
Nanostructured Porous Silicon for Bone Tissue Engineering: Kinetics of Particle Degradation and Si-Controlled Release

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Porous Silicon Particle Production
2.2. Optical Microscopy
2.3. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-rays Spectroscopy (EDX)
2.4. Induced Coupled Plasma—Mass Spectrometry (ICP-MS)
2.5. Raman Spectroscopy
2.6. Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.7. In Vivo Experiments
3. Results
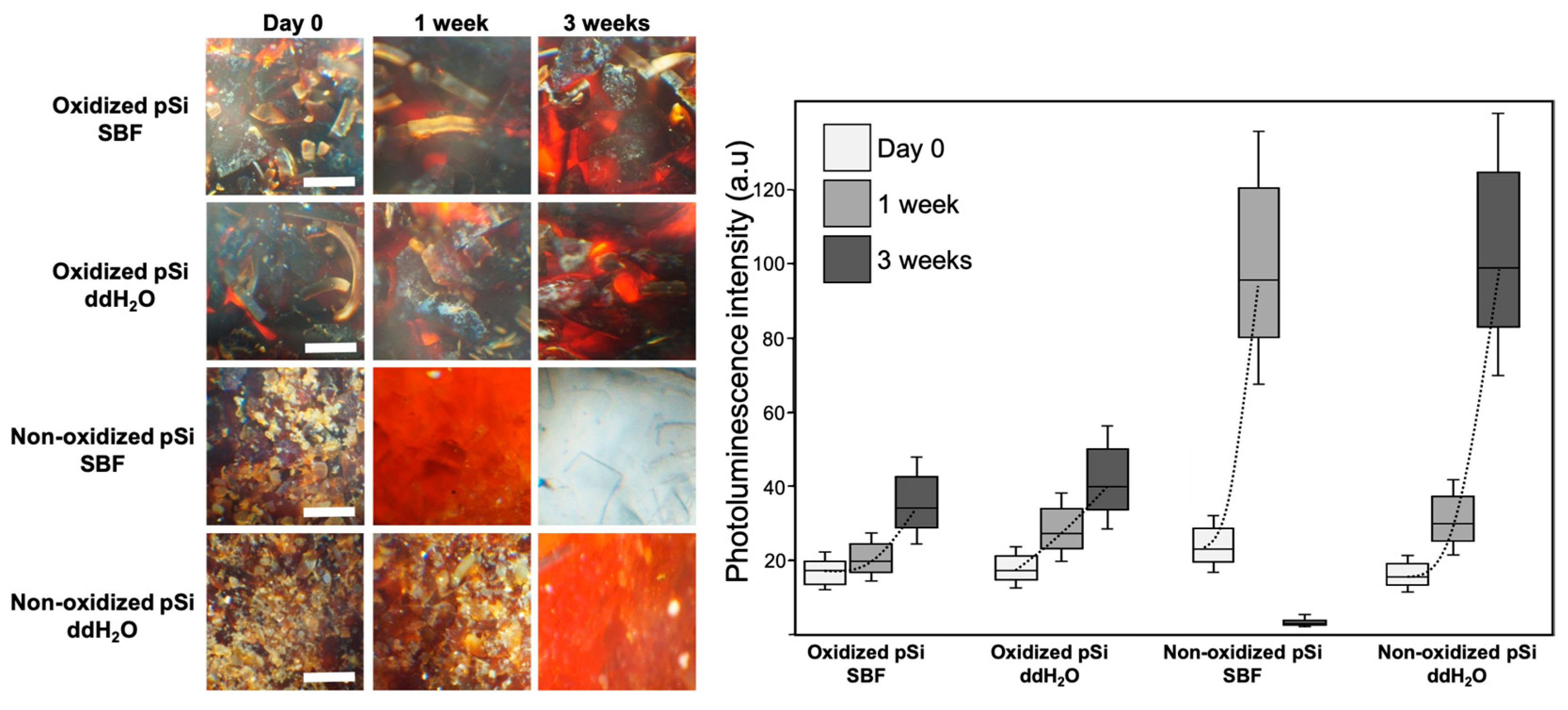
3.1. Particle Dissolution: Optical Microscopy and Si Release
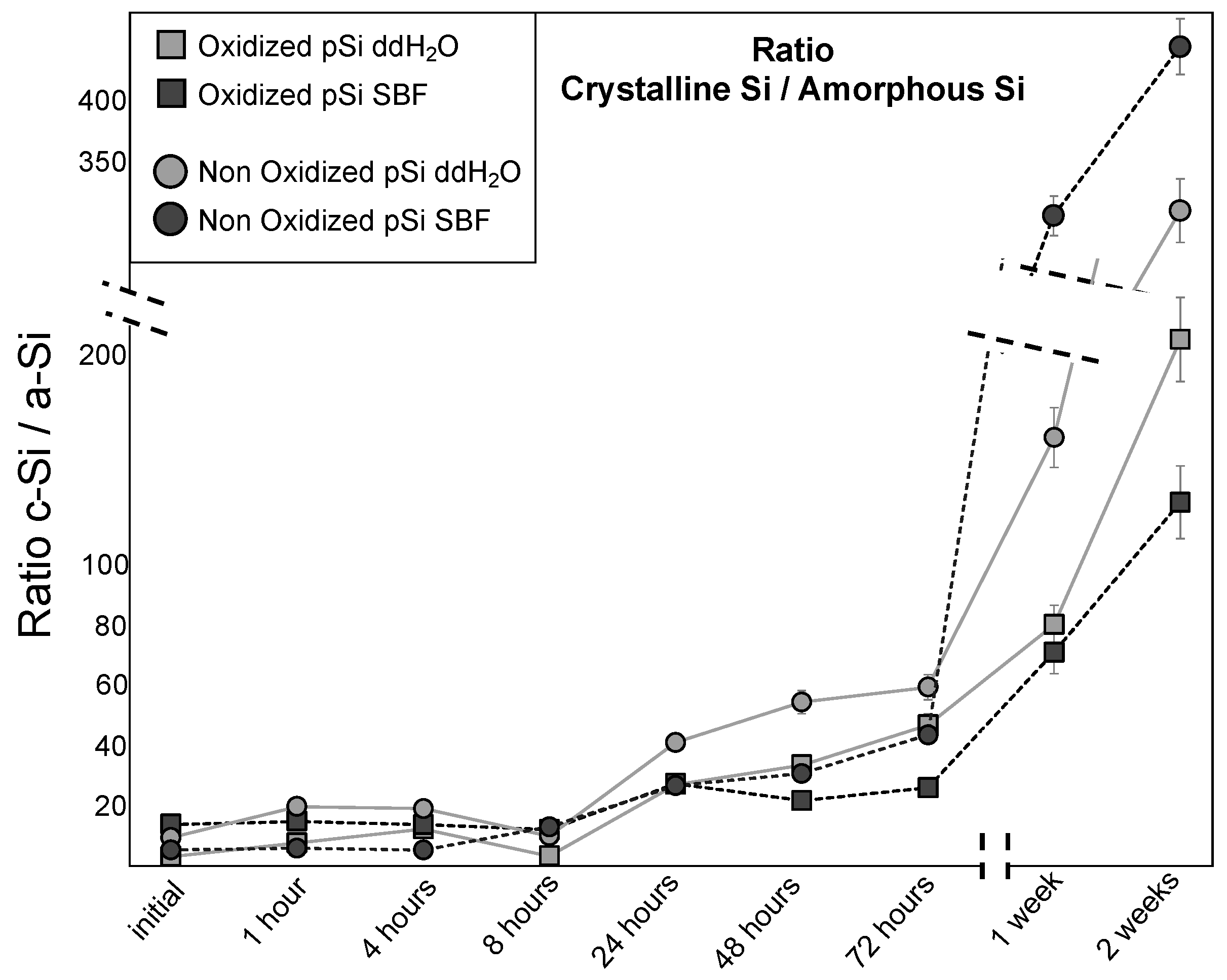
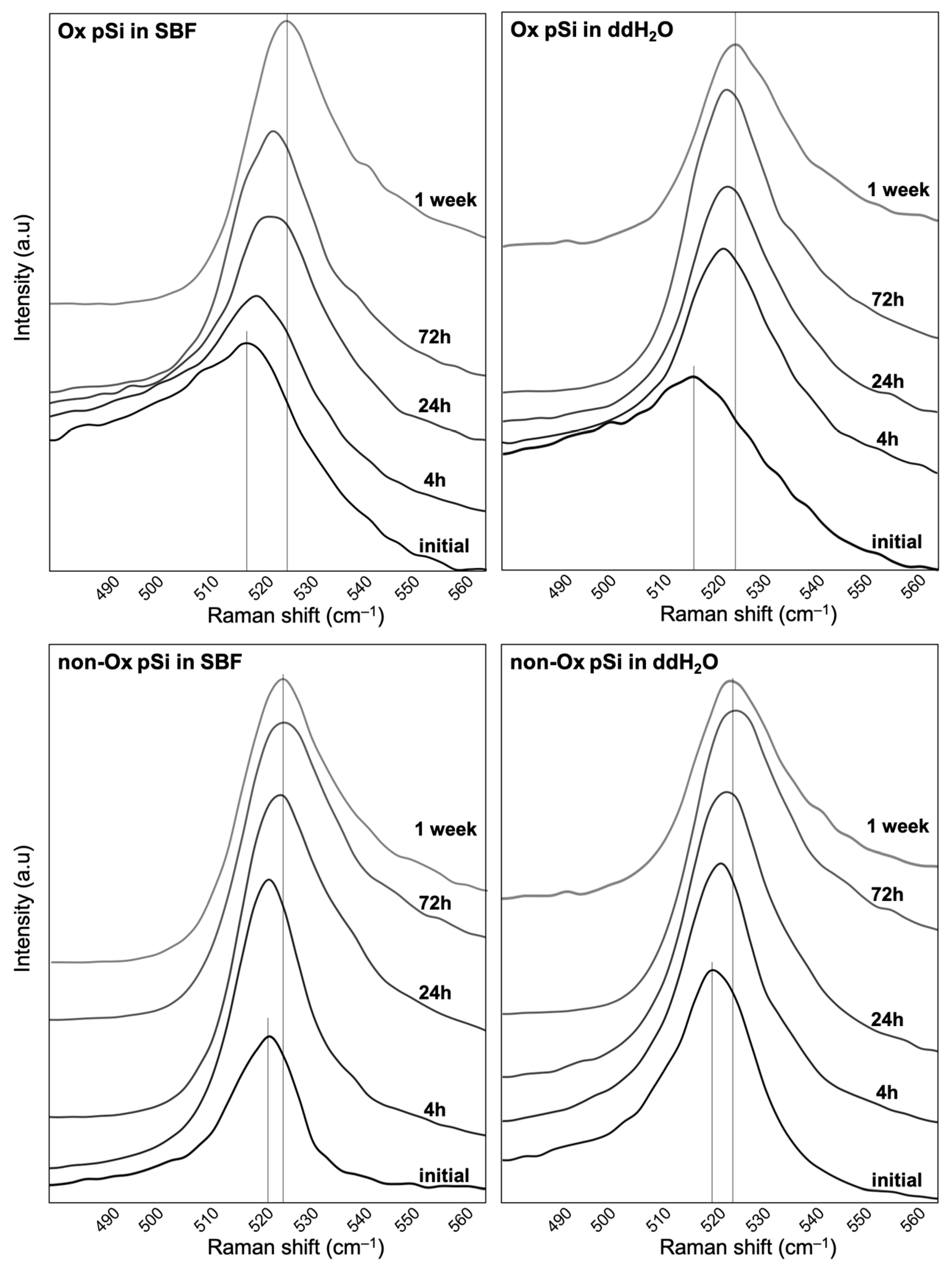
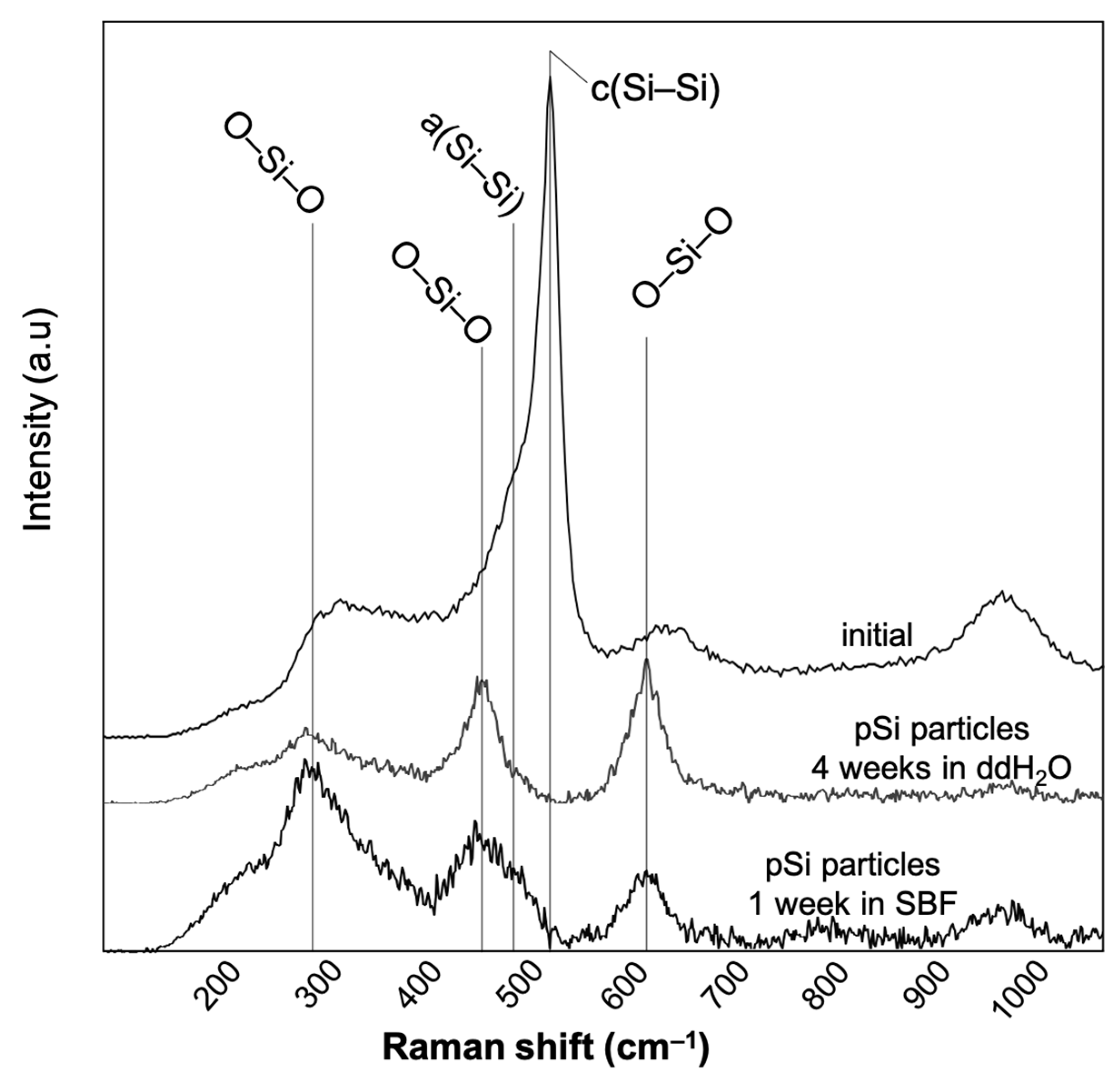
3.2. Raman Spectroscopy
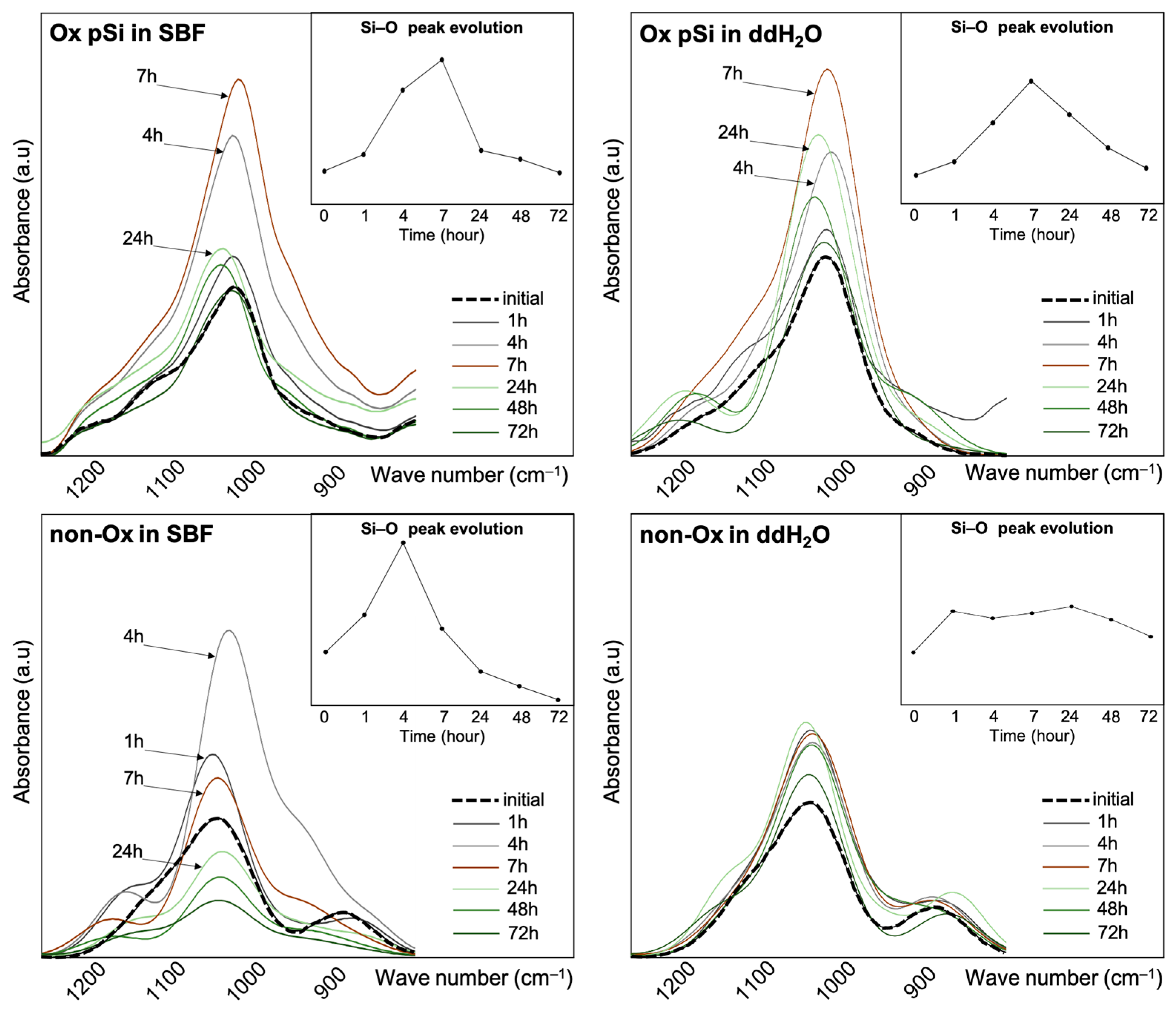
3.3. ATR-FTIR Spectroscopy
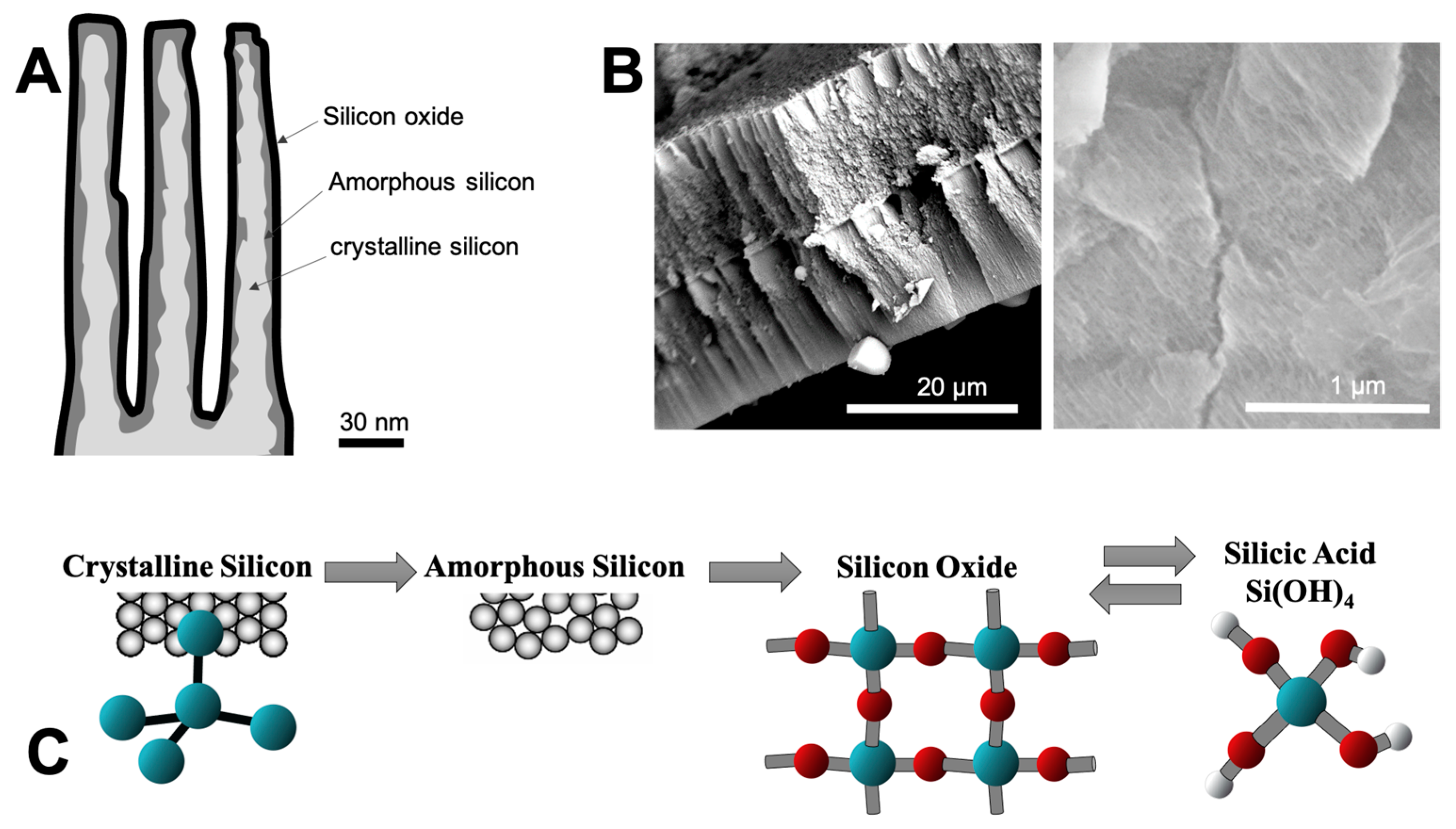
3.4. SEM and EDX Spectroscopy
3.5. In Vivo Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khokhlov, A.G.; Valiullin, R.R.; Kärger, J.; Zubareva, N.B.; Stepovich, M.A. Estimation of Pore Sizes in Porous Silicon by Scanning Electron Microscopy and NMR Cryoporometry. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2008, 2, 919–922. [Google Scholar] [CrossRef]
- Petrova, E.A.; Bogoslovskaya, K.N.; Balagurov, L.A.; Kochoradze, G.I. Room Temperature Oxidation of Porous Silicon in Air. Mater. Sci. Eng. B 2000, 69–70, 152–156. [Google Scholar] [CrossRef]
- Hernandez-Montelongo, J.; Munoz-Noval, A.; GarcÃia-RuÃiz, J.P.; Torres-Costa, V.; Martin-Palma, R.J.; Manso-Silvan, M. Nanostructured Porous Silicon: The Winding Road from Photonics to Cell Scaffolds—A Review. Front. Bioeng. Biotechnol. 2015, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Anglin, E.; Cheng, L.; Freeman, W.; Sailor, M. Porous Silicon in Drug Delivery Devices and Materials☆. Adv. Drug Deliv. Rev. 2008, 60, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Reffitt, D.M.; Jugdaohsingh, R.; Thompson, R.P.H.; Powell, J.J. Silicic Acid: Its Gastrointestinal Uptake and Urinary Excretion in Man and Effects on Aluminium Excretion. J. Inorg. Biochem. 1999, 76, 141–147. [Google Scholar] [CrossRef]
