Novel Tools towards Magnetic Guidance of Neurite Growth: (I) Guidance of Magnetic Nanoparticles into Neurite Extensions of Induced Human Neurons and In Vitro Functionalization with RAS Regulating Proteins

 , , , , ,
, , , , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
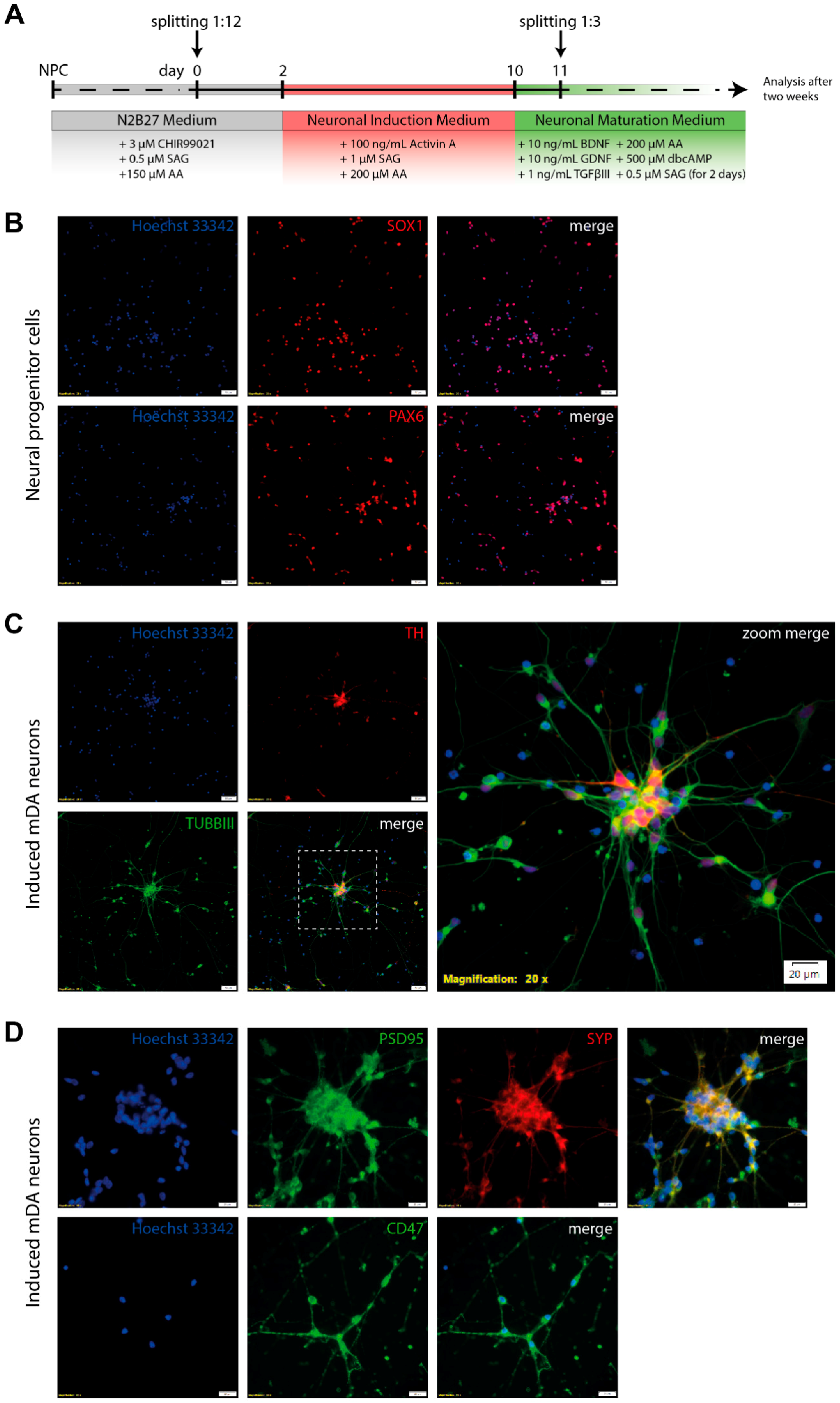
2.1. Culturing of Human Neural Progenitor Cells
2.2. Generation of mDA Neurons from NPCs
2.3. Immunocytochemistry
2.4. Patch-Clamp Recordings of Induced Dopaminergic Neurons
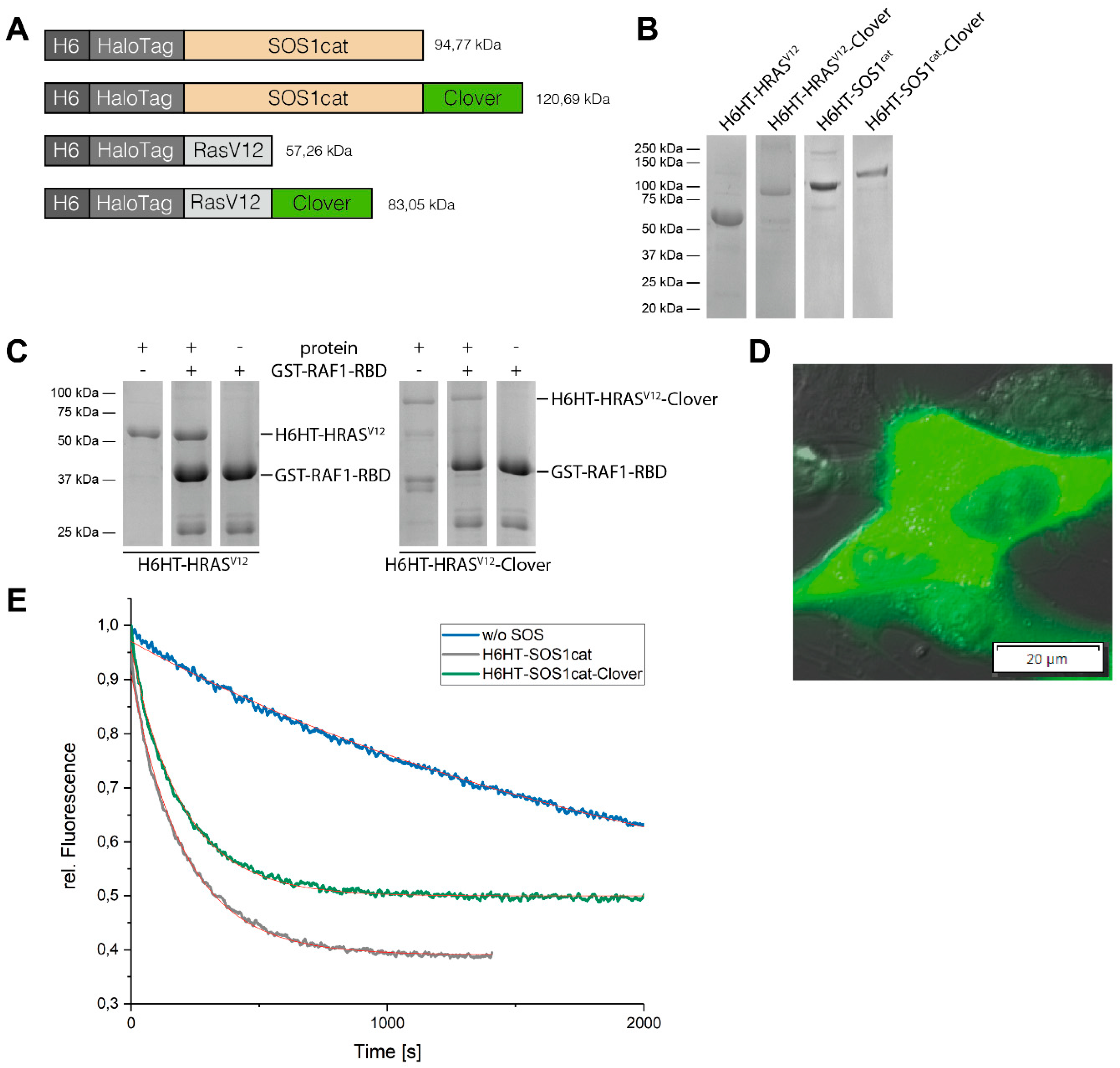
2.5. Protein Expression and Purification of HaloTag™ Fusion Proteins
2.6. GTP Exchange Factor Activity-Measurement of SOS1cat-Fusion Proteins
2.7. Pull-Down of H-RASV12-Fusion Proteins
2.8. Culturing of SH-SY5Y cells and Microinjection with H6HT-H-RASV12-Clover
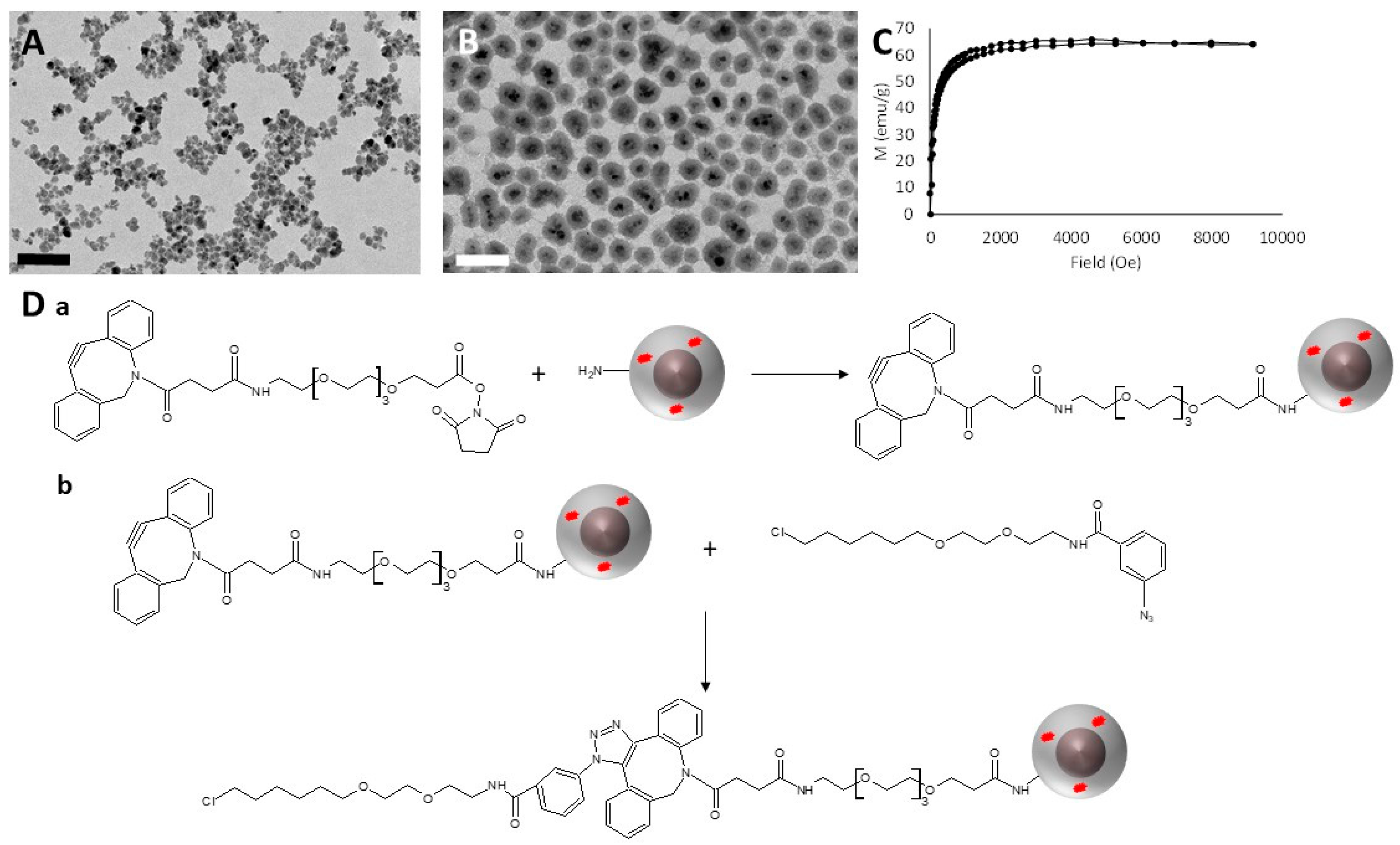
2.9. Synthesis and HaloTag™ Ligand Functionalization of γ-Fe2O3@SiO2 Core-Shell Nanoparticles (HTL-MNPs)
2.9.1. Synthesis of γ-Fe2O3 Cores
2.9.2. Synthesis of γ-Fe2O3@SiO2 Core-Shell Nanoparticles [18]
2.9.3. Functionalization of the γ-Fe2O3@SiO2 Core-Shell Nanoparticles with HaloTag™ Ligands [19]
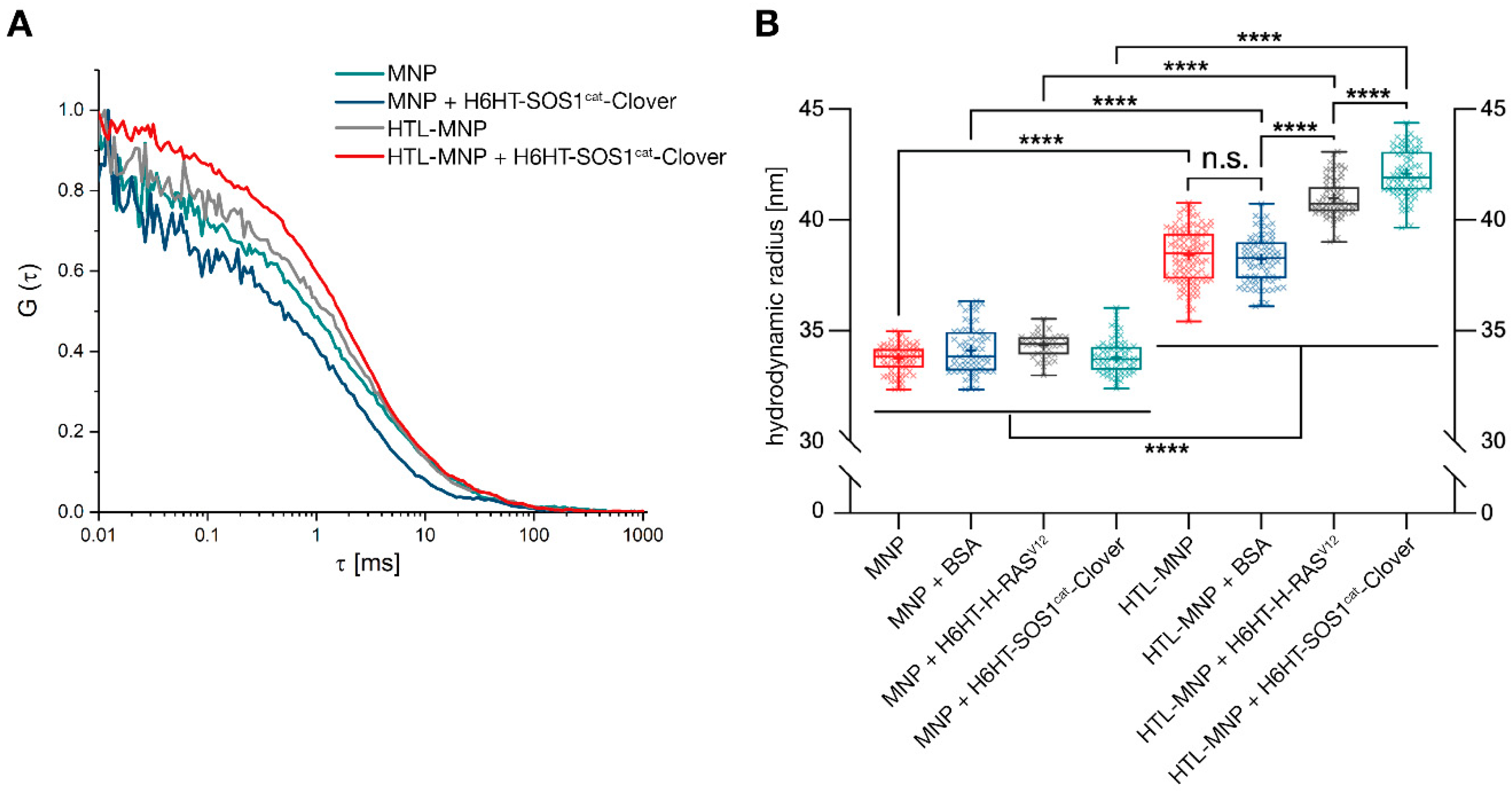
2.10. Protein-Functionalization of Magnetic Nanoparticles Demonstrated by Fluorescence Correlation Spectroscopy
2.11. Protein-Functionalization of Magnetic Nanoparticles Demonstrated by Multiangle Light Scattering
2.12. Microinjection and Remote Controlling of Magnetic Nanoparticles in NPCs and Induced DA Neurons
3. Results
3.1. Synthesis and HaloTag™ Ligand Functionalization of γ-Fe2O3@SiO2 Core-Shell Nanoparticles (HTL-MNPs)
3.2. Purification of HaloTag™-Fusion Proteins and their Characterization
3.3. Binding of HaloTag™-Fusion Proteins to HaloTag™ Ligand-Functionalized γ-Fe2O3@SiO2 Core-Shell Nanoparticles (HTL-MNPs)
3.4. Differentiation of Neural Progenitor Cells Into Midbrain Dopaminergic Neurons
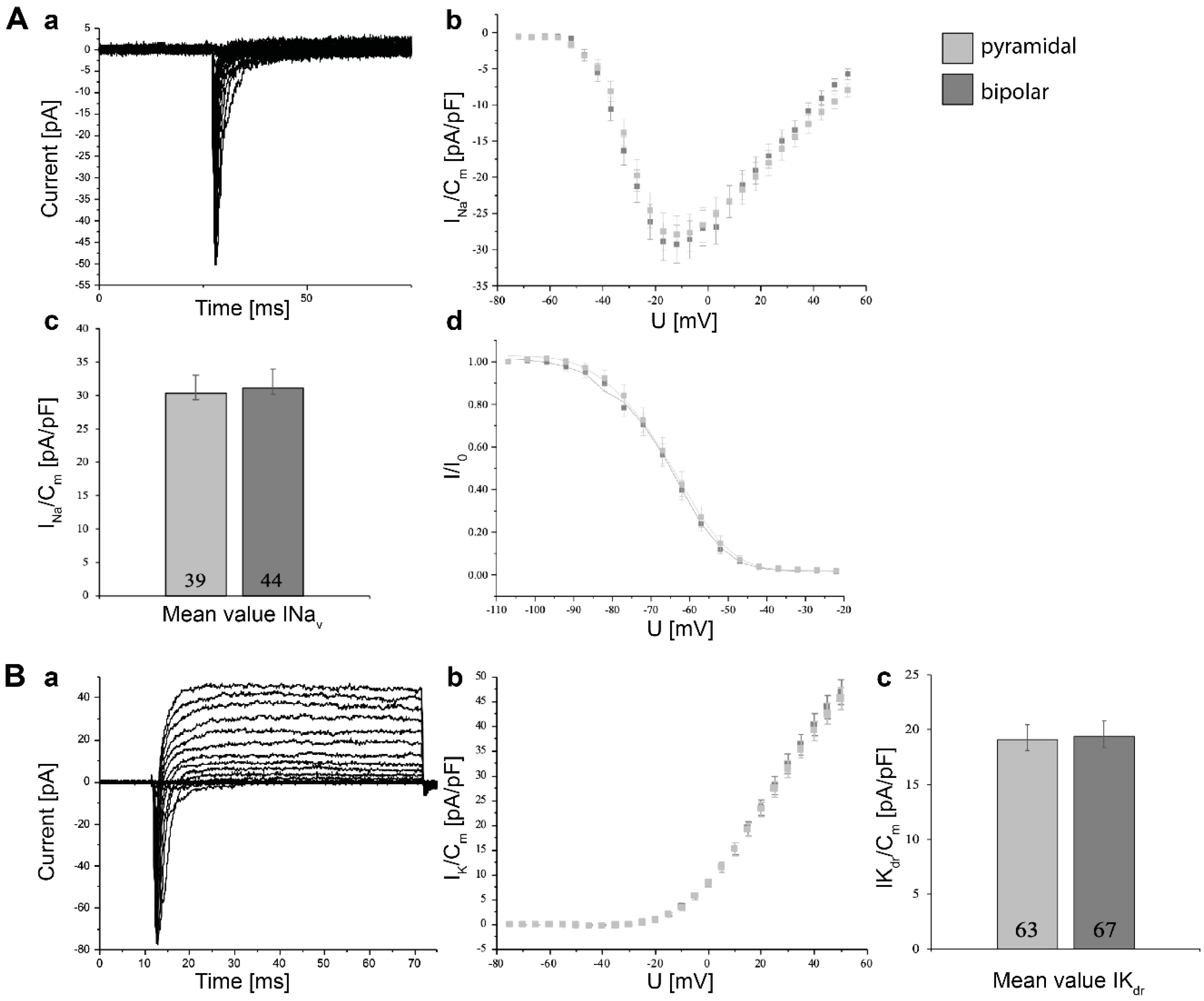
3.5. Electrophysiological Characterization of Induced Neurons
3.6. Accumulating Magnetic Nanoparticles in Fibers of Induced Neurons Upon Magnetic Stimulus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-Hydroxydopamine |
| AA | Ascorbic acid |
| AKT | Protein kinase B |
| BCL-2 | B-cell lymphoma 2 |
| BCL-xL | B-cell lymphoma-extra large |
| BDNF | Brain-derived neurotrophic factor |
| Brn2 | POU domain, class 3, transcription factor 2 |
| CD47 | Cluster of Differentiation 47 |
| CDC42 | Cell division cycle 42 |
| cDNA | Complementary DNA |
| DA | Dopaminergic |
| dbcAMP | Dibutyryl cyclic-AMP |
| DBS | Deep brain stimulation |
| DLS | Dynamic light scattering |
| DMEM | Dulbecco modified eagle medium |
