Sol-Gel Derived Tertiary Bioactive Glass–Ceramic Nanorods Prepared via Hydrothermal Process and Their Composites with Poly(Vinylpyrrolidone-Co-Vinylsilane)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sol-Gel Synthesis of Bioactive Glass
2.3. Hydrothermal Processing of Bioactive Glass
2.4. Synthesis of Co-Polymer
2.5. Synthesis of Bioactive Composite Biomaterials
2.6. Scanning Electron Microscopy (SEM), X-ray Diffraction (XRD), and Fourier Transform Infrared Spectroscopy (FTIR)
2.7. Solid-State Cross-Polarization Magic-Angle Spinning (CPMAS) 29Si NMR
2.8. Mechanical and Degradation Testing of Poly(VP-co-TEVS)/BG Composites
2.9. MC3T3-E1 Preosteoblast Culture
3. Results and Discussion
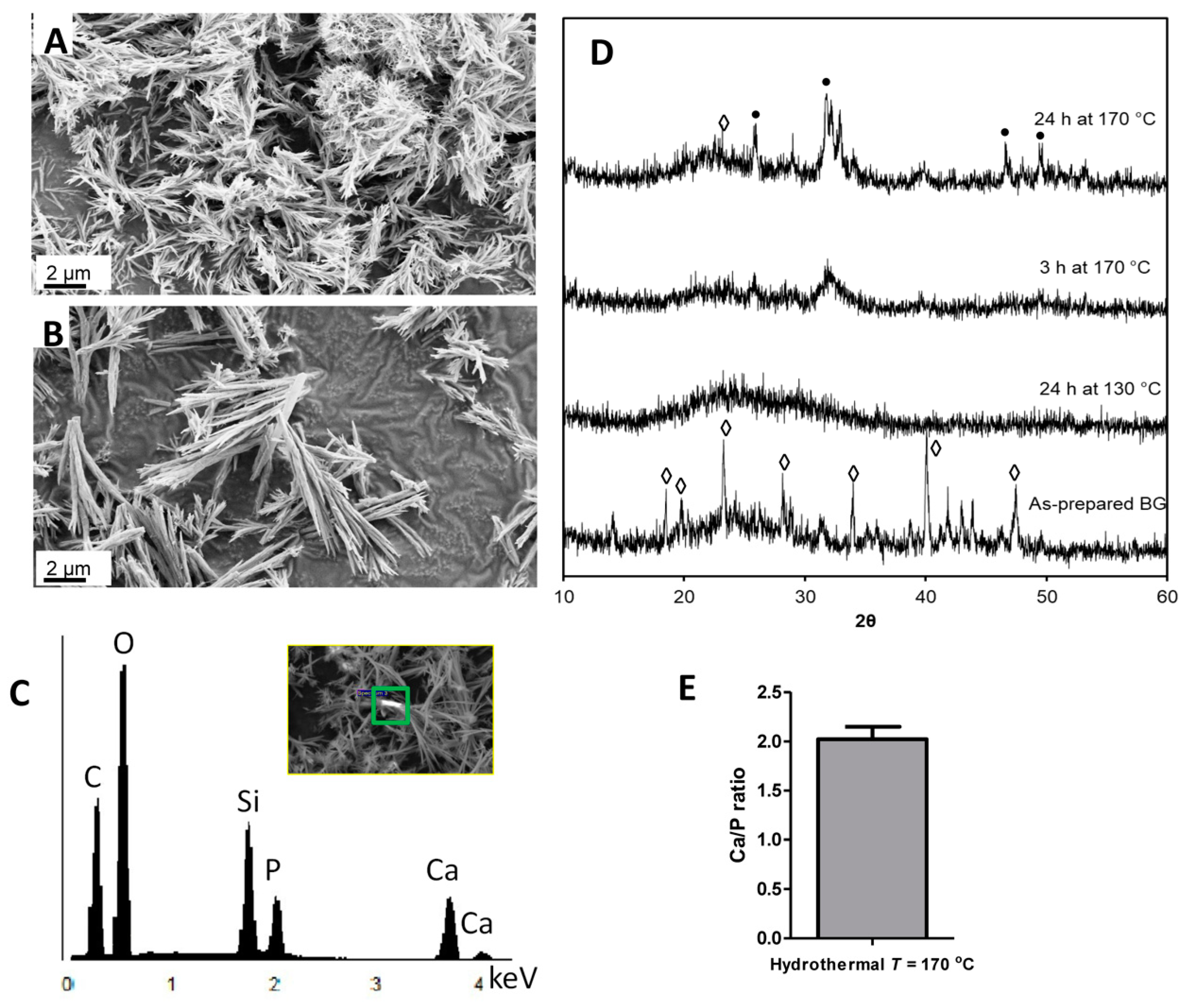
3.1. Hydrothermal Processing of Bioactive Glass
3.2. The Effect of Reaction Time on Morphology and Crystallinity
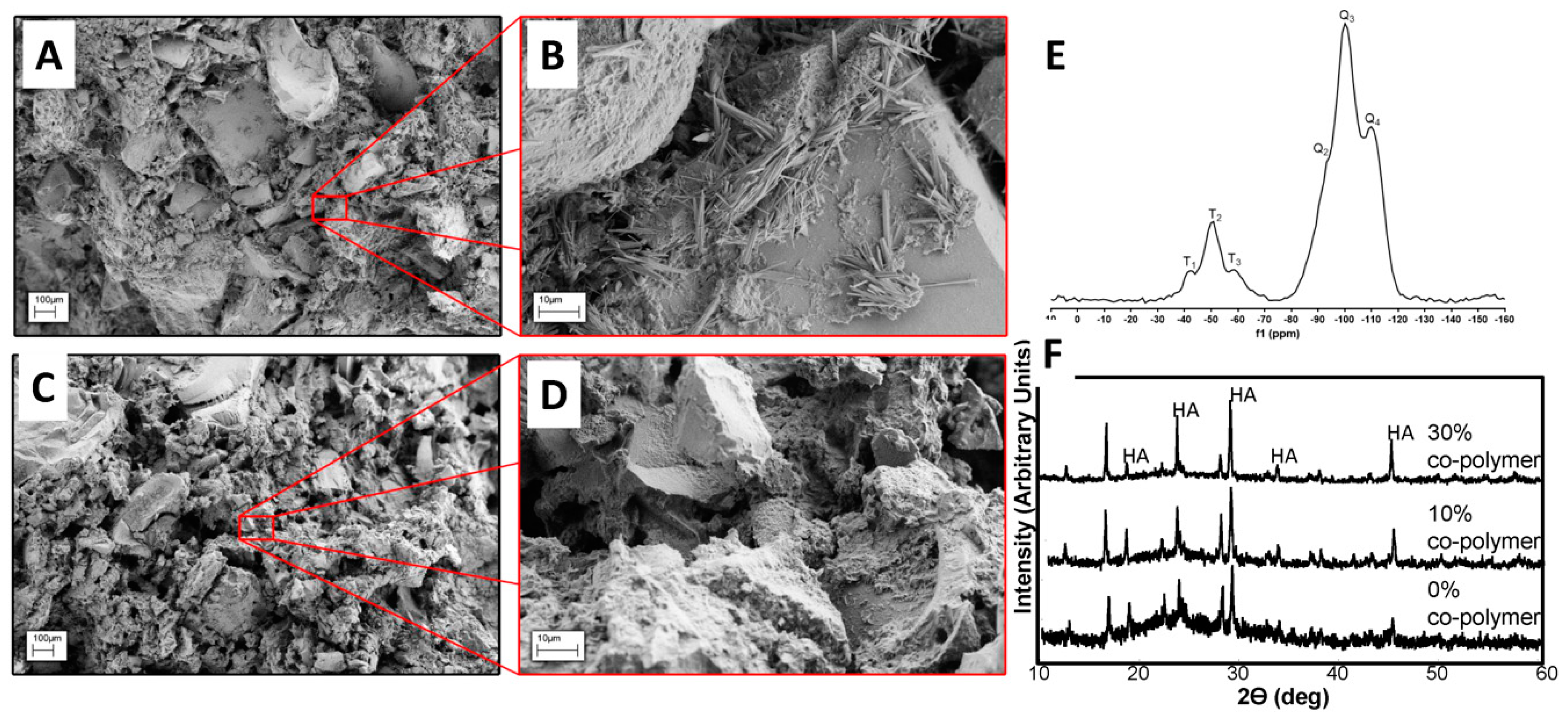
3.3. Characteristics of Chemical Network Formation as a Result of Hydrothermal Processing
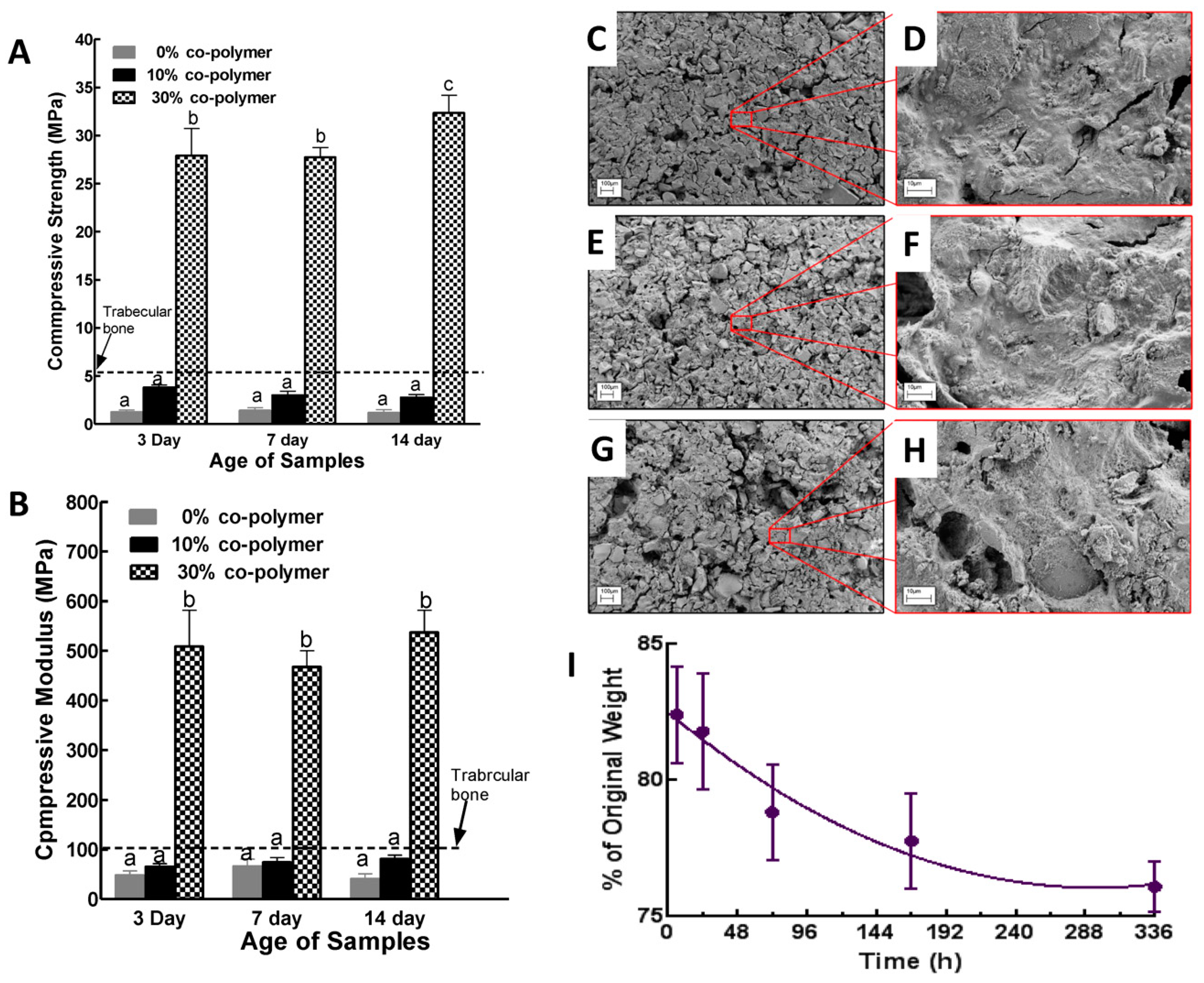
3.4. Properties of Composites Prepared with BG Nanorods and Synthesized Co-Polymers
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aslankoohi, N.; Mondal, D.; Rizkalla, A.S.; Mequanint, K. Bone repair and regenerative biomaterials: Towards recapitulating the microenvironment. Polymers 2019, 11, 1437. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L.; Splinter, R.J.; Allen, W.C.; Greenlee, T.K. Bonding mechanisms at the interface of ceramic prosthetic materials. J. Biomed. Mater. Res. 1971, 5, 117–141. [Google Scholar] [CrossRef]
- Kaur, G.; Pandey, O.P.; Singh, K.; Homa, D.; Scott, B.; Pickrell, G. A review of bioactive glasses: Their structure, properties, fabrication and apatite formation. J. Biomed. Mater. Res. Part A 2013, 102, 254–274. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.N.; Day, D.E.; Sonny Bal, B.; Fu, Q.; Jung, S.B.; Bonewald, L.F.; Tomsia, A.P. Bioactive glass in tissue engineering. Acta Biomater. 2011, 7, 2355–2373. [Google Scholar] [CrossRef]