- Collart Dutilleul, P.-Y.; Deville De Périère, D.; Cuisinier, F.J.; Cunin, F.; Gergely, C. Porous Silicon Scaffolds for Stem Cells Growth and Osteodifferentiation. In Porous Silicon for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2014; pp. 486–506. ISBN 978-0-85709-711-8. [Google Scholar]
- Collart-Dutilleul, P.-Y.; Secret, E.; Panayotov, I.; Deville de Périère, D.; Martín-Palma, R.J.; Torres-Costa, V.; Martin, M.; Gergely, C.; Durand, J.-O.; Cunin, F.; et al. Adhesion and Proliferation of Human Mesenchymal Stem Cells from Dental Pulp on Porous Silicon Scaffolds. ACS Appl. Mater. Interfaces 2014, 6, 1719–1728. [Google Scholar] [CrossRef]
- Renaud, M.; Bousquet, P.; Macias, G.; Rochefort, G.Y.; Durand, J.-O.; Marsal, L.F.; Cuisinier, F.; Cunin, F.; Collart-Dutilleul, P.-Y. Allogenic Stem Cells Carried by Porous Silicon Scaffolds for Active Bone Regeneration In Vivo. Bioengineering 2023, 10, 852. [Google Scholar] [CrossRef]
- Li, Y.Y.; Cunin, F.; Link, J.R.; Gao, T.; Betts, R.E.; Reiver, S.H.; Chin, V.; Bhatia, S.N.; Sailor, M.J. Polymer Replicas of Photonic Porous Silicon for Sensing and Drug Delivery Applications. Science 2003, 299, 2045–2047. [Google Scholar] [CrossRef]
- Wu, E.C.; Park, J.-H.; Park, J.; Segal, E.; Cunin, F.; Sailor, M.J. Oxidation-Triggered Release of Fluorescent Molecules or Drugs from Mesoporous Si Microparticles. ACS Nano 2008, 2, 2401–2409. [Google Scholar] [CrossRef]
- Chaix, A.; Cueto-Diaz, E.; Dominguez-Gil, S.; Spiteri, C.; Lichon, L.; Maynadier, M.; Dumail, X.; Aggad, D.; Delalande, A.; Bessière, A.; et al. Two-photon Light Trigger Sirna Transfection of Cancer Cells Using Non-Toxic Porous Silicon Nanoparticles. Adv. Healthc. Mater. 2023, 2301052. [Google Scholar] [CrossRef]
- Jeong, M.; Jung, Y.; Yoon, J.; Kang, J.; Lee, S.H.; Back, W.; Kim, H.; Sailor, M.J.; Kim, D.; Park, J.-H. Porous Silicon-Based Nanomedicine for Simultaneous Management of Joint Inflammation and Bone Erosion in Rheumatoid Arthritis. ACS Nano 2022, 16, 16118–16132. [Google Scholar] [CrossRef] [PubMed]
