglou, S. Regulation of lipid droplet and membrane biogenesis by the acidic tail of the phosphatidate phosphatase Pah1p. Mol. Biol. Cell 2013, 24, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Rajakumari, S.; Grillitsch, K.; Daum, G. Synthesis and turnover of non-polar lipids in yeast. Prog. Lipid Res. 2008, 47, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Pascual, F.; Carman, G.M. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta 2013, 1831, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Guerfal, M.; Claes, K.; Knittelfelder, O.; De Rycke, R.; Kohlwein, S.D.; Callewaert, N. Enhanced membrane protein expression by engineering increased intracellular membrane production. Microb. Cell. Fact. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.Z.; Amaral, P.F.F.; Belo, I. Yarrowia lipolytica: An industrial workhorse. Curr. Res. Technol. Educ. Top. Appl. Microbiol. Microbial Biotechnol. 2010, 2, 930–940. [Google Scholar]
- Kawahara, T.; Yanagi, H.; Yura, T.; Mori, K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Cell. Biol. 1997, 8, 1845–1862. [Google Scholar] [CrossRef]
- Neubig, R.; Siderovski, D. Regulators of G-protein signalling as new central nervous system drug targets. Nat. Rev. Drug. Discov. 2002, 1, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Tang, C.; Hahntow, I.; Michel, M.C. Impact of GPCRs in clinical medicine: Monogenic diseases, genetic variants and drug targets. Biochim. Biophys. Acta 2007, 1768, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Nallani, M.; Andreasson-Ochsner, M.; Tan, C.D.; Sinner, E.; Wisantoso, Y.; Geifman-Shochat, S.; Hunziker, W. Proteopolymersomes: In vitro production of a membrane protein in polymersome membranes. Biointerphases 2011, 6, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Germain, J.; Montemagno, C. Effects of different reconstitution procedures on membrane protein activities in proteopolymersomes. Nanotechnology 2006, 17, 1825–1830. [Google Scholar] [CrossRef]
- Martino, C.; Kim, S.; Horsfall, L.; Abbaspourrad, A.; Rosser, S.J.; Cooper, J.; Weitz, D.A. Protein expression, aggregation, and triggered release from polymersomes as artificial cell-like structures. Angew. Chem. Int. Ed. Engl. 2012, 51, 6416–6420. [Google Scholar] [CrossRef] [PubMed]
- Katzen, F.; Chang, G.; Kudlicki, W. The past, present and future of cell-free protein synthesis. Trends Biotechnol. 2005, 23, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.; Grinkova, Y.; Sligar, S. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.A.; Arroyo, E.S.; Kralj, J.M.; Hinz, A.K.; Kuhn, E.A.; Chromy, B.A.; Segelke, B.W.; Rothschild, K.J.; et al. Cell-free co-expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles. Mol. Cell. Proteomics 2008, 7, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Katzen, F.; Fletcher, J.E.; Yang, J.; Kang, D.; Peterson, T.C.; Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.; Chromy, B.A.; Hoeprich, P.D.; et al. Insertion of membrane proteins into discoidal membranes using a cell-free protein expression approach. J. Proteome Res. 2008, 7, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Katzen, F.; Peterson, T.C.; Kudlicki, W. Membrane protein expression: no cells required. Trends Biotechnol. 2009, 27, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.; Smith, H.M.; Dickson, J.; Austin, J.P. Interaction of apoprotein from porcine high-density lipoprotein with dimyristoyl lecithin. 1. The structure of the complexes. Eur. J. Biochem. 1976, 64, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, C.; Jones, J.; Ng, T.; Kercret, H.; Chung, B.; Segrest, J. Structural studies of apolipoprotein-A-I/phosphatidylcholine recombinants by high-field proton NMR, nondenaturing gradient gel-electrophoresis, and electron-microscopy. Biochemistry 1984, 23, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Jonas, A. Reconstitution of high-density lipoproteins. Methods Enzymol. 1986, 128, 553–582. [Google Scholar] [PubMed]