| ERK | Extracellular-signal-regulated kinase |
| FCS | Fluorescence correlation spectroscopy |
| FPLC | Fast protein liquid chromatography |
| GDNF | Glial cell line-derived neurotrophic factor |
| GDP | Guanosine diphosphate |
| GEF | Guanine nucleotide exchange factor |
| GST | Glutathione S-transferase |
| GTP | Guanosine triphosphate |
| GTPase | Guanosine triphosphatase |
| H6 | His6x-Tag |
| H6HT | H6 = His6x-Tag, HT = HaloTag™ |
| HEK293 | Human embryonic kidney 293 cell line |
| HeLa | Cervical cancer cell line derived from patient Henrietta Lacks |
| H-RAS | Harvey rat sarcoma |
| HT | HaloTag™ derived from bacterial haloalkane dehalogenase |
| HTL | HaloTag™ Ligand |
| HTL-MNP | HaloTag™ Ligand-functionalised MNP |
| iPSCs | Induced pluripotent stem cells |
| L-DOPA | Levodopa |
| Lmx1a | LIM homeobox transcription factor 1-alpha |
| MALS | Multiangle light scattering |
| MANT | N-Methylanthraniloyl |
| MAPK | Mitogen activated protein kinase |
| Mash1 | Achaete-scute homolog 1 |
| mDA | Midbrain dopaminergic |
| miRNA | Micro RNA |
| MNP | Magnetic nanoparticle |
| Myt1l | Myelin transcription factor 1-like protein |
| NdFeB | Neodymium-Ferrum-Boron |
| NGF | Nerve growth factor |
| NPC | Neural progenitor cell |
| Nurr1 | Nuclear receptor-related 1 protein |
| PAX6 | Paired box protein Pax-6 |
| PD | Parkinson’s disease |
| PEG | Polyethylene glycol |
| PI3K | Phosphoinositide-dependent kinase 1 |
| PSD95 | Post synaptic density 95 protein |
| RAB | RAS-like proteins in brain |
| RAC1 | RAS-related C3 botulinum toxin substrate |
| RAF1 | Rapidly accelerated fibrosarcoma |
| RAS | Rat sarcoma |
| RBD | RAS-binding domain |
| RHO | RAS homolog |
| SAG | Smoothened agonist |
| SEC | Size exclusion chromatography |
| SH-SY5Y | Human derived neuroblastoma cell line |
| SN | Substantia nigra |
| SOS1 | Son of Sevenless 1 |
| SOX1 | SRY-box containing gene 1 |
| SYP | Synaptophysin |
| TGF-β3 | Transforming growth factor, β 3 |
| TH | Tyrosine hydroxylase |
| TIAM | T-lymphoma invasion and metastasis-inducing protein |
| TRKA | Tropomyosin receptor kinase A |
| TRKB | Tropomyosin receptor kinase B |
| TUBBIII | Tubulin β-3 chainβ |
References
- Heumann, R.; Moratalla, R.; Herrero, M.T.; Chakrabarty, K.; Drucker-Colin, R.; Garcia-Montes, J.R.; Simola, N.; Morelli, M. Dyskinesia in parkinson’s disease: Mechanisms and current non-pharmacological interventions. J. Neurochem. 2014, 130, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Schöneborn, H.; Raudzus, F.; Coppey, M.; Neumann, S.; Heumann, R. Perspectives of ras and rheb gtpase signaling pathways in regenerating brain neurons. Int. J. Mol. Sci. 2018, 19, 4052. [Google Scholar] [CrossRef]