- Brink, M. The influence of alkali and alkaline earths on the working range for bioactive glasses. J. Biomed. Mater. Res. 1998, 36, 109–117. [Google Scholar] [CrossRef]
- Brovarone, C.V.; Verné, E.; Appendino, P. Macroporous bioactive glass-ceramic scaffolds for tissue engineering. J. Mater. Sci. Mater. Med. 2006, 17, 1069–1078. [Google Scholar] [CrossRef]
- Fu, Q.; Rahaman, M.N.; Sonny Bal, B.; Brown, R.F.; Day, D.E. Mechanical and in vitro performance of 13–93 bioactive glass scaffolds prepared by a polymer foam replication technique. Acta Biomater. 2008, 4, 1854–1864. [Google Scholar] [CrossRef]
- Hench, L.L.; West, J.K. The sol-gel process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Allo, B.A.; Rizkalla, A.S.; Mequanint, K. Synthesis and electrospinning of ε-polycaprolactone-bioactive glass hybrid biomaterials via a sol−gel process. Langmuir 2010, 26, 18340–18348. [Google Scholar] [CrossRef]
- Brinker, C.J. Hydrolysis and condensation of silicates: Effects on structure. J. Non-Cryst. Solids 1988, 100, 31–50. [Google Scholar] [CrossRef]
- Sepulveda, P.; Jones, J.R.; Hench, L.L. Characterization of melt-derived 45S5 and sol-gel–derived 58S bioactive glasses. J. Biomed. Mater. Res. 2002, 58, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Dziadek, M.; Zagrajczuk, B.; Jelen, P.; Olejniczak, Z.; Cholewa-Kowalska, K. Structural variations of bioactive glasses obtained by different synthesis routes. Ceram. Int. 2016, 42, 14700–14709. [Google Scholar] [CrossRef]
- Wajda, A.; Sitarz, M. Structural and microstructural comparison of bioactive melt-derived and gel-derived glasses from CaO-SiO2 binary system. Ceram. Int. 2018, 44, 8856–8863. [Google Scholar] [CrossRef]
