
Carbohydrate-Derived Amphiphilic Macromolecules: A Biophysical Structural Characterization and Analysis of Binding Behaviors to Model Membranes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

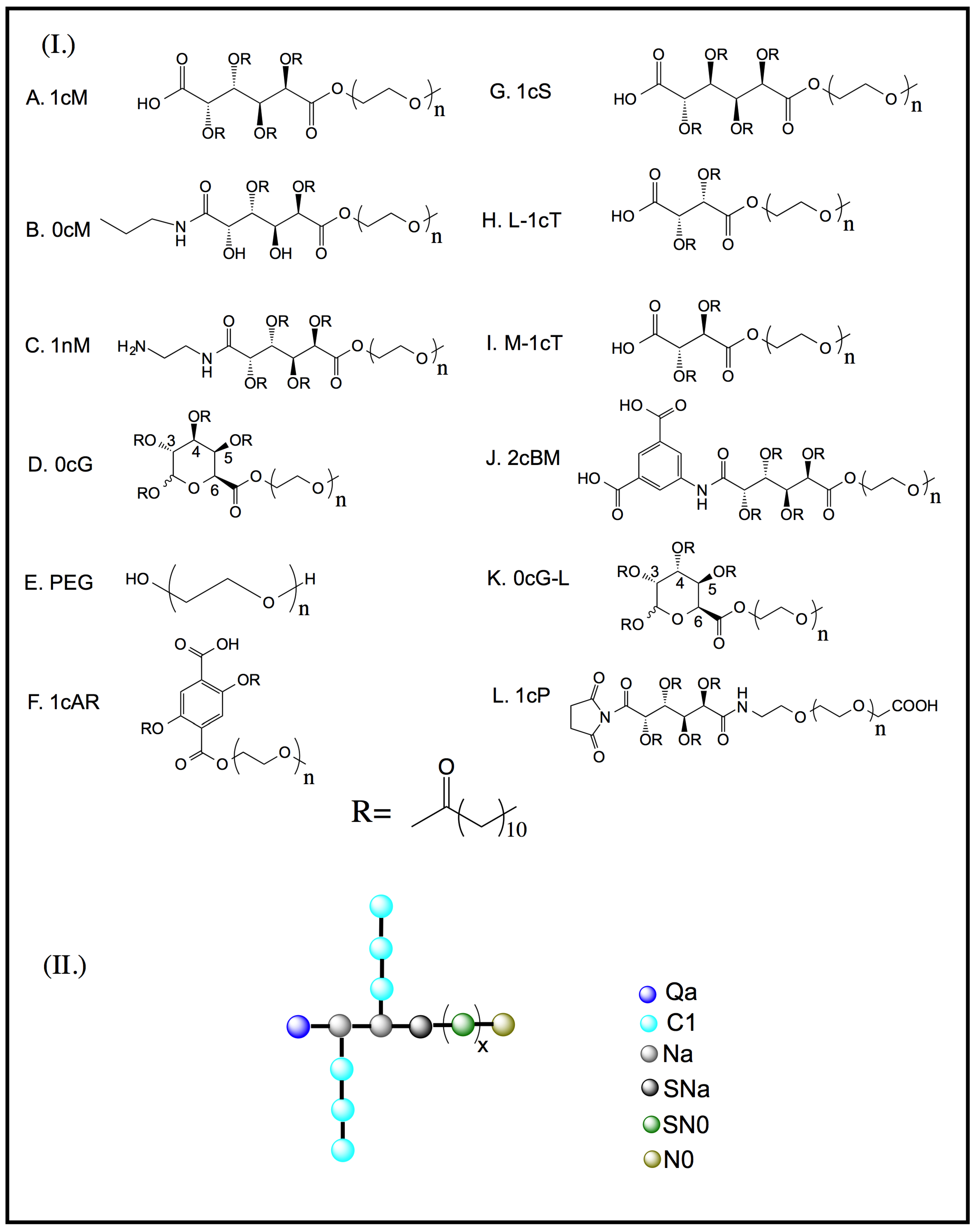
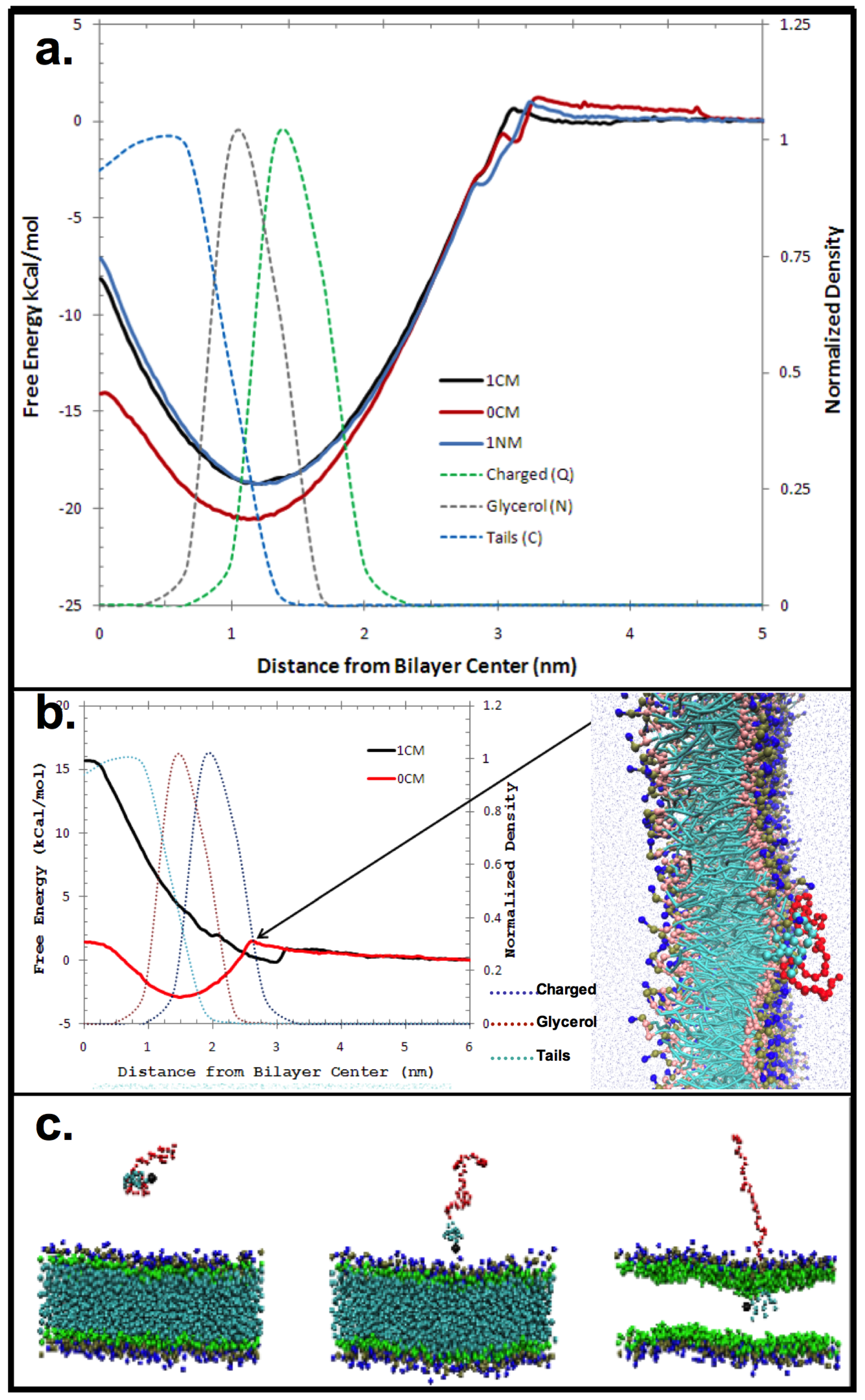
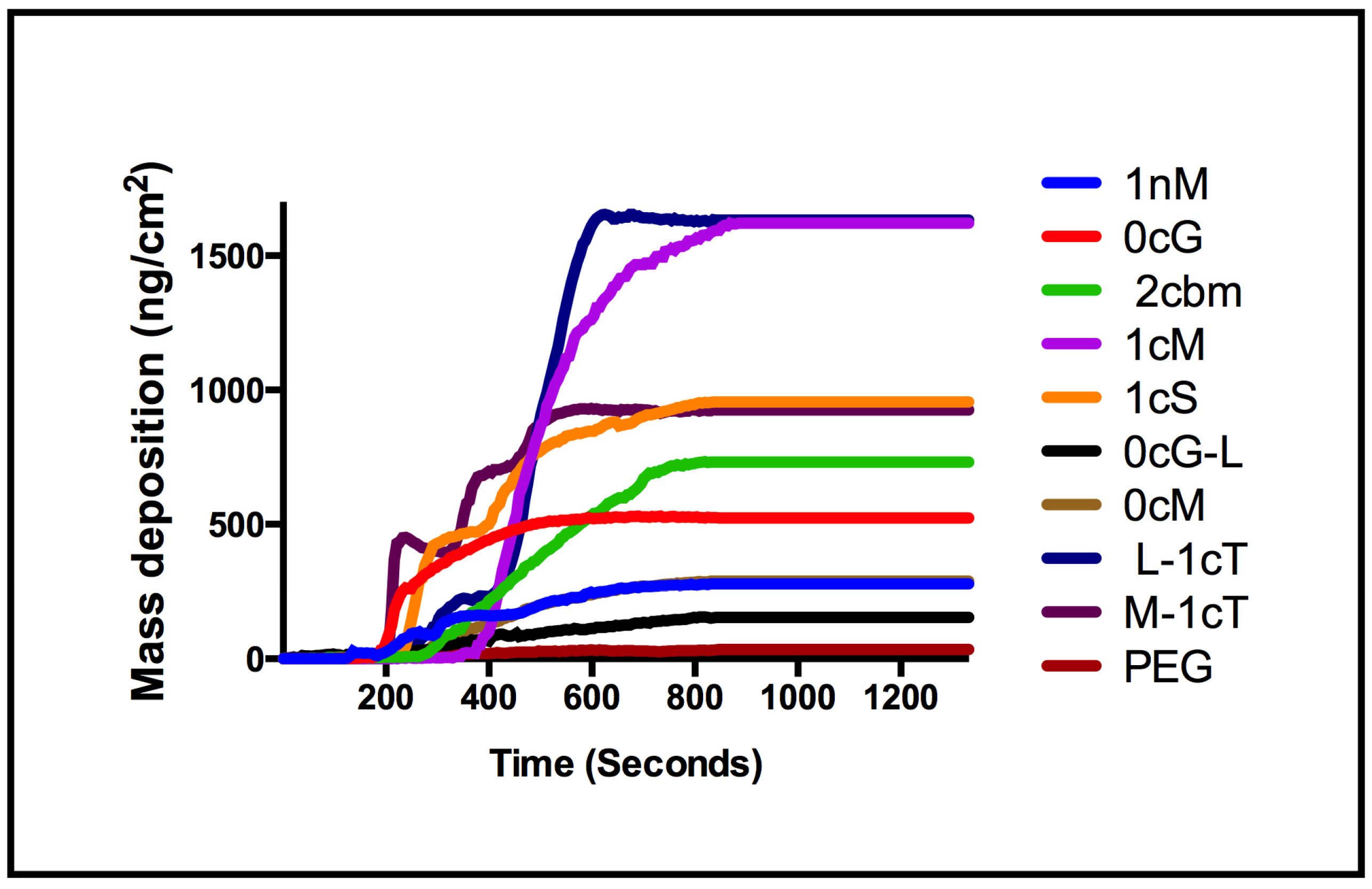
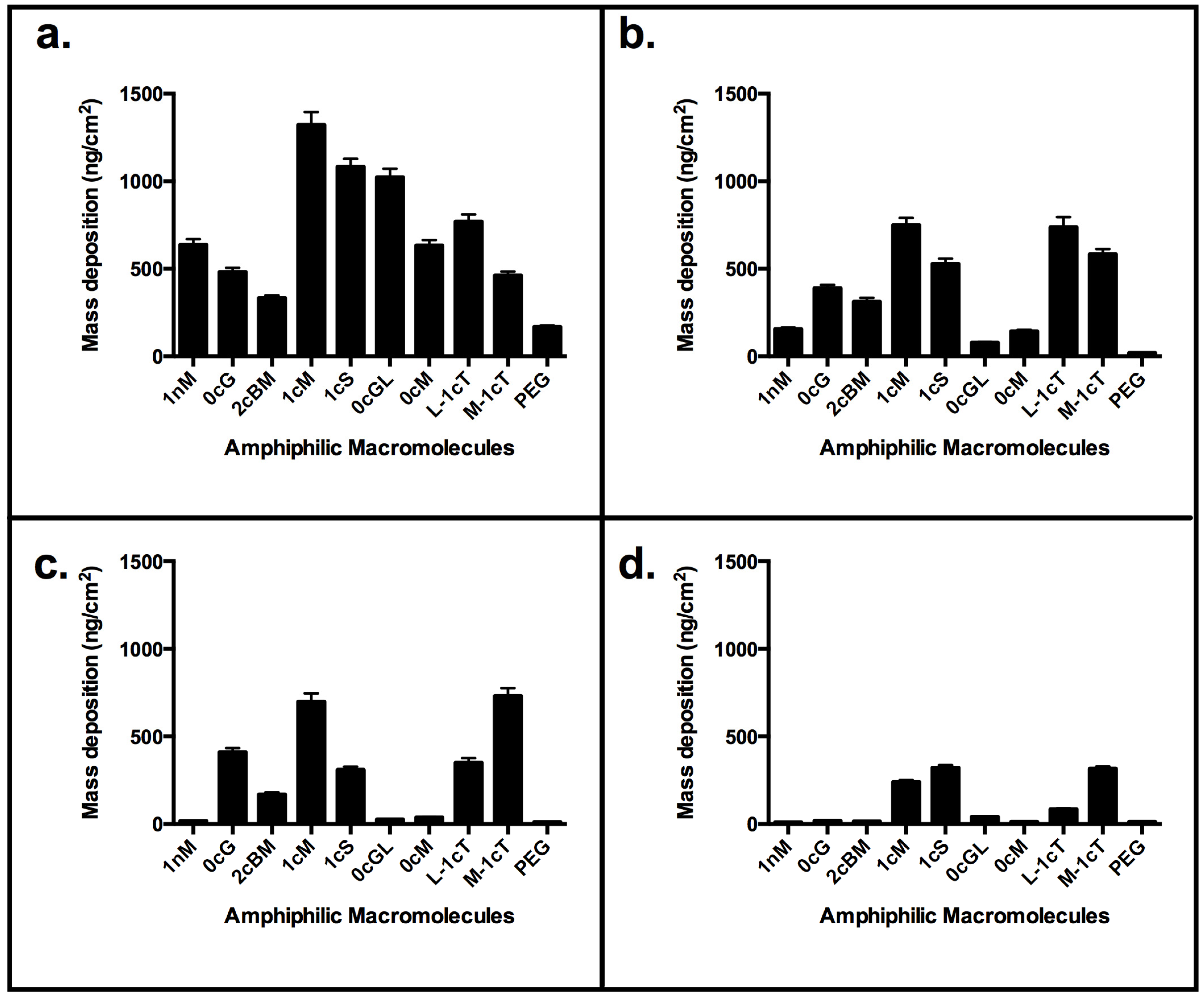
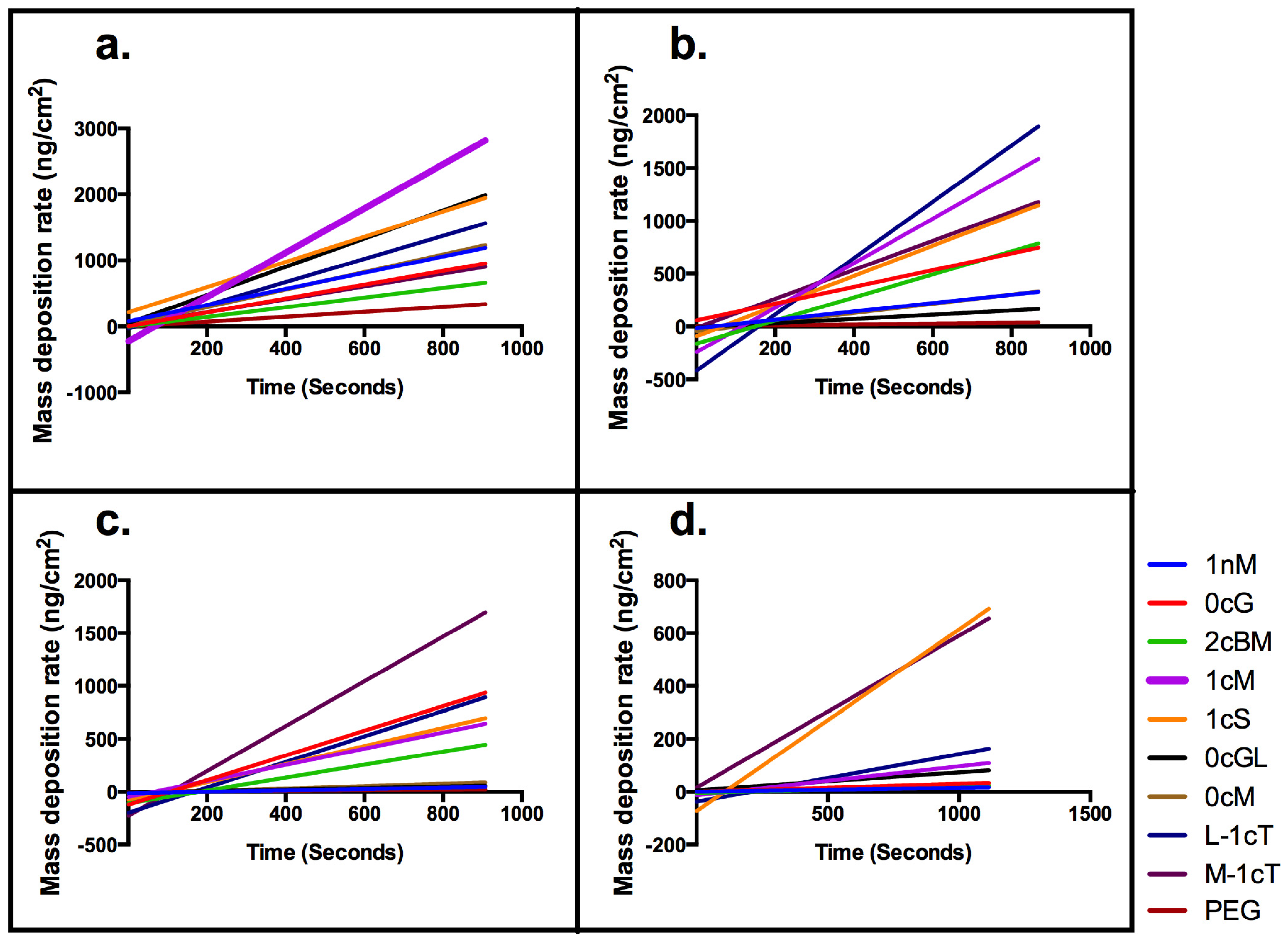
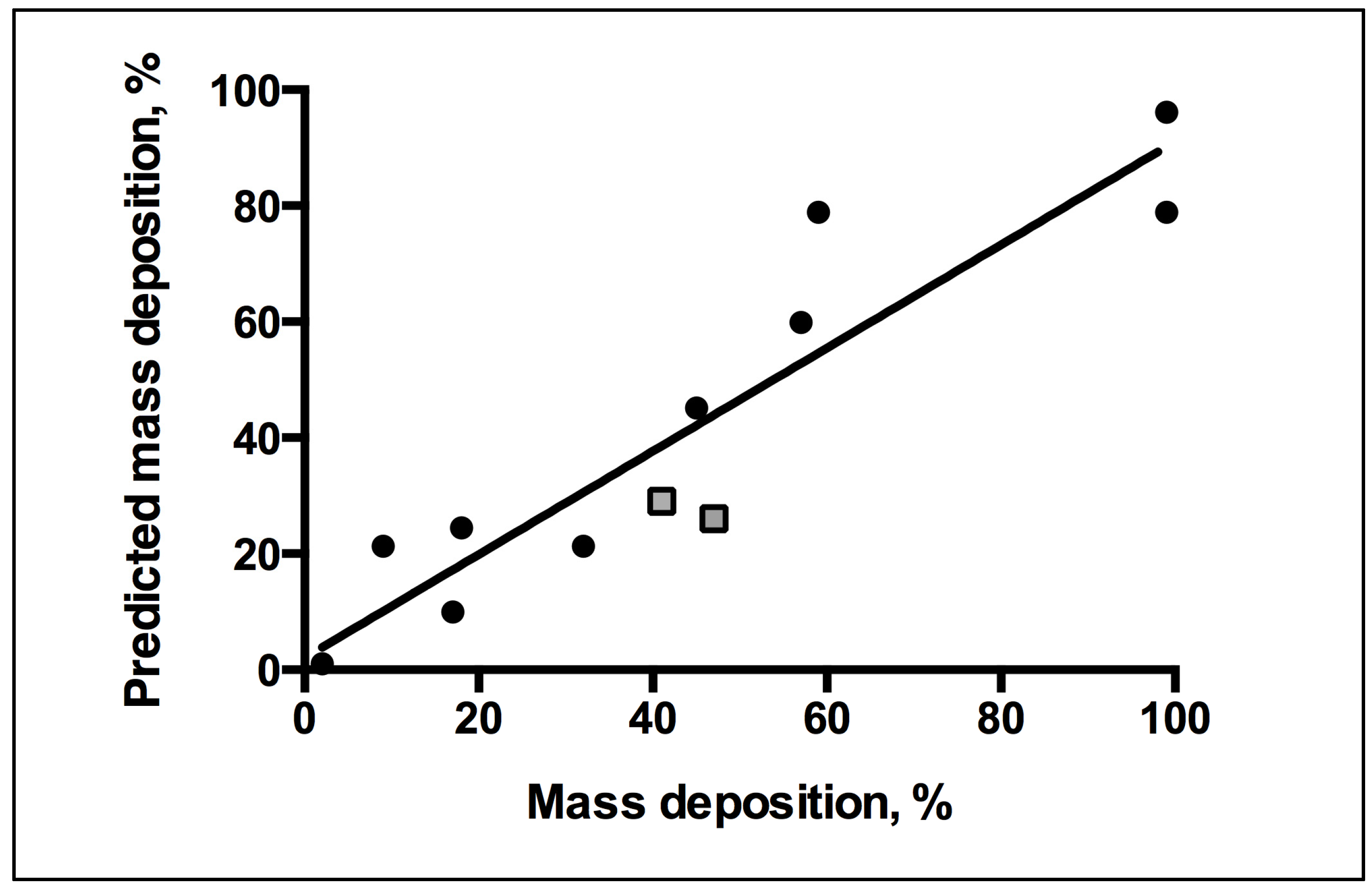
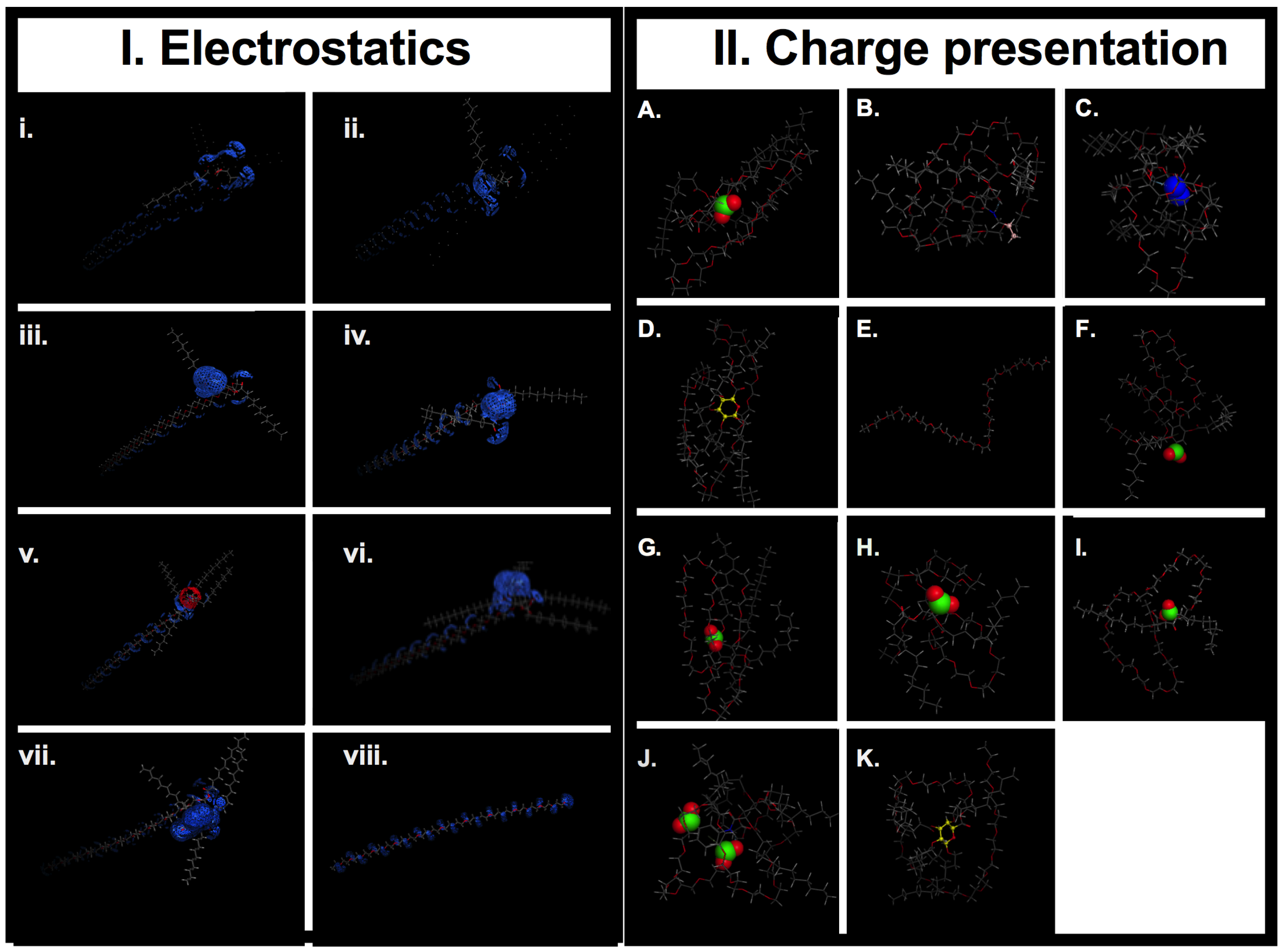
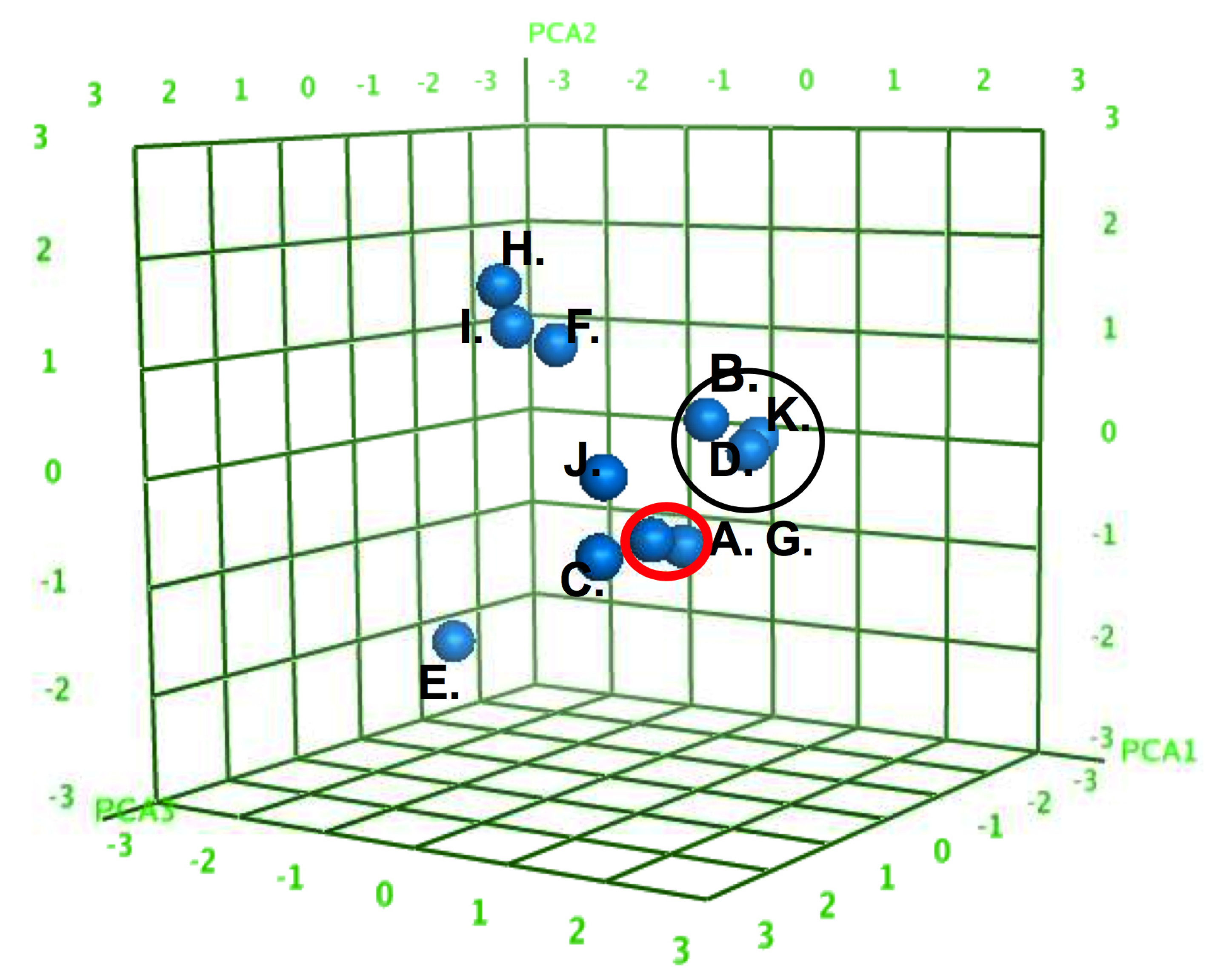
2. Results and Discussion
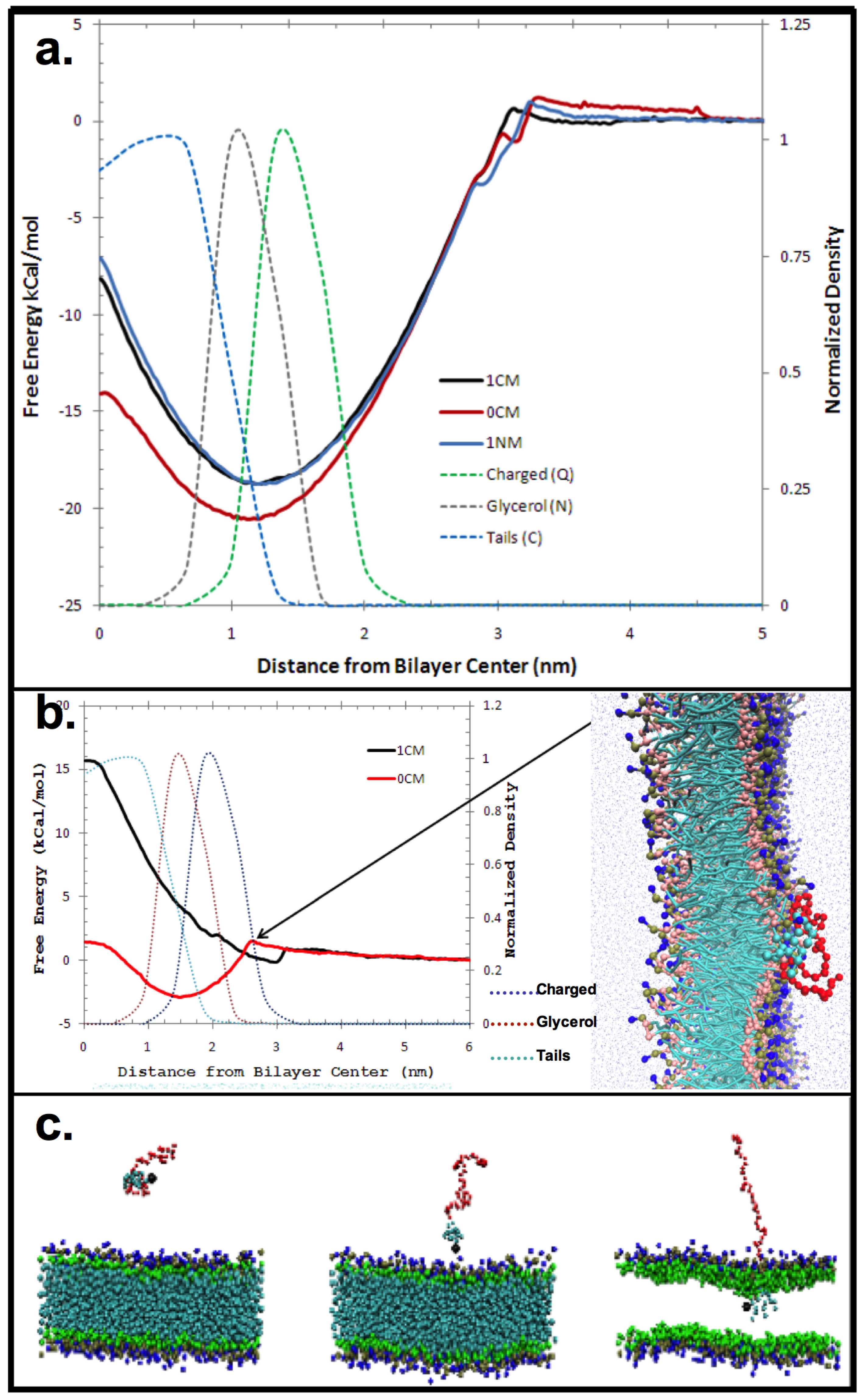
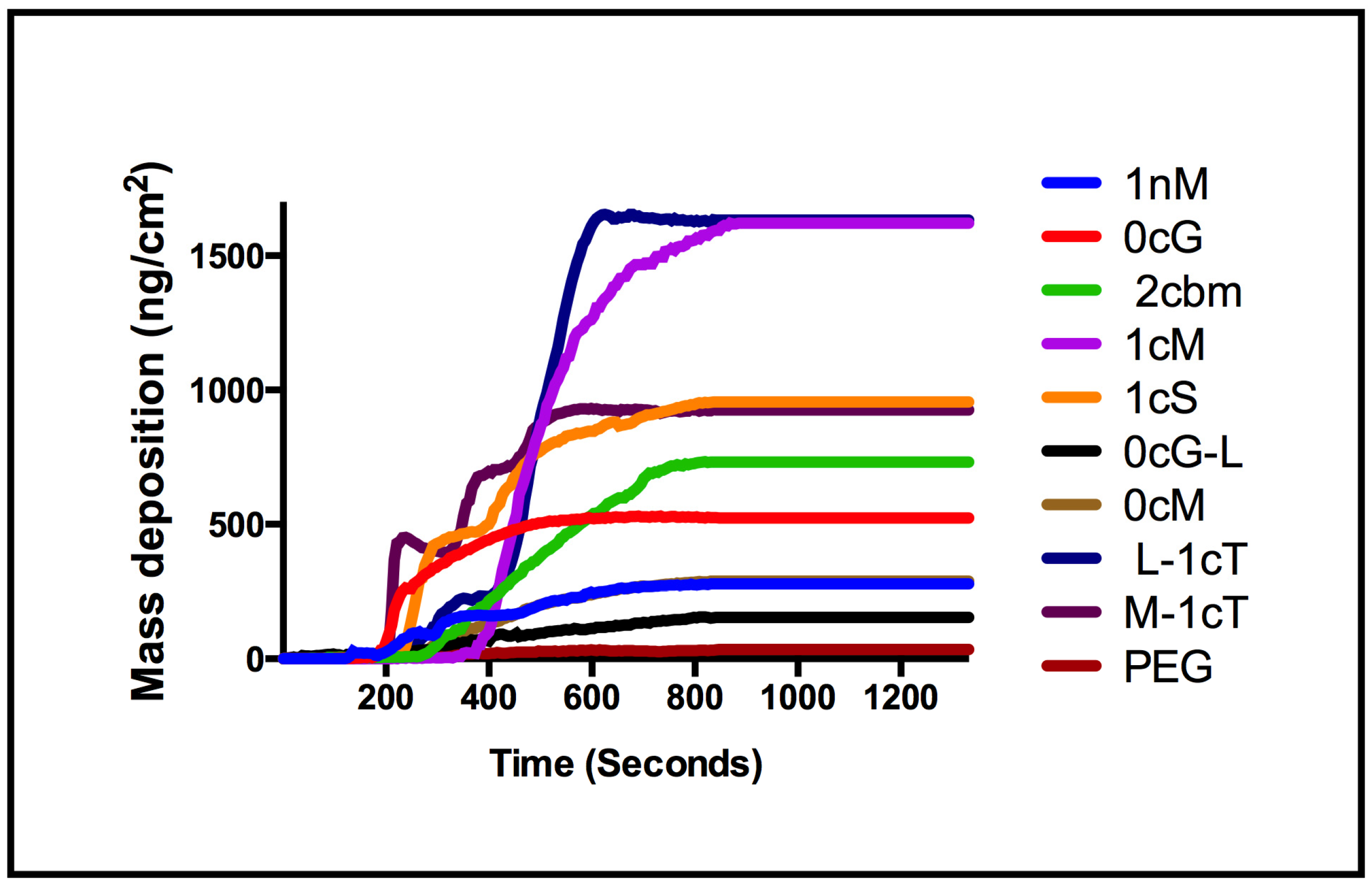
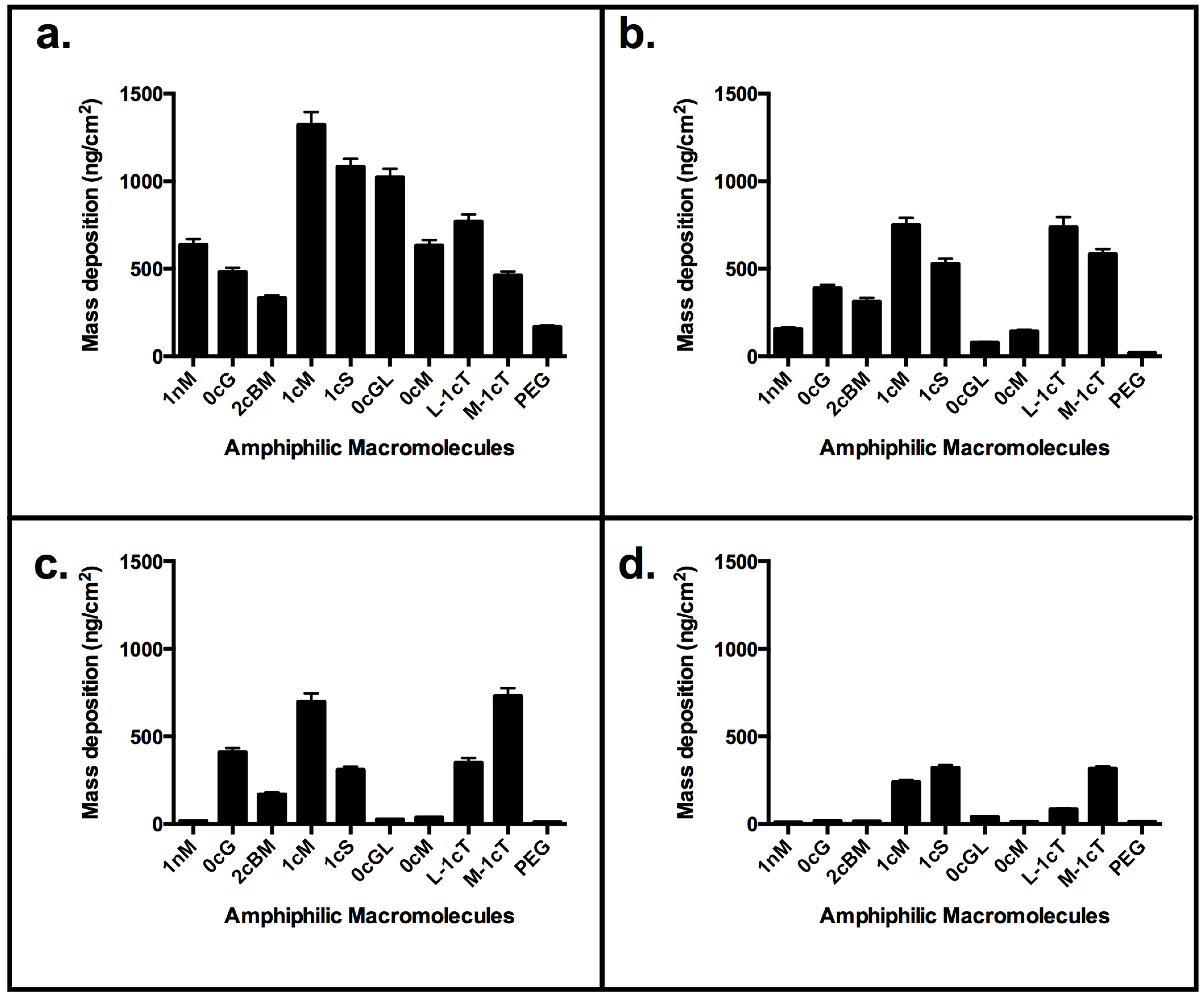
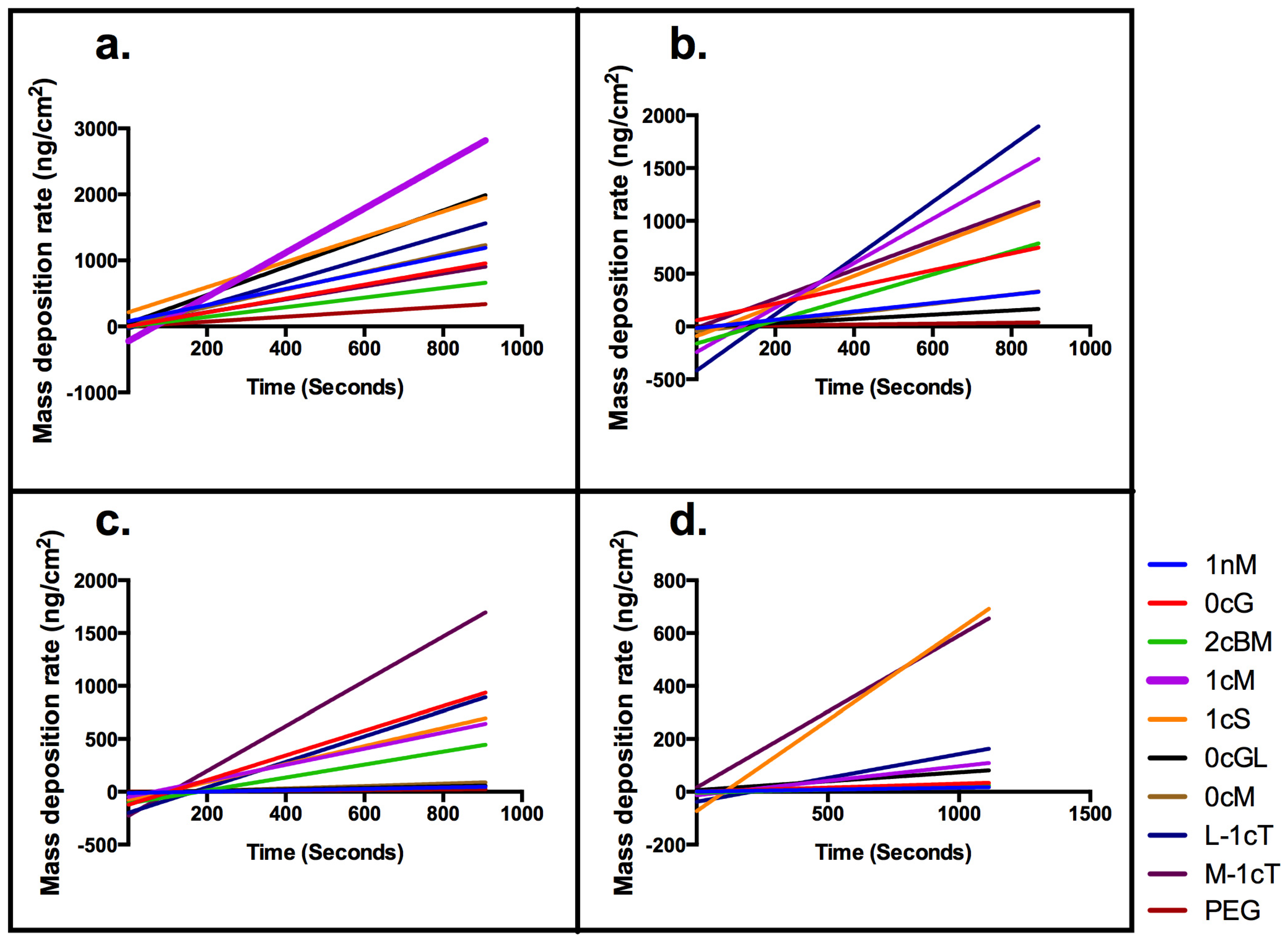
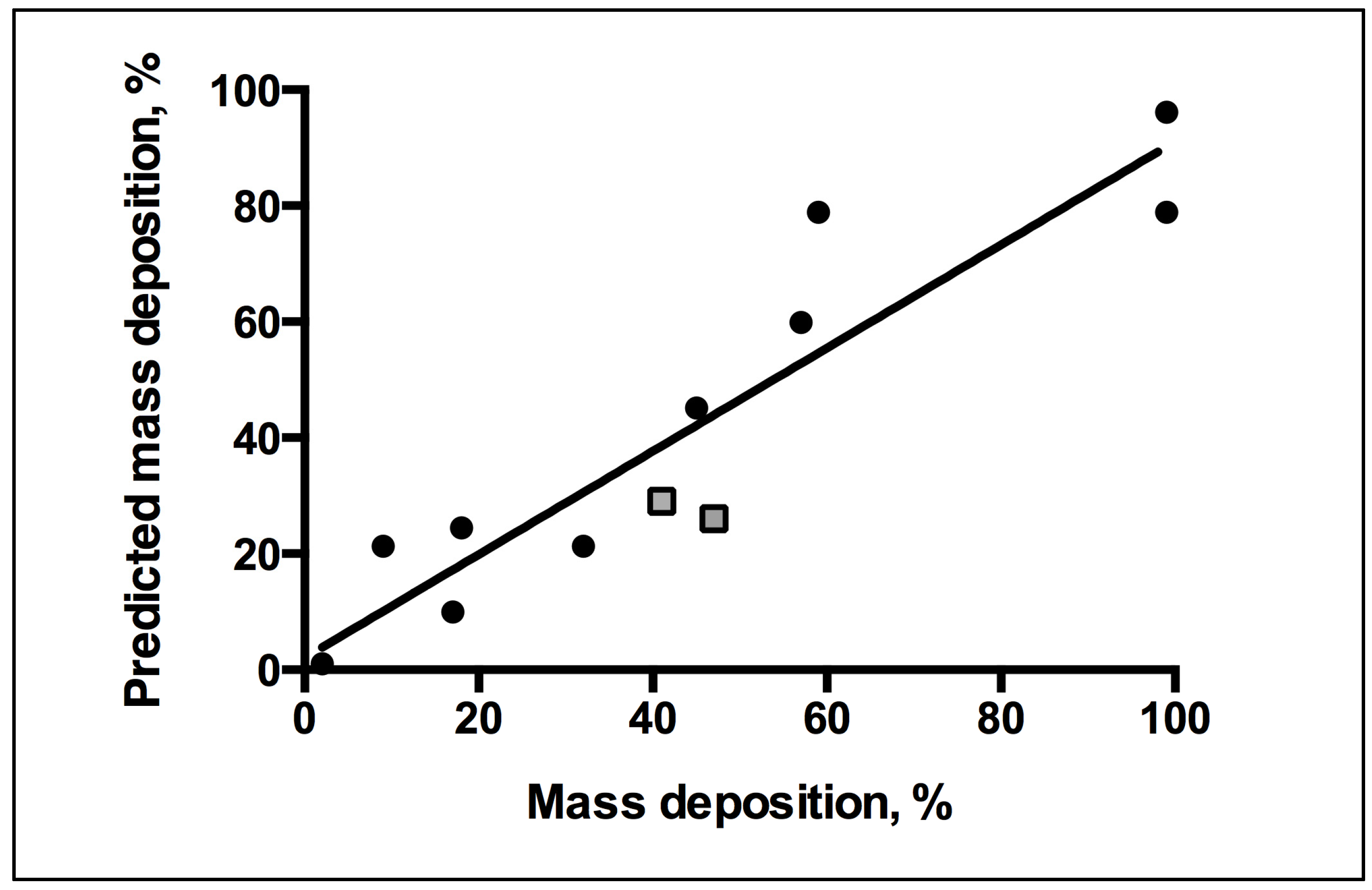
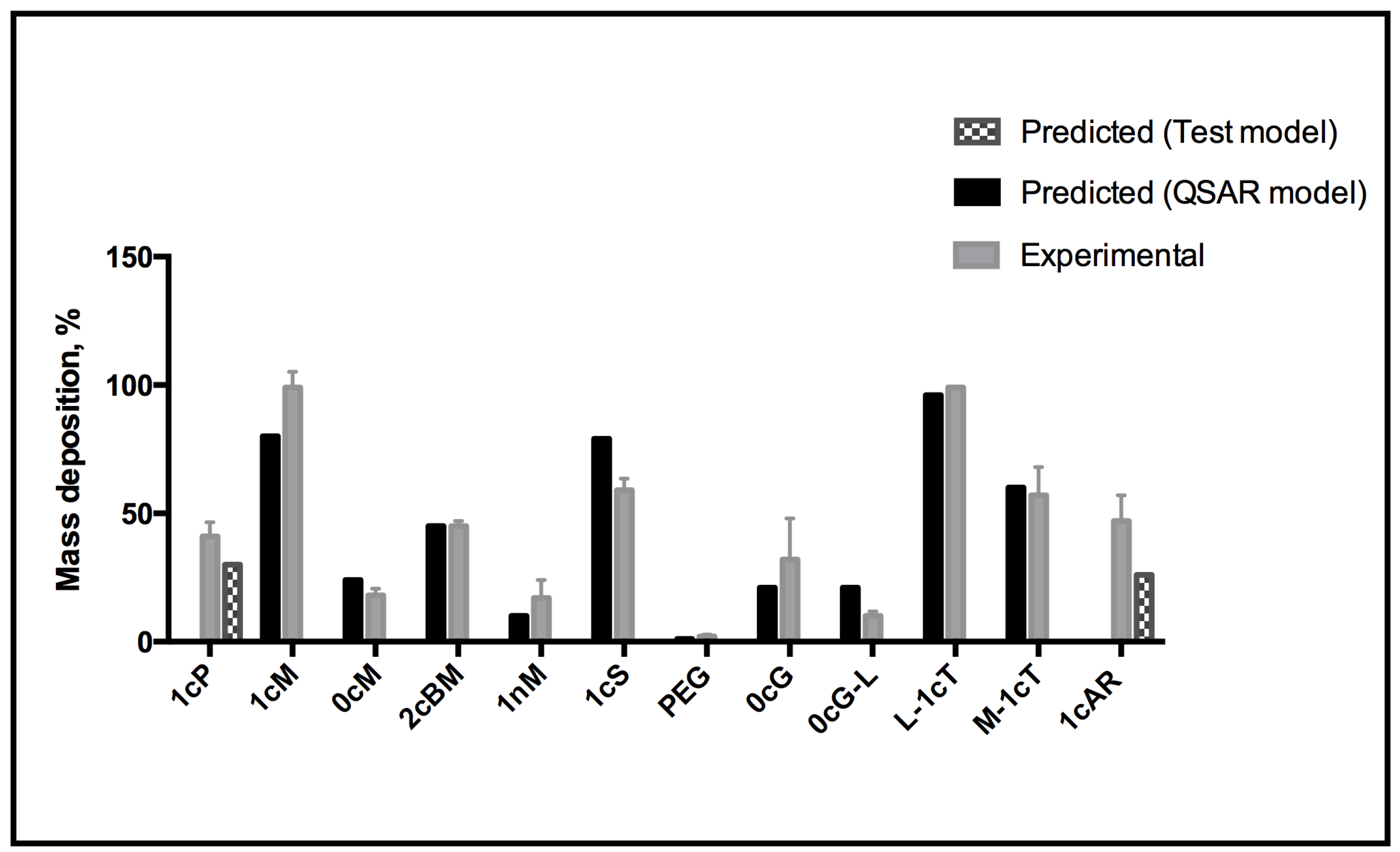
3. Experimental Section
3.1. Computational Modeling of AM Interactions with Model Membranes
3.1.1. Overview of the Modeling Workflow
3.1.2. CG MD Simulation System
3.1.3. CG MD Membrane-AM Interactions
3.1.4. Calculation of the Potential of Mean Force
3.1.5. Reverse Mapping of CG Structures to All-Atom AM Structures
3.1.6. All-Atom MD Simulations
3.1.7. Molecular Descriptor Generation
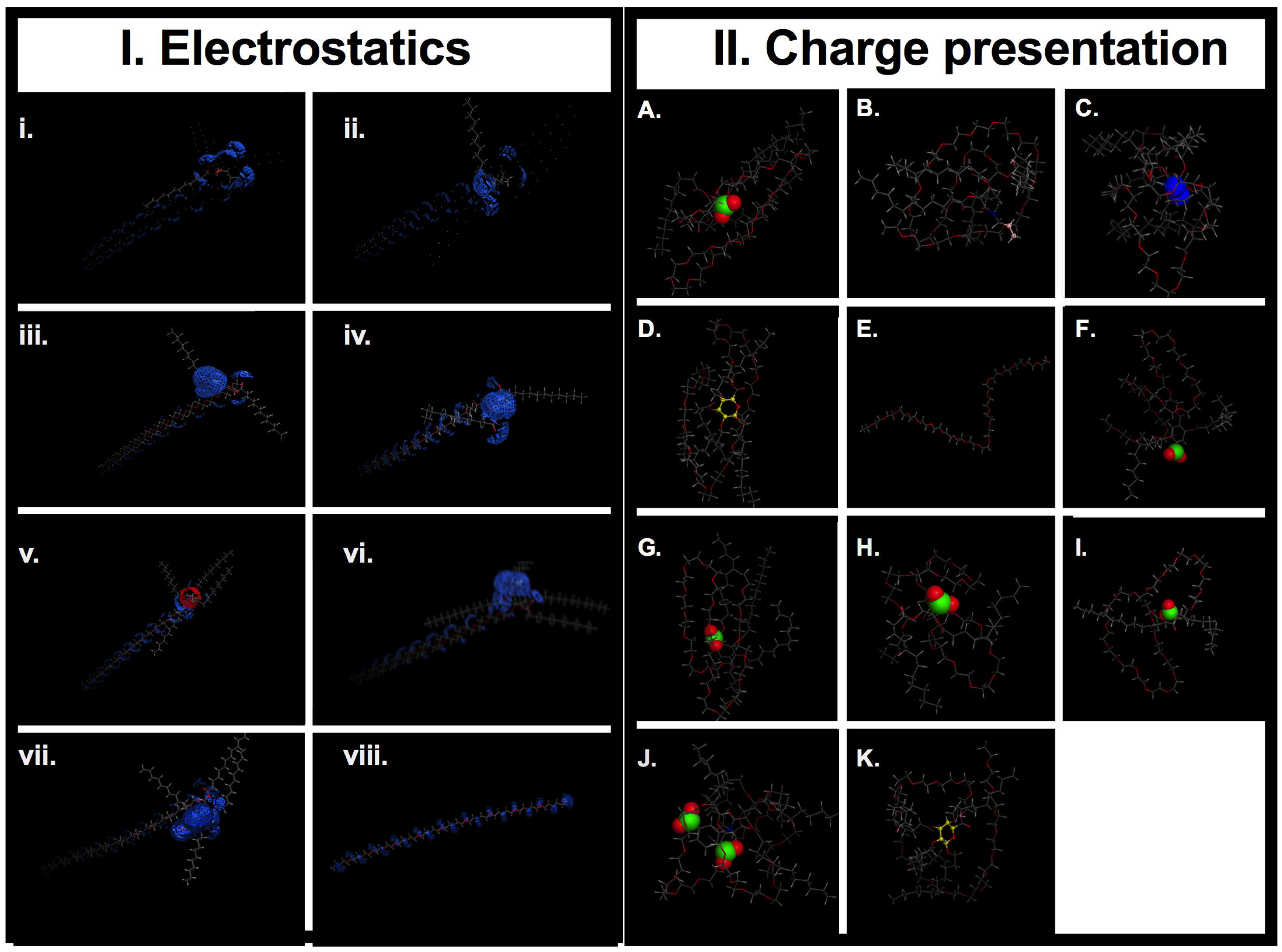
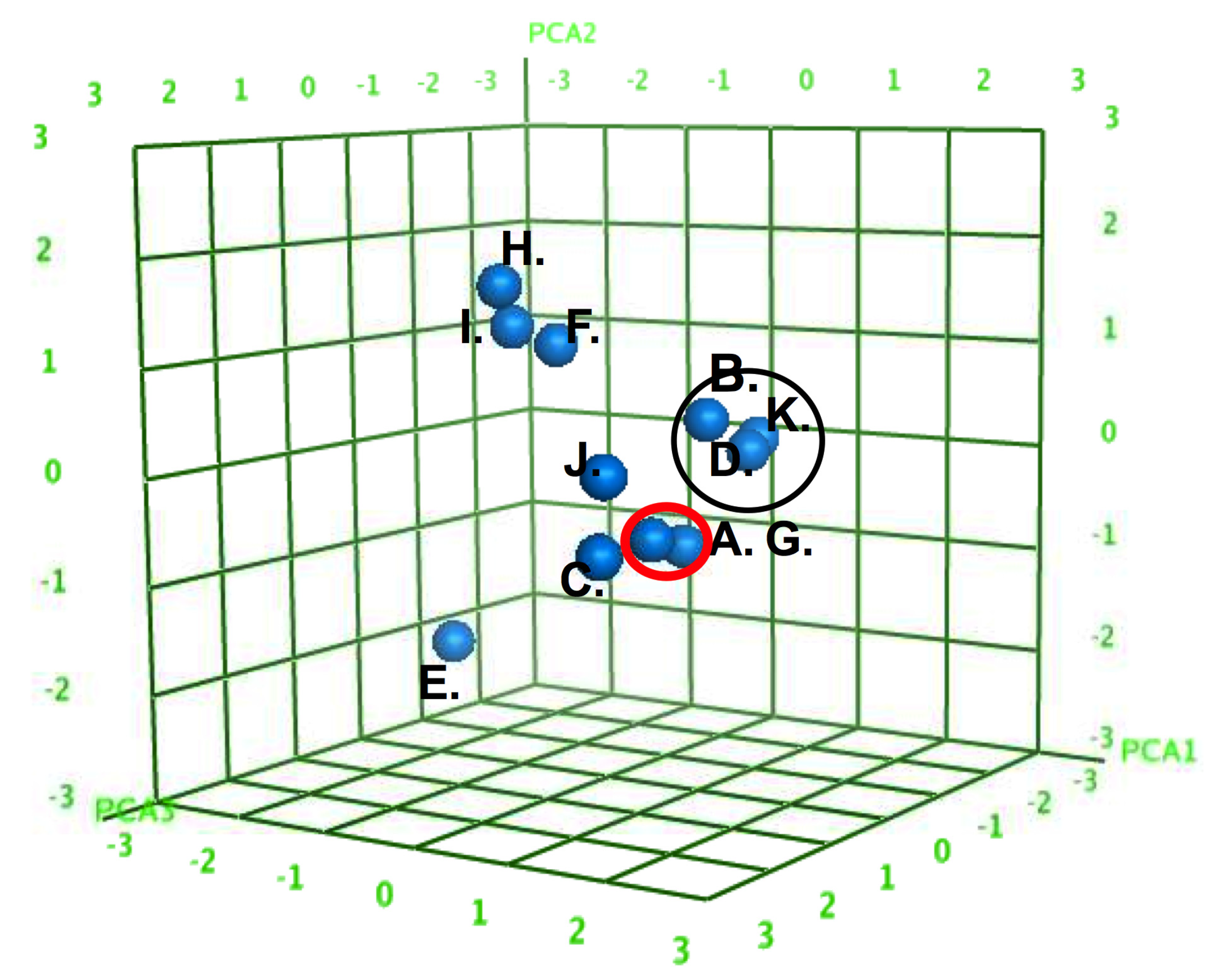
3.1.8. QSAR Modeling
3.2. Experimental Studies of AM Binding to Model Membranes
3.2.1. QCM-D Experimental Methods
3.2.2. Liposomes
3.2.3. Amphiphilic Macromolecules
3.2.4. Bilayer Formation and AM Exposure