- Borasio, G.D.; John, J.; Wittinghofer, A.; Barde, Y.A.; Sendtner, M.; Heumann, R. Ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron 1989, 2, 1087–1096. [Google Scholar] [CrossRef]
- Borasio, G.D.; Markus, A.; Wittinghofer, A.; Barde, Y.A.; Heumann, R. Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic neurons. J. Cell Biol. 1993, 121, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, U.; Alpár, A.; Seeger, G.; Heumann, R.; Arendt, T. Enhanced ras activity in pyramidal neurons induces cellular hypertrophy and changes in afferent and intrinsic connectivity in synras mice. Int. J. Dev. Neurosci. 2004, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Zhong, J.; Snider, W.D. Raf and akt mediate distinct aspects of sensory axon growth. Neuron 2002, 35, 65–76. [Google Scholar] [CrossRef]
- Hayashi, K.; Furuya, A.; Sakamaki, Y.; Akagi, T.; Shinoda, Y.; Sadakata, T.; Hashikawa, T.; Shimizu, K.; Minami, H.; Sano, Y.; et al. The brain-specific rasgef very-kind is required for normal dendritic growth in cerebellar granule cells and proper motor coordination. PLoS ONE 2017, 12, e0173175. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, H.K.; Takamiya, K.; Huganir, R.L. The role of synaptic gtpase-activating protein in neuronal development and synaptic plasticity. J. Neurosci. 2003, 23, 1119–1124. [Google Scholar] [CrossRef]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhohl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic activation of ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, K.; Serchov, T.; Mann, S.A.; Dietzel, I.D.; Heumann, R. Enhancement of dopaminergic properties and protection mediated by neuronal activation of ras in mouse ventral mesencephalic neurones. Eur. J. Neurosci. 2007, 25, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Etoc, F.; Lisse, D.; Bellaiche, Y.; Piehler, J.; Coppey, M.; Dahan, M. Subcellular control of rac-gtpase signalling by magnetogenetic manipulation inside living cells. Nat. Nanotechnol. 2013, 8, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Hargus, G.; Ehrlich, M.; Arauzo-Bravo, M.J.; Hemmer, K.; Hallmann, A.L.; Reinhardt, P.; Kim, K.P.; Adachi, K.; Santourlidis, S.; Ghanjati, F.; et al. Origin-dependent neural cell identities in differentiated human ipscs in vitro and after transplantation into the mouse brain. Cell Rep. 2014, 8, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Urh, M.; Rosenberg, M. Halotag, a platform technology for protein analysis. Curr. Chem. Genom. 2012, 6, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kanie, T.; Jackson, P.K. Guanine nucleotide exchange assay using fluorescent mant-gdp. Bio-Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Georgelin, T.; Bombard, S.; Siaugue, J.M.; Cabuil, V. Nanoparticle-mediated delivery of bleomycin. Angew. Chem. Int. Ed. Engl. 2010, 49, 8897–8901. [Google Scholar] [CrossRef]