- Lei, B.; Chen, X.; Wang, Y.; Zhao, N.; Du, C.; Fang, L. Surface nanoscale patterning of bioactive glass to support cellular growth and differentiation. J. Biomed. Mater. Res. Part A 2010, 94A, 1091–1099. [Google Scholar] [CrossRef]
- Azizian, M.; Rabiee, S.M. A Freestanding Sol-Gel Technique for Growth of Nanowire Arrays in SiO2-CaO-P2O5-ZrO2 System. Silicon 2016, 8, 233–237. [Google Scholar] [CrossRef]
- Rabiee, S.M.; Azizian, M. Effect of Zirconia Concentration on the Growth of Nanowires in Bioactive Glass–Ceramic Coatings. Int. J. Appl. Ceram. Technol. 2013, 10, 33–39. [Google Scholar] [CrossRef]
- Rahong, S.; Yasui, T.; Kaji, N.; Baba, Y. Recent developments in nanowires for bio-applications from molecular to cellular levels. Lab. Chip 2016, 16, 1126–1138. [Google Scholar] [CrossRef][Green Version]
- Zeimaran, E.; Pourshahrestani, S.; Shirazi, S.F.S.; Pingguan-Murphy, B.; Kadri, N.A.; Towler, M.R. Hydrothermal synthesis and characterisation of bioactive glass-ceramic nanorods. J. Non-Cryst. Solids 2016, 443, 118–124. [Google Scholar] [CrossRef]
- Hong, Z.; Merino, E.G.; Reis, R.L.; Mano, J.F. Novel Rice-shaped Bioactive Ceramic Nanoparticles. Adv. Eng. Mater. 2009, 11, B25–B29. [Google Scholar] [CrossRef]
- Qi, C.; Zhu, Y.-J.; Zhao, X.-Y.; Zhao, J.; Chen, F.; Cheng, G.-F.; Ruan, Y.J. High surface area carbonate apatite nanorod bundles: Surfactant-free sonochemical synthesis and drug loading and release properties. Mater. Res. Bull. 2013, 48, 1536–1540. [Google Scholar] [CrossRef]
- Byrappa, K.; Yoshimura, M. 1—Hydrothermal Technology—Principles and Applications. In Handbook of Hydrothermal Technology; Byrappa, K., Yoshimura, M., Eds.; William Andrew Publishing: Norwich, NY, USA, 2001; pp. 1–52. [Google Scholar]