- Klon, A.; Jones, M.; Segrest, J.; Harvey, S. Molecular belt models for the apolipoprotein A-I Paris and Milano mutations. Biophys. J. 2000, 79, 1679–1685. [Google Scholar] [CrossRef]
- Wlodawer, A.; Segrest, J.; Chung, B.; Chiovetti, R.; Weinstein, J. High-density lipoprotein recombinants—Evidence for a bicycle tire micelle structure obtained by neutron-scattering and electron-microscopy. FEBS Lett. 1979, 104, 231–235. [Google Scholar] [CrossRef]
- Denisov, I.G.; Sligar, S.G. Cytochromes P450 in nanodiscs. Biochim. Biophys. Acta 2011, 1814, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Shenkarev, Z.O.; Lyukmanova, E.N.; Butenko, I.O.; Petrovskaya, L.E.; Paramonov, A.S.; Shulepko, M.A.; Nekrasova, O.V.; Kirpichnikov, M.P.; Arseniev, A.S. Lipid-protein nanodiscs promote in vitro folding of transmembrane domains of multi-helical and multimeric membrane proteins. Biochim. Biophys. Acta 2013, 1828, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.T.; Wilcox, K.C.; Klein, W.L.; Sligar, S.G. Nanodisc-solubilized membrane protein library reflects the membrane proteome. Anal. Bioanal. Chem. 2013, 405, 4009–4016. [Google Scholar] [CrossRef] [PubMed]
- Proverbio, D.; Roos, C.; Beyermann, M.; Orban, E.; Doetsch, V.; Bernhard, F. Functional properties of cell-free expressed human endothelin A and endothelin B receptors in artificial membrane environments. Biochim. Biophys. Acta 2013, 1828, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Shadiac, N.; Nagarajan, Y.; Waters, S.; Hrmova, M. Close allies in membrane protein research: Cell-free synthesis and nanotechnology. Mol. Membr. Biol. 2013, 30, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Shenkarev, Z.O.; Paramonov, A.S.; Lyukmanova, E.N.; Gizatullina, A.K.; Zhuravleva, A.V.; Tagaev, A.A.; Yakimenko, Z.A.; Telezhinskaya, I.N.; Kirpichnikov, M.P.; Ovchinnikova, T.V.; et al. Peptaibol antiamoebin I: spatial structure, backbone dynamics, interaction with bicelles and lipid-protein nanodiscs, and pore formation in context of barrel-stave model. Chem. Biodivers. 2013, 10, 838–863. [Google Scholar] [CrossRef] [PubMed]
- Renthal, R. Helix insertion into bilayers and the evolution of membrane proteins. Cell. Mol. Life Sci. 2010, 67, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Barrera, F.N.; Weerakkody, D.; Anderson, M.; Andreev, O.A.; Reshetnyak, Y.K.; Engelman, D.M. Roles of carboxyl groups in the transmembrane insertion of peptides. J. Mol. Biol. 2011, 413, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.A.; Schiller, N.; von Heijne, G.; Deber, C.M. Hydrophobic blocks facilitate lipid compatibility and translocon recognition of transmembrane protein sequences. Biochemistry 2015, 54, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K. Chapter 2. Energy Conversion by Photosynthetic Organisms. In FAO Agricultural Services Bulletin: Food and Agriculture Organization of the United Nations; Miyamoto, K., Ed.; FAO Corporate Document Repository: Osaka, Japan, 1997. [Google Scholar]
- Key World Energy Statistics 2012; Organisation for Economic Co-operation and Development: Paris, France, 2013.