3.2.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sackmann, E. Supported membranes: Scientific and practical applications. Science 1996, 271, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Yaroslavov, A.A.; Sybachin, A.V.; Schrinner, M.; Ballauff, M.; Tsarkova, L.; Kesselman, E.; Schmidt, J.; Talmon, Y.; Menger, F.M. Liposomes remain intact when complexed with polycationic brushes. J. Am. Chem. Soc. 2010, 132, 5948–5949. [Google Scholar] [CrossRef] [PubMed]
- Inui, O.; Teramura, Y.; Iwata, H. Retention dynamics of amphiphilic polymers peg-lipids and pva-alkyl on the cell surface. ACS Appl. Mater. Interfaces 2010, 2, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Hehir, S.; Plourde, N.M.; Gu, L.; Poree, D.E.; Welsh, W.J.; Moghe, P.V.; Uhrich, K.E. Carbohydrate composition of amphiphilic macromolecules influences physicochemical properties and binding to atherogenic scavenger receptor A. Acta Biomater. 2012, 8, 3956–3962. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, J.; Barch, M.; Uhrich, K.E. Polymeric micelles based on amphiphilic scorpion-like macromolecules: Novel carriers for water-insoluble drugs. Pharm. Res. 2005, 22, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Uhrich, K.E. Novel amphiphilic macromolecules and their in vitro characterization as stabilized micellar drug delivery systems. J. Colloid Interface Sci. 2006, 298, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models of the cell surface. Nature 2005, 437, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The martini force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Poree, D.E.; Zablocki, K.; Faig, A.; Moghe, P.V.; Uhrich, K.E. Nanoscale amphiphilic macromolecules with