- Mondal, D.; Dixon, S.J.; Mequanint, K.; Rizkalla, A.S. Bioactivity, Degradation, and Mechanical Properties of Poly(vinylpyrrolidone-co-triethoxyvinylsilane)/Tertiary Bioactive Glass Hybrids. ACS Appl. Bio Mater. 2018, 1, 1369–1381. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef] [PubMed]
- Teodorescu, M.; Bercea, M. Poly(vinylpyrrolidone)—A Versatile Polymer for Biomedical and Beyond Medical Applications. Polym.-Plast. Technol. Eng. 2015, 54, 923–943. [Google Scholar] [CrossRef]
- Tang, J.; Alivisatos, A.P. Crystal Splitting in the Growth of Bi2S3. Nano Lett. 2006, 6, 2701–2706. [Google Scholar] [CrossRef] [PubMed]
- Sanosh, K.P.; Chu, M.C.; Balakrishnan, A.; Kim, T.N.; Cho, S.J. Preparation and characterization of nano-hydroxyapatite powder using sol-gel technique. B Mater. Sci. 2009, 32, 465–470. [Google Scholar] [CrossRef]
- Gentile, P.; Wilcock, C.J.; Miller, C.A.; Moorehead, R.; Hatton, P.V. Process Optimisation to Control the Physico-Chemical Characteristics of Biomimetic Nanoscale Hydroxyapatites Prepared Using Wet Chemical Precipitation. Materials 2015, 8, 2297–2310. [Google Scholar] [CrossRef]
- Ma, J.; Chen, C.Z.; Wang, D.G.; Meng, X.G.; Shi, J.Z. Influence of the sintering temperature on the structural feature and bioactivity of sol–gel derived SiO2–CaO–P2O5 bioglass. Ceram. Int. 2010, 36, 1911–1916. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Renella, R.A.; Papale, F. Sol–gel synthesis of SiO2–CaO–P2O5 glasses: Influence of the heat treatment on their bioactivity and biocompatibility. Ceram. Int. 2015, 41, 12578–12588. [Google Scholar] [CrossRef]
- Gunawidjaja, P.N.; Mathew, R.; Lo, A.Y.H.; Izquierdo-Barba, I.; García, A.; Arcos, D.; Vallet-Regí, M.; Edén, M. Local structures of mesoporous bioactive glasses and their surface alterations in vitro: Inferences from solid-state nuclear magnetic resonance. Philos. Trans. A Math. Phys. Eng. Sci. 2012, 370, 1376–1399. [Google Scholar] [CrossRef]