- Kruse, O.; Rupprecht, J.; Mussgnug, J.H.; Dismukes, G.C.; Hankamer, B. Photosynthesis: A blueprint for solar energy capture and biohydrogen production technologies. Photochem. Photobiol. Sci. 2005, 4, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.W.; Lewis, N.S. Solar energy conversion. Phys. Today 2007, 60, 37–42. [Google Scholar] [CrossRef]
- Kosourov, S.; Seibert, M.; Ghirardi, M.L. Effects of extracellular pH on the metabolic pathways in sulfur-deprived, H2-producing Chlamydomonas reinhardtii cultures. Plant. Cell. Physiol. 2003, 44, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, J.; Lomas, M.; LaRoche, J.; Falkowski, P.G. Light intensity regulation of cab gene transcription is signaled by the redox state of the plastoquinone pool. Proc. Natl. Acad. Sci. USA 1995, 92, 10237–10241. [Google Scholar] [CrossRef] [PubMed]
- Flachmann, R.; Kühlbrandt, W. Accumulation of plant antenna complexes is regulated by post-transcriptional mechanisms in tobacco. Plant Cell. 1995, 7, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Yang, D.; Andersson, B. Regulatory proteolysis of the major light-harvesting chlorophyll a/b protein of photosystem II by a light-induced membrane-associated enzymic system. Eur. J. Biochem. 1995, 231, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Durnford, D.G.; Price, J.A.; McKim, S.M.; Sarchfield, M.L. Light-harvesting complex gene expression is controlled by both transcriptional and post-transcriptional mechanisms during photoacclimation in Chlamydomonas reinhardtii. Physiol. Plantarum 2003, 118, 193–205. [Google Scholar] [CrossRef]
- Tokutsu, R.; Teramoto, H.; Takahashi, Y.; Ono, T.; Minagawa, J. The light-harvesting complex of photosystem I in Chlamydomonas reinhardtii: Protein composition, gene structures and phylogenic implications. Plant. Cell. Physiol. 2004, 45, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Mussgnug, J.H.; Wobbe, L.; Elles, I.; Claus, C.; Hamilton, M.; Fink, A.; Kahmann, U.; Kapazoglou, A.; Mullineaux, C.W.; Hippler, M.; et al. NAB1 is an RNA binding protein involved in the light-regulated differential expression of the light-harvesting antenna of Chlamydomonas reinhardtii. Plant Cell 2005, 17, 3409–3421. [Google Scholar] [CrossRef] [PubMed]
- Dall’Osto, L.; Bressan, M.; Bassi, R. Biogenesis of light harvesting proteins. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Adir, N.; Zer, H.; Shochat, S.; Ohad, I. Photoinhibition—A historical perspective. Photosynth. Res. 2003, 76, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Li, X.; Niyogi, K.K. Non-photochemical quenching. A response to excess light energy. Plant. Physiol. 2001, 125, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Polle, J.E.; Kanakagiri, S.; Jin, E.; Masuda, T.; Melis, A. Truncated chlorophyll antenna size of the photosystems—a practical method to improve microalgal productivity and hydrogen production in mass culture. Int. J. Hydrogen Energ. 2002, 27, 1257–1264. [Google Scholar] [CrossRef]
- Polle, J.E.; Kanakagiri, S.; Melis, A. Tla1, a DNA insertional transformant of the green alga Chlamydomonas reinhardtii with a truncated light-harvesting chlorophyll antenna size. Planta 2003, 217, 49–59. [Google Scholar] [PubMed]
- Prince, R.C.; Kheshgi, H.S. The photobiological production of hydrogen: Potential efficiency and effectiveness as a renewable fuel. Crit. Rev. Microbiol. 2005, 31, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mussgnug, J.H.; Thomas-Hall, S.; Rupprecht, J.; Foo, A.; Klassen, V.; McDowall, A.; Schenk, P.M.; Kruse, O.; Hankamer, B. Engineering photosynthetic light capture: Impacts on improved solar energy to biomass conversion. Plant Biotechnol. J. 2007, 5, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Oey, M.; Ross, I.L.; Stephens, E.; Steinbeck, J.; Wolf, J.; Radzun, K.A.; Kügler, J.; Ringsmuth, A.K.; Kruse, O.; Hankamer, B. RNAi knock-down of LHCBM1, 2 and 3 increases photosynthetic H2 production efficiency of the green alga Chlamydomonas reinhardtii. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Radakovits, R.; Jinkerson, R.E.; Darzins, A.; Posewitz, M.C. Genetic engineering of algae for enhanced biofuel production. Eukaryot. Cell 2010, 9, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Islam, K.; Pease, R.J. Mobilisation of triacylglycerol stores. Biochim. Biophys. Acta 2000, 1483, 37–57. [Google Scholar] [CrossRef]