variable lipophilicity and stereochemistry modulate inhibition of oxidized low-density lipoprotein uptake. Biomacromolecules 2013, 14, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, E.L.; Caramori, G.F.; Rambo, C.R. Nanoparticle translocation through a lipid bilayer tuned by surface chemistry. Phys. Chem. Chem. Phys. PCCP 2013, 15, 2282–2290. [Google Scholar]
- Reviakine, I.; Johannsmann, D.; Richter, R.P. Hearing what you cannot see and visualizing what you hear: Interpreting quartz crystal microbalance data from solvated interfaces. Anal. Chem. 2011, 83, 8838–8848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, N. Thermodynamics of charged nanoparticle adsorption on charge-neutral membranes: A simulation study. J. Phys. Chem. B 2010, 114, 2749–2754. [Google Scholar] [CrossRef] [PubMed]
- Stalgren, J.J.; Claesson, P.M.; Warnheim, T. Adsorption of liposomes and emulsions studied with a quartz crystal microbalance. Adv. Colloid Interface Sci. 2001, 89–90, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A.; Kinnunen, P.K. Biophysical properties of lipids and dynamic membranes. Trends Cell Biol. 2006, 16, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.R.; Kholodovych, V.; Tomasini, M.D.; Abdelhamid, D.; Petersen, L.K.; Welsh, W.J.; Uhrich, K.E.; Moghe, P.V. In silico design of anti-atherogenic biomaterials. Biomaterials 2013, 34, 7950–7959. [Google Scholar] [CrossRef] [PubMed]