- Martin, R.A.; Twyman, H.L.; Rees, G.J.; Smith, J.M.; Barney, E.R.; Smith, M.E.; Hanna, J.V.; Newport, R.J. A structural investigation of the alkali metal site distribution within bioactive glass using neutron diffraction and multinuclear solid state NMR. Phys. Chem. Chem. Phys. 2012, 14, 12105–12113. [Google Scholar] [CrossRef]
- Pedone, A.; Charpentier, T.; Malavasi, G.; Menziani, M.C. New Insights into the Atomic Structure of 45S5 Bioglass by Means of Solid-State NMR Spectroscopy and Accurate First-Principles Simulations. Chem. Mater. 2010, 22, 5644–5652. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Schnitzler, V.; Tancret, F.; Bouler, J.M. Calcium phosphate cements for bone substitution: Chemistry, handling and mechanical properties. Acta Biomater. 2014, 10, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Gil, F.J.; Ginebra, M.P.; Driessens, F.C.; Planell, J.A.; Best, S.M. Calcium phosphate bone cements for clinical applications. Part II: Precipitate formation during setting reactions. J. Mater. Sci. Mater. Med. 1999, 10, 177–183. [Google Scholar] [CrossRef]
- Mondal, D.; Rizkalla, A.S.; Mequanint, K. Bioactive borophosphosilicate-polycaprolactone hybrid biomaterials via a non--aqueous sol gel process. RSC Adv. 2016, 6, 92824–92832. [Google Scholar] [CrossRef]
- Mondal, D.; Dixon, S.J.; Mequanint, K.; Rizkalla, A.S. Mechanically-competent and cytocompatible polycaprolactone-borophosphosilicate hybrid biomaterials. J. Mech. Behav. Biomed. Mater. 2017, 75, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.X.; Vo, M.Q.; Nguyen, T.A.; Bui, H.T. Investigation of bioactive glass-cermaic 60SiO2-30CaO-10P2O5 prepared by hydrothermal method. Adv. Mater. Sci. Eng. 2019, 2019, 1528326. [Google Scholar]