- Etoc, F.; Vicario, C.; Lisse, D.; Siaugue, J.M.; Piehler, J.; Coppey, M.; Dahan, M. Magnetogenetic control of protein gradients inside living cells with high spatial and temporal resolution. Nano Lett. 2015, 15, 3487–3494. [Google Scholar] [CrossRef]
- Bacia, K.; Haustein, E.; Schwille, P. Fluorescence correlation spectroscopy: Principles and applications. Cold Spring Harb. Protoc. 2014, 2014, 709–725. [Google Scholar] [CrossRef]
- Rigler, R.; Mets, Ü.; Widengren, J.; Kask, P. Fluorescence correlation spectroscopy with high count rate and low background: Analysis of translational diffusion. Eur. Biophys. J. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Wang, R.; Brattain, M.G. The maximal size of protein to diffuse through the nuclear pore is larger than 60kda. FEBS Lett. 2007, 581, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. ‘Reprogrammed’ Stem Cells Implanted into Patient with Parkinson’s Disease. Available online: https://www.nature.com/articles/d41586-018-07407-9 (accessed on 18 April 2019).
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human trials of stem cell-derived dopamine neurons for parkinson’s disease: Dawn of a new era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.N.; Tolkovsky, A.M. A role for mapk/erk in sympathetic neuron survival: Protection against a p53-dependent, jnk-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999, 19, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Xifro, X.; Minano-Molina, A.J.; Saura, C.A.; Rodriguez-Alvarez, J. Ras protein activation is a key event in activity-dependent survival of cerebellar granule neurons. J. Biol. Chem. 2014, 289, 8462–8472. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Feramisco, J.R. Microinjection of the ras oncogene protein into pc12 cells induces morphological differentiation. Cell 1985, 42, 841–848. [Google Scholar] [CrossRef]
- Noda, M.; Ko, M.; Ogura, A.; Liu, D.G.; Amano, T.; Takano, T.; Ikawa, Y. Sarcoma viruses carrying ras oncogenes induce differentiation-associated properties in a neuronal cell line. Nature 1985, 318, 73–75. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Noda, M.; Kitayama, H.; Ikawa, Y. Possible involvement of two signaling pathways in induction of neuron-associated properties by v-ha-ras gene in pc12 cells. J. Biol. Chem. 1988, 263, 12102–12108. [Google Scholar]
- Fivaz, M.; Bandara, S.; Inoue, T.; Meyer, T. Robust neuronal symmetry breaking by ras-triggered local positive feedback. Curr. Biol. CB 2008, 18, 44–50. [Google Scholar] [CrossRef]