- Pontinha, A.D.R.; Moreira, B.B.; Melo, B.L.; Melo-Diogo, D.D.; Correia, I.J.; Alves, P. Silica Aerogel-Polycaprolactone Scaffolds for Bone Tissue Engineering. Int. J. Mol. Sci. 2023, 24, 10128. [Google Scholar] [CrossRef] [PubMed]
- Coffer, J.L.; Whitehead, M.A.; Nagesha, D.K.; Mukherjee, P.; Akkaraju, G.; Totolici, M.; Saffie, R.S.; Canham, L.T. Porous Silicon-Based Scaffolds for Tissue Engineering and Other Biomedical Applications. Phys. Status Solidi A 2005, 202, 1451–1455. [Google Scholar] [CrossRef]
- Canham, L.T.; Houlton, M.R.; Leong, W.Y.; Pickering, C.; Keen, J.M. Atmospheric Impregnation of Porous Silicon at Room Temperature. J. Appl. Phys. 1991, 70, 422–431. [Google Scholar] [CrossRef]
- McMillan, N.K.; Coffer, J.L. Porous Silicon and Related Composites as Functional Tissue Engineering Scaffolds. In Porous Silicon for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2021; pp. 493–516. ISBN 978-0-12-821677-4. [Google Scholar]
- Coffer, J.L. Functional Semiconducting Silicon Nanowires and Their Composites as Tissue Scaffolds. In Semiconducting Silicon Nanowires for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2022; pp. 137–152. ISBN 978-0-12-821351-3. [Google Scholar]
- Sailor, M.J. Porous Silicon in Practice: Preparation, Characterization and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 978-3-527-64190-1. [Google Scholar]
- Buriak, J.M.; Stewart, M.P.; Geders, T.W.; Allen, M.J.; Choi, H.C.; Smith, J.; Raftery, D.; Canham, L.T. Lewis Acid Mediated Hydrosilylation on Porous Silicon Surfaces. J. Am. Chem. Soc. 1999, 121, 11491–11502. [Google Scholar] [CrossRef]
- Kilian, K.A.; Böcking, T.; Gooding, J.J. The Importance of Surface Chemistry in Mesoporous Materials: Lessons from Porous Silicon Biosensors. Chem. Commun. 2009, 6, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Pacholski, C. Photonic Crystal Sensors Based on Porous Silicon. Sensors 2013, 13, 4694–4713. [Google Scholar] [CrossRef]