- Lehner, R.; Vance, D. Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem. J. 1999, 343, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tietge, U.J.; Bakillah, A.; Maugeais, C.; Tsukamoto, K.; Hussain, M.; Rader, D.J. Hepatic overexpression of microsomal triglyceride transfer protein (MTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J. Lipid Res. 1999, 40, 2134–2139. [Google Scholar] [PubMed]
- Tietge, U.J.; Maugeais, C.; Cain, W.; Grass, D.; Glick, J.M.; de Beer, F.C.; Rader, D.J. Overexpression of secretory phospholipase A(2) causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A-I. J. Biol. Chem. 2000, 275, 10077–10084. [Google Scholar] [CrossRef] [PubMed]
- Pighin, J.A.; Zheng, H.; Balakshin, L.J.; Goodman, I.P.; Western, T.L.; Jetter, R.; Kunst, L.; Samuels, A.L. Plant cuticular lipid export requires an ABC transporter. Science 2004, 306, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Mentewab, A.; Stewart, C.N. Overexpression of an Arabidopsis thaliana ABC transporter confers kanamycin resistance to transgenic plants. Nat. Biotechnol. 2005, 23, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gupta, S.; Xu, F.; Liverman, A.D.; Moschetta, A.; Mangelsdorf, D.J.; Repa, J.J.; Hobbs, H.H.; Cohen, J.C. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J. Biol. Chem. 2005, 280, 8742–8747. [Google Scholar] [CrossRef] [PubMed]
- Panikashvili, D.; Savaldi-Goldstein, S.; Mandel, T.; Yifhar, T.; Franke, R.B.; Höfer, R.; Schreiber, L.; Chory, J.; Aharoni, A. The Arabidopsis DESPERADO/AtWBC11 transporter is required for cutin and wax secretion. Plant Physiol. 2007, 145, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, G. Challenges in engineering microbes for biofuels production. Science 2007, 315, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Charbit, A.; Boulain, J.C.; Ryter, A.; Hofnung, M. Probing the topology of a bacterial membrane protein by genetic insertion of a foreign epitope; expression at the cell surface. EMBO J. 1986, 5, 3029–3037. [Google Scholar] [PubMed]
- Wu, C.H.; Mulchandani, A.; Chen, W. Versatile microbial surface-display for environmental remediation and biofuels production. Trends Microbiol. 2008, 16, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Shigechi, H.; Koh, J.; Fujita, Y.; Matsumoto, T.; Bito, Y.; Ueda, M.; Satoh, E.; Fukuda, H.; Kondo, A. Direct production of ethanol from raw corn starch via fermentation by use of a novel surface-engineered yeast strain codisplaying glucoamylase and α-amylase. Appl. Environ. Microbiol. 2004, 70, 5037–5040. [Google Scholar] [CrossRef] [PubMed]
- Shigechi, H.; Uyama, K.; Fujita, Y.; Matsumoto, T.; Ueda, M.; Tanaka, A.; Fukuda, H.; Kondo, H. Efficient ethanol production from starch through development of novel flocculent yeast strains displaying glucoamylase and co-displaying or secreting α-amylase. J. Molec. Catal. B 2002, 17, 179–187. [Google Scholar] [CrossRef]
- Kondo, A.; Shigechi, H.; Abe, M.; Uyama, K.; Matsumoto, T.; Takahashi, S.; Ueda, M.; Tanaka, A.; Kishimoto, M.; Fukuda, H. High-level ethanol production from starch by a flocculent Saccharomyces cerevisiae strain displaying cell-surface glucoamylase. Appl. Microbiol. Biotechnol. 2002, 58, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Katahira, S.; Ueda, M.; Tanaka, A.; Okada, H.; Morikawa, Y.; Fukuda, H.; Kondo, A. Construction of whole-cell biocatalyst for xylan degradation through cell-surface xylanase display in Saccharomyces cerevisiae. J. Molec. Catal. B 2002, 17, 189–195. [Google Scholar] [CrossRef]
- Fujita, Y.; Takahashi, S.; Ueda, M.; Tanaka, A.; Okada, H.; Morikawa, Y.; Kawaguchi, T.; Arai, M.; Fukuda, H.; Kondo, A. Direct and efficient production of ethanol from cellulosic material with a yeast strain displaying cellulolytic enzymes. Appl. Environ. Microbiol. 2002, 68, 5136–5141. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, G.; Hausman, J.F.; Strauss, J.; Ertan, H.; Siddiqui, K.S. Destructuring plant biomass: Focus on fungal and extremophilic cell wall hydrolases. Plant Sci. 2015, 234, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; van Zyl, W.H.; McBride, J.E.; Laser, M. Consolidated bioprocessing of cellulosic biomass: an update. Curr. Opin. Biotechnol. 2005, 16, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Xie, D.; Li, F.; Hu, Y.; Wei, C.; Feng, C. Microbial fuel cell as a biocapacitor by using pseudo-capacitive anode materials. J. Power Sourc. 2014, 246, 642–649. [Google Scholar] [CrossRef]