- Makwana, M.; Serchov, T.; Hristova, M.; Bohatschek, M.; Gschwendtner, A.; Kalla, R.; Liu, Z.; Heumann, R.; Raivich, G. Regulation and function of neuronal gtp-ras in facial motor nerve regeneration. J. Neurochem. 2009, 108, 1453–1463. [Google Scholar] [CrossRef]
- Zhong, J. Ras and downstream raf-mek and pi3k-akt signaling in neuronal development, function and dysfunction. Biol. Chem. 2016, 397, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bradke, F.; Dotti, C.G. The role of local actin instability in axon formation. Science 1999, 283, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Kakumoto, T.; Nakata, T. Optogenetic control of pip3: Pip3 is sufficient to induce the actin-based active part of growth cones and is regulated via endocytosis. PLoS ONE 2013, 8, e70861. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Cornejo, B.J.; Kuhn, T.B.; Bamburg, J.R. Cdc42 stimulates neurite outgrowth and formation of growth cone filopodia and lamellipodia. J. Neurobiol. 2000, 43, 352–364. [Google Scholar] [CrossRef]
- Lamoureux, P.; Altun-Gultekin, Z.F.; Lin, C.; Wagner, J.A.; Heidemann, S.R. Rac is required for growth cone function but not neurite assembly. J. Cell Sci. 1997, 110 Pt 5, 635–641. [Google Scholar]
- Ng, J.; Nardine, T.; Harms, M.; Tzu, J.; Goldstein, A.; Sun, Y.; Dietzl, G.; Dickson, B.J.; Luo, L. Rac gtpases control axon growth, guidance and branching. Nature 2002, 416, 442–447. [Google Scholar] [CrossRef]
- Yamashita, T.; Tucker, K.L.; Barde, Y.A. Neurotrophin binding to the p75 receptor modulates rho activity and axonal outgrowth. Neuron 1999, 24, 585–593. [Google Scholar] [CrossRef]
- Villarroel-Campos, D.; Bronfman, F.C.; Gonzalez-Billault, C. Rab gtpase signaling in neurite outgrowth and axon specification. Cytoskeleton (Hoboken N.J.) 2016, 73, 498–507. [Google Scholar] [CrossRef]
- Jaiswal, M.; Dvorsky, R.; Amin, E.; Risse, S.L.; Fansa, E.K.; Zhang, S.C.; Taha, M.S.; Gauhar, A.R.; Nakhaei-Rad, S.; Kordes, C.; et al. Functional cross-talk between ras and rho pathways: A ras-specific gtpase-activating protein (p120rasgap) competitively inhibits the rhogap activity of deleted in liver cancer (dlc) tumor suppressor by masking the catalytic arginine finger. J. Biol. Chem. 2014, 289, 6839–6849. [Google Scholar] [CrossRef]
- Nienhaus, G.U.; Maffre, P.; Nienhaus, K. Studying the protein corona on nanoparticles by fcs. Methods Enzym. 2013, 519, 115–137. [Google Scholar] [CrossRef]