- Renaud, M.; Farkasdi, S.; Pons, C.; Panayotov, I.; Collart-Dutilleul, P.-Y.; Taillades, H.; Desoutter, A.; Bousquet, P.; Varga, G.; Cuisinier, F.; et al. A New Rat Model for Translational Research in Bone Regeneration. Tissue Eng. Part C Methods 2016, 22, 125–131. [Google Scholar] [CrossRef]
- Collart-Dutilleul, P.-Y.; Panayotov, I.; Secret, E.; Cunin, F.; Gergely, C.; Cuisinier, F.; Martin, M. Initial Stem Cell Adhesion on Porous Silicon Surface: Molecular Architecture of Actin Cytoskeleton and Filopodial Growth. Nanoscale Res. Lett. 2014, 9, 564. [Google Scholar] [CrossRef]
- Sui, Z.; Leong, P.P.; Herman, I.P.; Higashi, G.S.; Temkin, H. Raman Analysis of Light-emitting Porous Silicon. Appl. Phys. Lett. 1992, 60, 2086–2088. [Google Scholar] [CrossRef]
- Popovic, D.M.; Milosavljevic, V.; Zekic, A.; Romcevic, N.; Daniels, S. Raman Scattering Analysis of Silicon Dioxide Single Crystal Treated by Direct Current Plasma Discharge. Appl. Phys. Lett. 2011, 98, 051503. [Google Scholar] [CrossRef]
- Borowicz, P.; Taube, A.; Rzodkiewicz, W.; Latek, M.; Gierałtowska, S. Raman Spectra of High-Kappa Dielectric Layers Investigated with Micro-Raman Spectroscopy Comparison with Silicon Dioxide. Sci. World J. 2013, 2013, 208081. [Google Scholar] [CrossRef]
- Wang, W.; Favors, Z.; Ionescu, R.; Ye, R.; Bay, H.H.; Ozkan, M.; Ozkan, C.S. Monodisperse Porous Silicon Spheres as Anode Materials for Lithium Ion Batteries. Sci. Rep. 2015, 5, 8781. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, D.J.; Cho, K.M.; Kim, S.J.; Park, J.-K.; Jung, H.-T. Complete Magnesiothermic Reduction Reaction of Vertically Aligned Mesoporous Silica Channels to Form Pure Silicon Nanoparticles. Sci. Rep. 2015, 5, 9014. [Google Scholar] [CrossRef] [PubMed]
- Sorieul, S.; Costantini, J.-M.; Gosmain, L.; Thomé, L.; Grob, J.-J. Raman Spectroscopy Study of Heavy-Ion-Irradiated α-SiC. J. Phys. Condens. Matter 2006, 18, 5235–5251. [Google Scholar] [CrossRef]
- Mayerhöfer, T.G.; Shen, Z.; Leonova, E.; Edén, M.; Kriltz, A.; Popp, J. Consolidated Silica Glass from Nanoparticles. J. Solid State Chem. 2008, 181, 2442–2447. [Google Scholar] [CrossRef]