- Gubskaya, A.V.; Kholodovych, V.; Knight, D.; Kohn, J.; Welsh, W.J. Prediction of fibrinogen adsorption for biodegradable polymers: Integration of molecular dynamics and surrogate modeling. Polymer 2007, 48, 5788–5801. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Knight, D.; Kohn, J.; Rasheed, K.; Weber, N.; Kholodovych, V.; Welsh, W.J. Using surrogate modeling in the prediction of fibrinogen adsorption onto polymer surfaces. J. Chem. Inf. Comput. Sci. 2004, 44, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Li, S.; Ai, N.; Hu, L.; Welsh, W.J.; You, G. Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: Discovery and molecular characterization. Mol. Pharm. 2012, 9, 3340–3346. [Google Scholar] [CrossRef] [PubMed]
- Fomovska, A.; Wood, R.D.; Mui, E.; Dubey, J.P.; Ferreira, L.R.; Hickman, M.R.; Lee, P.J.; Leed, S.E.; Auschwitz, J.M.; Welsh, W.J.; et al. Salicylanilide inhibitors of toxoplasma gondii. J. Med. Chem. 2012, 55, 8375–8391. [Google Scholar] [CrossRef] [PubMed]
- Seelig, J.; Gally, G.U.; Wohlgemuth, R. Orientation and flexibility of the choline head group in phosphatidylcholine bilayers. Biochim. Biophys. Acta 1977, 467, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Uhrikova, D.; Kucerka, N.; Teixeira, J.; Gordeliy, V.; Balgavy, P. Structural changes in dipalmitoylphosphatidylcholine bilayer promoted by Ca2+ ions: A small-angle neutron scattering study. Chem. Phys. Lipids 2008, 155, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H.; Pascher, I.; Pearson, R.H.; Sundell, S. Preferred conformation and molecular packing of phosphatidylethanolamine and phosphatidylcholine. Biochim. Biophys. Acta 1981, 650, 21–51. [Google Scholar] [CrossRef] [PubMed]