- Shang, L.; Nienhaus, G.U. In situ characterization of protein adsorption onto nanoparticles by fluorescence correlation spectroscopy. Acc. Chem. Res. 2017, 50, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Lorber, B.; Fischer, F.; Bailly, M.; Roy, H.; Kern, D. Protein analysis by dynamic light scattering: Methods and techniques for students. Biochem. Mol. Biol. Educ. A Bimon. Publ. Int. Union Biochem. Mol. Biol. 2012, 40, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Reid, J.C.; Yang, Y.P. Utilizing dynamic light scattering as a process analytical technology for protein folding and aggregation monitoring in vaccine manufacturing. J. Pharm. Sci. 2013, 102, 4284–4290. [Google Scholar] [CrossRef] [PubMed]
- Karow, A.R.; Gotzl, J.; Garidel, P. Resolving power of dynamic light scattering for protein and polystyrene nanoparticles. Pharm. Dev. Technol. 2015, 20, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Chauhan, N.; Othman, S.F.; Khalilzad-Sharghi, V.; Ebeling, M.C.; Khan, S.; Jaggi, M.; Chauhan, S.C. Implications of protein corona on physico-chemical and biological properties of magnetic nanoparticles. Biomaterials 2015, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Satzer, P.; Svec, F.; Sekot, G.; Jungbauer, A. Protein adsorption onto nanoparticles induces conformational changes: Particle size dependency, kinetics, and mechanisms. Eng. Life Sci. 2016, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Lehnen, D.; Barral, S.; Cardoso, T.; Grealish, S.; Heuer, A.; Smiyakin, A.; Kirkeby, A.; Kollet, J.; Cremer, H.; Parmar, M.; et al. Iap-based cell sorting results in homogeneous transplantable dopaminergic precursor cells derived from human pluripotent stem cells. Stem Cell Rep. 2017, 9, 1207–1220. [Google Scholar] [CrossRef]
- Wei, M.; Li, S.; Yang, Z.; Zheng, W.; Le, W. Gold nanoparticles enhance the differentiation of embryonic stem cells into dopaminergic neurons via mtor/p70s6k pathway. Nanomedicine (Lond. Engl.) 2017, 12, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Lee, E.; Kim, H.; Youn, D.-H.; Jung, J.; Kim, H.; Chang, Y.; Lee, W.; Shin, J.; Baek, S.; et al. Electromagnetized gold nanoparticles mediate direct lineage reprogramming into induced dopamine neurons in vivo for parkinson’s disease therapy. Nat. Nanotechnol. 2017, 12, 1006–1014. [Google Scholar] [CrossRef]
- Chang, J.-H.; Tsai, P.-H.; Wang, K.-Y.; Wei, Y.-T.; Chiou, S.-H.; Mou, C.-Y. Generation of functional dopaminergic neurons from reprogramming fibroblasts by nonviral-based mesoporous silica nanoparticles. Sci. Rep. 2018, 8, 11. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Bjorklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, Z.; Zhong, P.; Ren, Y.; Liang, G.; Schilling, H.A.; Hu, Z.; Zhang, Y.; Wang, X.; Chen, S.; et al. Cell cycle and p53 gate the direct conversion of human fibroblasts to dopaminergic neurons. Nat. Commun. 2015, 6, 10100. [Google Scholar] [CrossRef] [PubMed]