- Yang, L.Y.; Li, H.Z.; Liu, J.; Sun, Z.Q.; Tang, S.S.; Lei, M. Dual Yolk-Shell Structure of Carbon and Silica-Coated Silicon for High-Performance Lithium-Ion Batteries. Sci. Rep. 2015, 5, 10908. [Google Scholar] [CrossRef]
- Xie, Y.H.; Wilson, W.L.; Ross, F.M.; Mucha, J.A.; Fitzgerald, E.A.; Macaulay, J.M.; Harris, T.D. Luminescence and Structural Study of Porous Silicon Films. J. Appl. Phys. 1992, 71, 2403–2407. [Google Scholar] [CrossRef]
- Massoud, M.; Canut, B.; Newby, P.; Frechette, L.; Chapuis, P.O.; Bluet, J.M. Swift Heavy Ion Irradiation Reduces Porous Silicon Thermal Conductivity. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2014, 341, 27–31. [Google Scholar] [CrossRef]
- Weldon, M.K.; Queeney, K.T.; Gurevich, A.B.; Stefanov, B.B.; Chabal, Y.J.; Raghavachari, K. Si–H Bending Modes as a Probe of Local Chemical Structure: Thermal and Chemical Routes to Decomposition of H2O on Si(100)-(2 × 1). J. Chem. Phys. 2000, 113, 2440–2446. [Google Scholar] [CrossRef]
- Bisi, O.; Ossicini, S.; Pavesi, L. Porous Silicon: A Quantum Sponge Structure for Silicon Based Optoelectronics. Surf. Sci. Rep. 2000, 38, 1–126. [Google Scholar] [CrossRef]
- Mawhinney, D.B.; Glass, J.A.; Yates, J.T. FTIR Study of the Oxidation of Porous Silicon. J. Phys. Chem. B 1997, 101, 1202–1206. [Google Scholar] [CrossRef]
- Vasquez-Agustin, M.-A.; Rodríguez, Á.; García-Salgado, G.; Romero-Paredes, G.; Peña-Sierra, R. FTIR and Photoluminescence Studies of Porous Silicon Layers Oxidized in Controlled Water Vapor Conditions. Rev. Mex. Física 2007, 53, 431–435. [Google Scholar]
- Pong, K.L.; Chen, S.C.; Cheah, K.W. Photoluminescence of Laser Ablated Silicon. Solid State Commun. 1996, 99, 887–890. [Google Scholar] [CrossRef]
- Szili, E.J.; Jane, A.; Low, S.P.; Sweetman, M.; Macardle, P.; Kumar, S.; Smart, R.S.C.; Voelcker, N.H. Interferometric Porous Silicon Transducers Using an Enzymatically Amplified Optical Signal. Sens. Actuators B Chem. 2011, 160, 341–348. [Google Scholar] [CrossRef]
- Glass, J.A.; Wovchko, E.A.; Yates, J.T. Reaction of Atomic Hydrogen with Hydrogenated Porous Silicon—Detection of Precursor to Silane Formation. Surf. Sci. 1996, 348, 325–334. [Google Scholar] [CrossRef]