- Park, D.H.; Kim, S.K.; Shin, I.H.; Jeong, Y.J. Electricity production in biofuel cell using modified graphite electrode with neutral red. Biotechnol. Lett. 2000, 22, 1301–1304. [Google Scholar] [CrossRef]
- Gao, F.; Viry, L.; Maugey, M.; Poulin, P.; Mano, N. Engineering hybrid nanotube wires for high-power biofuel cells. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Aelterman, P.; Verstraete, W. Bioanode performance in bioelectrochemical systems: Recent improvements and prospects. Trends Biotechnol. 2009, 27, 168–178. [Google Scholar]
- Kim, H.J.; Park, H.S.; Hyun, M.S.; Chang, I.S.; Kim, M.; Kim, B.H. A mediator-less microbial fuel cell using a metal reducing bacterium, Shewanella putrefaciens. Enz. Microb. Technol. 2002, 30, 145–152. [Google Scholar] [CrossRef]
- Trinh, N.T.; Park, J.H.; Kim, B. Increased generation of electricity in a microbial fuel cell using Geobacter sulfurreducens. Korean J. Chem. Eng. 2009, 26, 748–753. [Google Scholar] [CrossRef]
- Joya, K.S.; Joya, Y.F.; Ocakoglu, K.; van de Krol, R. Water-splitting catalysis and solar fuel devices: Artificial leaves on the move. Angew. Chem. Int. Ed. Engl. 2013, 52, 10426–10437. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking photosynthetic solar energy transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wasielewski, M.R. Energy, charge, and spin transport in molecules and self-assembled nanostructures inspired by photosynthesis. J. Org. Chem. 2006, 71, 5051–5066. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, S.; Piletsky, S.; Turner, A. Application of natural receptors in sensors and assays. Anal. Chem. 2002, 74, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Kintzios, S.; Pistola, E.; Panagiotopoulos, P.; Bomsel, M.; Alexandropoulos, N.; Bem, F.; Ekonomou, G.; Biselis, J.; Levin, R. Bioelectric recognition assay (BERA). Biosens. Bioelectron. 2001, 16, 325–336. [Google Scholar] [CrossRef]
- Varelas, V.; Sanvicens, N.; M-Pilar-Marco; Kintzios, S. Development of a cellular biosensor for the detection of 2,4,6-trichloroanisole (TCA). Talanta 2011, 84, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Moschopoulou, G.; Vitsa, K.; Bem, F.; Vassilakos, N.; Perdikaris, A.; Blouhos, P.; Yialouris, C.; Frosyniotis, D.; Anthopoulos, I.; Mangana, O.; et al. Engineering of the membrane of fibroblast cells with virus-specific antibodies: a novel biosensor tool for virus detection. Biosens. Bioelectron. 2008, 24, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Rider, T.; Petrovick, M.; Nargi, F.; Harper, J.; Schwoebel, E.; Mathews, R.; Blanchard, D.J.; Bortolin, L.T.; Young, A.M.; Chen, J.; et al. A B cell-based sensor for rapid identification of pathogens. Science 2003, 301, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.; Naal, R.M.; Batt, C.; Tabb, J.; Holowka, D. Development of a mast cell-based biosensor. Biosens. Bioelectron. 2008, 23, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Bhunia, A.K. Cell-based biosensor for rapid screening of pathogens and toxins. Biosens. Bioelectron. 2010, 26, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.; Spellman, P.; Brown, P.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- O'Shaughnessy, T.J.; Liu, J.L.; Ma, W. Passaged neural stem cell-derived neuronal networks for a portable biosensor. Biosens. Bioelectron. 2009, 24, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Rhoades, B.; Azzazy, H.; Wu, M. The use of neuronal networks on multielectrode arrays as biosensors. Biosens. Bioelectron. 1995, 10, 553–567. [Google Scholar] [CrossRef]
- Massobrio, G.; Massobrio, A.; Massobrio, L.; Massobrio, P. Silicon-based biosensor functionalised with carbon nanotubes to investigate neuronal electrical activity in pH-stimulated environment: A modelling approach. Micro Nano Lett. 2011, 6, 689–693. [Google Scholar] [CrossRef]