- Mirakhori, F.; Zeynali, B.; Rassouli, H.; Salekdeh, G.H.; Baharvand, H. Direct conversion of human fibroblasts into dopaminergic neural progenitor-like cells using tat-mediated protein transduction of recombinant factors. Biochem. Biophys. Res. Commun. 2015, 459, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Paliga, D.; Raudzus, F.; Leppla, S.H.; Heumann, R.; Neumann, S. Lethal factor domain-mediated delivery of nurr1 transcription factor enhances tyrosine hydroxylase activity and protects from neurotoxin-induced degeneration of dopaminergic cells. Mol. Neurobiol. 2019, 56, 3393–3403. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an alzheimer’s disease model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef]
- Di Val Cervo, P.R.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Chen, W.; Huang, Q.; Ma, S.; Li, M. Progress in dopaminergic cell replacement and regenerative strategies for parkinson’s disease. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef]
- Khan, F.A.; Almohazey, D.; Alomari, M.; Almofty, S.A. Impact of nanoparticles on neuron biology: Current research trends. Int. J. Nanomed. 2018, 13, 2767–2776. [Google Scholar] [CrossRef]
- Li, Y.; Ju, D. The role of autophagy in nanoparticles-induced toxicity and its related cellular and molecular mechanisms. Adv. Exp. Med. Biol. 2018, 1048, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. Pegylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Etoc, F.; Balloul, E.; Vicario, C.; Normanno, D.; Liße, D.; Sittner, A.; Piehler, J.; Dahan, M.; Coppey, M. Non-specific interactions govern cytosolic diffusion of nanosized objects in mammalian cells. Nat. Mater. 2018, 17, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Duce, J.A.; Rascol, O.; Devedjian, J.C.; Berg, D.; Dexter, D.; Cabantchik, Z.I.; Bush, A.I.; Devos, D. Iron as a therapeutic target for parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Kan, H.; Sakurai, K.; Arai, N.; Kato, D.; Kawashima, S.; Ueki, Y.; Matsukawa, N. Voxel-based quantitative susceptibility mapping in parkinson’s disease with mild cognitive impairment. Mov. Disord. Off. J. Mov. Disord. Soc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Monzel, C. Magnetic control of cellular processes using biofunctional nanoparticles. Chem. Sci. 2017, 8, 7330–7338. [Google Scholar] [CrossRef] [PubMed]
- Rost, B.R.; Schneider-Warme, F.; Schmitz, D.; Hegemann, P. Optogenetic tools for subcellular applications in neuroscience. Neuron 2017, 96, 572–603. [Google Scholar] [CrossRef]