- Culler, S.R.; Ishida, H.; Koenig, J.L. Structure of Silane Coupling Agents Adsorbed on Silicon Powder. J. Colloid Interface Sci. 1985, 106, 334–346. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Ishida, H.; Koenig, J.L. The Structure of γ-Aminopropyltriethoxysilane on Glass Surfaces. J. Colloid Interface Sci. 1980, 74, 396–404. [Google Scholar] [CrossRef]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Aqueous and Thermal Oxidation of Porous Silicon Microparticles: Implications on Molecular Interactions. Langmuir 2008, 24, 14222–14226. [Google Scholar] [CrossRef]
- Errien, N.; Joubert, P.; Chaillou, A.; Mahric, C.; Godon, C.; Louarn, G.; Froyer, G. Electrochemical Growth of Poly(3-Dodecylthiophene) into Porous Silicon: A Nanocomposite with Tubes or Wires? Mater. Sci. Eng. B 2003, 100, 259–262. [Google Scholar] [CrossRef]
- Iqbal, Z.; Vepřek, S.; Webb, A.P.; Capezzuto, P. Raman Scattering from Small Particle Size Polycrystalline Silicon. Solid State Commun. 1981, 37, 993–996. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Iwagami, K.; Morita, N.; Matsunobe, T.; Ishida, H. Characterization of Fluorine-Doped Silicon Dioxide Film by Raman Spectroscopy. Thin Solid Films 1997, 310, 167–170. [Google Scholar] [CrossRef]
- Bao, Z.; Weatherspoon, M.R.; Shian, S.; Cai, Y.; Graham, P.D.; Allan, S.M.; Ahmad, G.; Dickerson, M.B.; Church, B.C.; Kang, Z.; et al. Chemical Reduction of Three-Dimensional Silica Micro-Assemblies into Microporous Silicon Replicas. Nature 2007, 446, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Hai, N.H.; Grigoriants, I.; Gedanken, A. Converting Stöber Silica and Mediterranean Sand to High Surface Area Silicon by a Reaction under Autogenic Pressure at Elevated Temperatures. J. Phys. Chem. C 2009, 113, 10521–10526. [Google Scholar] [CrossRef]
- Borowicz, P.; Latek, M.; Rzodkiewicz, W.; Łaszcz, A.; Czerwinski, A.; Ratajczak, J. Deep-Ultraviolet Raman Investigation of Silicon Oxide: Thin Film on Silicon Substrate versus Bulk Material. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 045003. [Google Scholar] [CrossRef]