- Taguchi, Y.; Yamamuro, T.; Nakamura, T.; Nishimura, N.; Kokubo, T.; Takahata, E.; Yoshihara, S. A bioactive glass powder-ammonium hydrogen phosphate composite for repairing bone defects. J. Appl. Biomater. 1990, 1, 217–223. [Google Scholar] [CrossRef]
- Nishimura, N.; Yamamuro, T.; Taguchi, Y.; Ikenaga, M.; Nakamura, T.; Kokubo, T.; Yoshihara, S. A new bioactive bone cement: Its histological and mechanical characterization. J. Appl. Biomater. 1991, 2, 219–229. [Google Scholar] [CrossRef]
- Kokubo, T.; Yoshihara, S.; Nishimura, N.; Yamamuro, T.; Nakamura, T. Bioactive Bone Cement Based on Ca-SiO2-P2O5 Glass. J. Am. Ceram. Soc. 1991, 74, 1739–1741. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondal, D.; Zaharia, A.; Mequanint, K.; Rizkalla, A.S. Sol-Gel Derived Tertiary Bioactive Glass–Ceramic Nanorods Prepared via Hydrothermal Process and Their Composites with Poly(Vinylpyrrolidone-Co-Vinylsilane). J. Funct. Biomater. 2020, 11, 35. https://doi.org/10.3390/jfb11020035
Mondal D, Zaharia A, Mequanint K, Rizkalla AS. Sol-Gel Derived Tertiary Bioactive Glass–Ceramic Nanorods Prepared via Hydrothermal Process and Their Composites with Poly(Vinylpyrrolidone-Co-Vinylsilane). Journal of Functional Biomaterials. 2020; 11(2):35. https://doi.org/10.3390/jfb11020035
Chicago/Turabian StyleMondal, Dibakar, Andrei Zaharia, Kibret Mequanint, and Amin S. Rizkalla. 2020. "Sol-Gel Derived Tertiary Bioactive Glass–Ceramic Nanorods Prepared via Hydrothermal Process and Their Composites with Poly(Vinylpyrrolidone-Co-Vinylsilane)" Journal of Functional Biomaterials 11, no. 2: 35. https://doi.org/10.3390/jfb11020035
APA StyleMondal, D., Zaharia, A., Mequanint, K., & Rizkalla, A. S. (2020). Sol-Gel Derived Tertiary Bioactive Glass–Ceramic Nanorods Prepared via Hydrothermal Process and Their Composites with Poly(Vinylpyrrolidone-Co-Vinylsilane). Journal of Functional Biomaterials, 11(2), 35. https://doi.org/10.3390/jfb11020035