- Toettcher, J.E.; Weiner, O.D.; Lim, W.A. Using optogenetics to interrogate the dynamic control of signal transmission by the ras/erk module. Cell 2013, 155, 1422–1434. [Google Scholar] [CrossRef]
- Zhang, K.; Duan, L.; Ong, Q.; Lin, Z.; Varman, P.M.; Sung, K.; Cui, B. Light-mediated kinetic control reveals the temporal effect of the raf/mek/erk pathway in pc12 cell neurite outgrowth. PLoS ONE 2014, 9, e92917. [Google Scholar] [CrossRef]
- Goglia, A.G.; Wilson, M.Z.; DiGiorno, D.B.; Toettcher, J.E. Optogenetic control of ras/erk signaling using the phy-pif system. Methods Mol. Biol. 2017, 1636, 3–20. [Google Scholar] [CrossRef]
- Chen, S.; Weitemier, A.Z.; Zeng, X.; He, L.; Wang, X.; Tao, Y.; Huang, A.J.Y.; Hashimotodani, Y.; Kano, M.; Iwasaki, H.; et al. Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science 2018, 359, 679–684. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schöneborn, H.; Raudzus, F.; Secret, E.; Otten, N.; Michel, A.; Fresnais, J.; Ménager, C.; Siaugue, J.-M.; Zaehres, H.; Dietzel, I.D.; et al. Novel Tools towards Magnetic Guidance of Neurite Growth: (I) Guidance of Magnetic Nanoparticles into Neurite Extensions of Induced Human Neurons and In Vitro Functionalization with RAS Regulating Proteins. J. Funct. Biomater. 2019, 10, 32. https://doi.org/10.3390/jfb10030032
Schöneborn H, Raudzus F, Secret E, Otten N, Michel A, Fresnais J, Ménager C, Siaugue J-M, Zaehres H, Dietzel ID, et al. Novel Tools towards Magnetic Guidance of Neurite Growth: (I) Guidance of Magnetic Nanoparticles into Neurite Extensions of Induced Human Neurons and In Vitro Functionalization with RAS Regulating Proteins. Journal of Functional Biomaterials. 2019; 10(3):32. https://doi.org/10.3390/jfb10030032
Chicago/Turabian StyleSchöneborn, Hendrik, Fabian Raudzus, Emilie Secret, Nils Otten, Aude Michel, Jérome Fresnais, Christine Ménager, Jean-Michel Siaugue, Holm Zaehres, Irmgard D. Dietzel, and et al. 2019. "Novel Tools towards Magnetic Guidance of Neurite Growth: (I) Guidance of Magnetic Nanoparticles into Neurite Extensions of Induced Human Neurons and In Vitro Functionalization with RAS Regulating Proteins" Journal of Functional Biomaterials 10, no. 3: 32. https://doi.org/10.3390/jfb10030032
APA StyleSchöneborn, H., Raudzus, F., Secret, E., Otten, N., Michel, A., Fresnais, J., Ménager, C., Siaugue, J.-M., Zaehres, H., Dietzel, I. D., & Heumann, R. (2019). Novel Tools towards Magnetic Guidance of Neurite Growth: (I) Guidance of Magnetic Nanoparticles into Neurite Extensions of Induced Human Neurons and In Vitro Functionalization with RAS Regulating Proteins. Journal of Functional Biomaterials, 10(3), 32. https://doi.org/10.3390/jfb10030032