- Liang, Y.; Miranda, C.R.; Scandolo, S. Infrared and Raman Spectra of Silica Polymorphs from an Ab Initio Parametrized Polarizable Force Field. J. Chem. Phys. 2006, 125, 194524. [Google Scholar] [CrossRef]
- Meier, C.; Lüttjohann, S.; Kravets, V.G.; Nienhaus, H.; Lorke, A.; Wiggers, H. Raman Properties of Silicon Nanoparticles. Phys. E Low-Dimens. Syst. Nanostruct. 2006, 32, 155–158. [Google Scholar] [CrossRef]
- Richter, H.; Wang, Z.P.; Ley, L. The One Phonon Raman Spectrum in Microcrystalline Silicon. Solid State Commun. 1981, 39, 625–629. [Google Scholar] [CrossRef]
- Ivanda, M. Raman Spectroscopy of Porous Silicon. In Handbook of Porous Silicon; Canham, L., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 611–620. ISBN 978-3-319-71381-6. [Google Scholar]
- Campbell, I.H.; Fauchet, P.M. The Effects of Microcrystal Size and Shape on the One Phonon Raman Spectra of Crystalline Semiconductors. Solid State Commun. 1986, 58, 739–741. [Google Scholar] [CrossRef]
- Irmer, G. Raman Scattering of Nanoporous Semiconductors. J. Raman Spectrosc. 2007, 38, 634–646. [Google Scholar] [CrossRef]
- Tolstik, E.; Osminkina, L.A.; Matthäus, C.; Burkhardt, M.; Tsurikov, K.E.; Natashina, U.A.; Timoshenko, V.Y.; Heintzmann, R.; Popp, J.; Sivakov, V. Studies of Silicon Nanoparticles Uptake and Biodegradation in Cancer Cells by Raman Spectroscopy. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Gongalsky, M.B.; Sviridov, A.P.; Bezsudnova, Y.I.; Osminkina, L.A. Biodegradation Model of Porous Silicon Nanoparticles. Colloids Surf. B Biointerfaces 2020, 190, 110946. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber (cm−1) | Raman Assignment | References |
|---|---|---|
| 480 | Amorphous silicon | [24] |
| 508–510 | Porous silicon | [24] |
| 518 | Light emitting porous silicon | [24] |
| 520/521 | Crystalline Silicon | [24,25,26,27,28] |
| 535–540 | Crystalline silicon (TO) | [29] |
| 600/620 (D2) | 3-member ring in SiO2 structure (Densified bulk SiO2) | [30] |
| 958/967 | Si–OH | [30,31] |
| 1097 | Si–O | [31] |