- Parvez, S.; Venkataraman, C.; Mukherji, S. A review on advantages of implementing luminescence inhibition test (Vibrio fischeri) for acute toxicity prediction of chemicals. Environ. Int. 2006, 32, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lei, C.; Shen, Q.; Li, L.; Wang, M.; Guo, M.; Huang, Y.; Nie, Z.; Yao, S. Analysis of copper nanoparticles toxicity based on a stress-responsive bacterial biosensor array. Nanoscale 2013, 5, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Karube, I.; Matsunaga, T.; Mitsuda, S.; Suzuki, S. Microbial electrode BOD sensors. Biotechnol. Bioeng. 2009, 102, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Dhall, P.; Siddiqi, T.O.; Ahmad, A.; Kumar, R.; Kumar, A. Selection of an apt support for the immobilization of microbes for the development of a BOD biosensor. Anal. Meth. 2013, 5, 1533–1541. [Google Scholar] [CrossRef]
- Kara, S.; Keskinler, B.; Erhan, E. A novel microbial BOD biosensor developed by the immobilization of P. syringae in micro-cellular polymers. J. Chem. Technol. Biotechnol. 2009, 84, 511–518. [Google Scholar] [CrossRef]
- Akay, G.; Erhan, E.; Keskinler, B. Bioprocess intensification in flow-through monolithic microbioreactors with immobilized bacteria. Biotechnol. Bioeng. 2005, 90, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, N.J.; Johnson, T.W.; Oh, S.H. High-density arrays of submicron spherical supported lipid bilayers. Anal. Chem. 2012, 84, 8207–8213. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, N.J.; Johnson, T.W.; Jordan, L.R.; Xu, X.; Warrington, A.E.; Rodriguez, M.; Oh, S.H. Formation of biomembrane microarrays with a squeegee-based assembly method. J. Vis. Exp. 2014, 87. [Google Scholar] [CrossRef] [PubMed]
- Malmstadt, N.; Nash, M.A.; Purnell, R.F.; Schmidt, J.J. Automated formation of lipid-bilayer membranes in a microfluidic device. Nano Lett. 2006, 6, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Baaken, G.; Sondermann, M.; Schlemmer, C.; Rühe, J.; Behrends, J.C. Planar microelectrode-cavity array for high-resolution and parallel electrical recording of membrane ionic currents. Lab. Chip 2008, 8, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Suzuki, H.; Le Pioufle, B.; Takeuchi, S. Multichannel simultaneous measurements of single-molecule translocation in alpha-hemolysin nanopore array. Anal. Chem. 2009, 81, 9866–9870. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Geng, J.; Ahn, C.H.; Guo, P. Formation of lipid bilayers inside microfluidic channel array for monitoring membrane-embedded nanopores of phi29 DNA packaging nanomotor. Biomed. Microdevices 2012, 14, 921–928. [Google Scholar] [CrossRef] [PubMed]
- del Rio Martinez, J.M.; Zaitseva, E.; Petersen, S.; Baaken, G.; Behrends, J.C. Automated formation of lipid membrane microarrays for ionic single-molecule sensing with protein nanopores. Small 2015, 11, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Villar, G.; Graham, A.D.; Bayley, H. A tissue-like printed material. Science 2013, 340, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Villar, G.; Heron, A.J.; Bayley, H. Formation of droplet networks that function in aqueous environments. Nat. Nanotechnol. 2011, 6, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Vercoutere, W.; Winters-Hilt, S.; Olsen, H.; Deamer, D.; Haussler, D.; Akeson, M. Rapid discrimination among individual DNA hairpin molecules at single-nucleotide resolution using an ion channel. Nat. Biotechnol. 2001, 19, 248–252. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeui, C.T.; Mathew, M.P.; Liu, L.; Urias, E.; Yarema, K.J. Cell Surface and Membrane Engineering: Emerging Technologies and Applications. J. Funct. Biomater. 2015, 6, 454-485. https://doi.org/10.3390/jfb6020454
Saeui CT, Mathew MP, Liu L, Urias E, Yarema KJ. Cell Surface and Membrane Engineering: Emerging Technologies and Applications. Journal of Functional Biomaterials. 2015; 6(2):454-485. https://doi.org/10.3390/jfb6020454
Chicago/Turabian StyleSaeui, Christopher T., Mohit P. Mathew, Lingshui Liu, Esteban Urias, and Kevin J. Yarema. 2015. "Cell Surface and Membrane Engineering: Emerging Technologies and Applications" Journal of Functional Biomaterials 6, no. 2: 454-485. https://doi.org/10.3390/jfb6020454
APA StyleSaeui, C. T., Mathew, M. P., Liu, L., Urias, E., & Yarema, K. J. (2015). Cell Surface and Membrane Engineering: Emerging Technologies and Applications. Journal of Functional Biomaterials, 6(2), 454-485. https://doi.org/10.3390/jfb6020454