- Ninham, B.W.; Parsegian, V.A. Electrostatic potential between surfaces bearing ionizable groups in ionic equilibrium with physiologic saline solution. J. Theor. Biol. 1971, 31, 405–428. [Google Scholar] [CrossRef] [PubMed]
- Chnari, E.; Lari, H.B.; Tian, L.; Uhrich, K.E.; Moghe, P.V. Nanoscale anionic macromolecules for selective retention of low-density lipoproteins. Biomaterials 2005, 26, 3749–3758. [Google Scholar] [CrossRef] [PubMed]
- York, A.W.; Zablocki, K.R.; Lewis, D.R.; Gu, L.; Uhrich, K.E.; Prud’homme, R.K.; Moghe, P.V. Kinetically assembled nanoparticles of bioactive macromolecules exhibit enhanced stability and cell-targeted biological efficacy. Adv. Mater. 2012, 24, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Del Rosario, L.S.; Demirdirek, B.; Harmon, A.; Orban, D.; Uhrich, K.E. Micellar nanocarriers assembled from doxorubicin-conjugated amphiphilic macromolecules (DOX-AM). Macromol. Biosci. 2010, 10, 415–423. [Google Scholar]
- Wilhelm, C.; Billotey, C.; Roger, J.; Pons, J.N.; Bacri, J.C.; Gazeau, F. Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials 2003, 24, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Messina, R. Electrostatics in soft matter. J. Phys. Condens. Matter 2009, 21. [Google Scholar] [CrossRef]
- Zheng, C.; Vanderkooi, G. Molecular origin of the internal dipole potential in lipid bilayers: Calculation of the electrostatic potential. Biophys. J. 1992, 63, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; biomolecular simulation programs; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Lee, H.; de Vries, A.H.; Marrink, S.J.; Pastor, R.W. A coarse-grained model for polyethylene oxide and polyethylene glycol: Conformation and hydrodynamics. J. Phys. Chem. B 2009, 113, 13186–13194. [Google Scholar] [CrossRef] [PubMed]
- Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of lennard-jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Molecular dynamics simulations of hydrophilic pores in lipid bilayers. Biophys. J. 2004, 86, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.K.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.P.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. Gromacs 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Rzepiela, A.J.; Schäfer, L.V.; Goga, N.; Risselada, H.J.; De Vries, A.H.; Marrink, S.J. Reconstruction of atomistic details from coarse-grained structures. J. Comput. Chem. 2010, 31, 1333–1343. [Google Scholar] [PubMed]
- Sousa da Silva, A.; Vranken, W. ACPYPE—AnteChamber PYthon parser interfacE. BMC Res. Notes 2012, 5. [Google Scholar] [CrossRef]
- Allen, M.P.T.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Labute, P.; Chemical Computing Group Inc., Montreal, Quebec, Canada. Moe logp(octanol/ water) model. Source code in $MOE/lib/svl/quasar.svl/q_logp.svl. Unpublished work. 1998. [Google Scholar]
- Labute, P.; Chemical Computing Group Inc, Montreal, Quebec, Canada. Moe molar refractivity model. Source code in $MOE/lib/svl/quasar.svlq_mref.svl. Unpublished work. 1998. [Google Scholar]
- McFarland, J.W.; Berger, C.M.; Froshauer, S.A.; Hayashi, S.F.; Hecker, S.J.; Jaynes, B.H.; Jefson, M.R.; Kamicker, B.J.; Lipinski, C.A.; Lundy, K.M.; et al. Quantitative structure-activity relationships among macrolide antibacterial agents: In vitro and in vivo potency against pasteurella multocida. J. Med. Chem. 1997, 40, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. Qsar applicabilty domain estimation by projection of the training set descriptor space: A review. Altern. Lab. Anim. ATLA 2005, 33, 445–459. [Google Scholar]
- Sauerbrey, G. Verwendung von schwingquarzen zur waging dunner schicten und zur mikrowagung. Z. Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Keller, C.A.; Kasemo, B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 1998, 75, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.M.; Lash, M.H.; Tishbi, N.; Lent, D.; Mintzer, E.A.; Uhrich, K.E. Thermodynamic and physical interactions between novel polymeric surfactants and lipids: Toward designing stable polymer-lipid complexes. Langmuir 2011, 27, 9131–9138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Plourde, N.M.; Iverson, N.; Moghe, P.V.; Uhrich, K.E. Nanoscale amphiphilic macromolecules as lipoprotein inhibitors: The role of charge and architecture. Int. J. Nanomed. 2007, 2, 697–705. [Google Scholar]
- Iverson, N.M.; Plourde, N.M.; Sparks, S.M.; Wang, J.; Patel, E.N.; Shah, P.S.; Lewis, D.R.; Zablocki, K.R.; Nackman, G.B.; Uhrich, K.E.; et al. Dual use of amphiphilic macromolecules as cholesterol efflux triggers and inhibitors of macrophage athero-inflammation. Biomaterials 2011, 32, 8319–8327. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.R.; Petersen, L.K.; Zablocki, K.; York, A.W.; Uhrich, K.E.; Prud’homme, R.K.; Moghe, P.V. Sugar based amphiphilic nanoparticles arrest atherosclerosis in vivo. PNAS 2015, 112, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, A.A.T.; Tomasini, M.; Kholodovych, V.; Gu, L.; Sommerfeld, S.D.; Uhrich, K.E.; Murthy, N.S.; Welsh, W.J.; Moghe, P.V. Carbohydrate-Derived Amphiphilic Macromolecules: A Biophysical Structural Characterization and Analysis of Binding Behaviors to Model Membranes. J. Funct. Biomater. 2015, 6, 171-191. https://doi.org/10.3390/jfb6020171
Martin AAT, Tomasini M, Kholodovych V, Gu L, Sommerfeld SD, Uhrich KE, Murthy NS, Welsh WJ, Moghe PV. Carbohydrate-Derived Amphiphilic Macromolecules: A Biophysical Structural Characterization and Analysis of Binding Behaviors to Model Membranes. Journal of Functional Biomaterials. 2015; 6(2):171-191. https://doi.org/10.3390/jfb6020171
Chicago/Turabian StyleMartin, Adriana A. T., Michael Tomasini, Vladyslav Kholodovych, Li Gu, Sven Daniel Sommerfeld, Kathryn E. Uhrich, N. Sanjeeva Murthy, William J. Welsh, and Prabhas V. Moghe. 2015. "Carbohydrate-Derived Amphiphilic Macromolecules: A Biophysical Structural Characterization and Analysis of Binding Behaviors to Model Membranes" Journal of Functional Biomaterials 6, no. 2: 171-191. https://doi.org/10.3390/jfb6020171
APA StyleMartin, A. A. T., Tomasini, M., Kholodovych, V., Gu, L., Sommerfeld, S. D., Uhrich, K. E., Murthy, N. S., Welsh, W. J., & Moghe, P. V. (2015). Carbohydrate-Derived Amphiphilic Macromolecules: A Biophysical Structural Characterization and Analysis of Binding Behaviors to Model Membranes. Journal of Functional Biomaterials, 6(2), 171-191. https://doi.org/10.3390/jfb6020171