| Absorbance (cm−1) | Species | Vibrations | References |
|---|---|---|---|
| 640 | –SiHx (x = 3,2,1) | δ(Si–H) wagging | [32] |
| 880 | OySiHx | δ(O–Si–H) bending | [33] |
| 905 | Si–H | δ(Si–H) deformation | [33] |
| 938 | Si–H | δ(Si–H) deformation | [27] |
| 1038 | Si–O | v(Si–O) stretching | [34] |
| 1056 | Si–O | v(Si–O) stretching | [35] |
| 1065 | Si–O–Si | v(Si–O) stretching | [36,37] |
| 1105, 1150, 1100 | Si–O–Si | v(Si–O–Si) asymmetric stretching | [35,36,38,39] |
| 1300 | Si–O–Si | Deformation mode | [40] |
| 1411 | Si–CH2 | Deformation mode | [41] |
| 1486, 1567, 1600 | NH2 | Asymmeteric and symmeteric stretching | [41,42] |
| 2080 | Si3Si–H | Stretching vibration | [43] |
| 2187 | O3Si–H | Stretching vibrations | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, N.; Salehi, H.; Cueto-Díaz, E.J.; Desoutter, A.; Cuisinier, F.; Cunin, F.; Collart-Dutilleul, P.-Y. Nanostructured Porous Silicon for Bone Tissue Engineering: Kinetics of Particle Degradation and Si-Controlled Release. J. Funct. Biomater. 2023, 14, 493. https://doi.org/10.3390/jfb14100493
Fatima N, Salehi H, Cueto-Díaz EJ, Desoutter A, Cuisinier F, Cunin F, Collart-Dutilleul P-Y. Nanostructured Porous Silicon for Bone Tissue Engineering: Kinetics of Particle Degradation and Si-Controlled Release. Journal of Functional Biomaterials. 2023; 14(10):493. https://doi.org/10.3390/jfb14100493
Chicago/Turabian StyleFatima, Naveen, Hamideh Salehi, Eduardo J. Cueto-Díaz, Alban Desoutter, Frédéric Cuisinier, Frédérique Cunin, and Pierre-Yves Collart-Dutilleul. 2023. "Nanostructured Porous Silicon for Bone Tissue Engineering: Kinetics of Particle Degradation and Si-Controlled Release" Journal of Functional Biomaterials 14, no. 10: 493. https://doi.org/10.3390/jfb14100493
APA StyleFatima, N., Salehi, H., Cueto-Díaz, E. J., Desoutter, A., Cuisinier, F., Cunin, F., & Collart-Dutilleul, P.-Y. (2023). Nanostructured Porous Silicon for Bone Tissue Engineering: Kinetics of Particle Degradation and Si-Controlled Release. Journal of Functional Biomaterials, 14(10), 493. https://doi.org/10.3390/jfb14100493