The Reasonable Effectiveness of Randomness in Scalable and Integrative Gene Regulatory Network Inference and Beyond



Abstract
1. Introduction
“… So there are very complex different ways that genes are regulated. I kind of look at it as playing music: You have chords on a guitar, or you play with a right and a left hand on the piano. It depends what strings you push down and what strings you strum, or what keys are up and what keys are down, that determine what the profile of the gene expression will be or the sound that you hear.”David M. Bodine [1]
2. The Molecular Biology of Gene Regulation and the Technologies Used to Study It
3. A Primer on Randomness and Randomized Algorithms
3.1. A Mathematical Theory of Randomness
The Theory of Computation and Kolmogorov’s Definition of Randomness
3.2. Randomized Algorithms
4. On the History and Current Applications of Randomness and Randomized Methods in Computational Biology and Gene Regulation Inference
4.1. Markov Chain Monte Carlo Based Sampling for Gene Regulatory Network Structure Selection
Integration of Prior Knowledge on Network Structures
4.2. Random Forest Regression Based Gene Regulatory Network Inference from Transcriptomic Data
Extension to Prior Knowledge Integration
4.3. Random Walks for Gene Module Detection in Biological Networks
| Algorithm 1 Markov Clustering |
| Input: Graph adjacency matrix A, Expansion parameter e, inflation parameter i Output: Transition matrix M // add self-loops to graph adjacency matrix A // initialize canonical transition matrix M while M has not converged do // Expansion for each do for each do // Inflation end for end for end while |
Regularized Markov Clustering and Random Walks with Restart
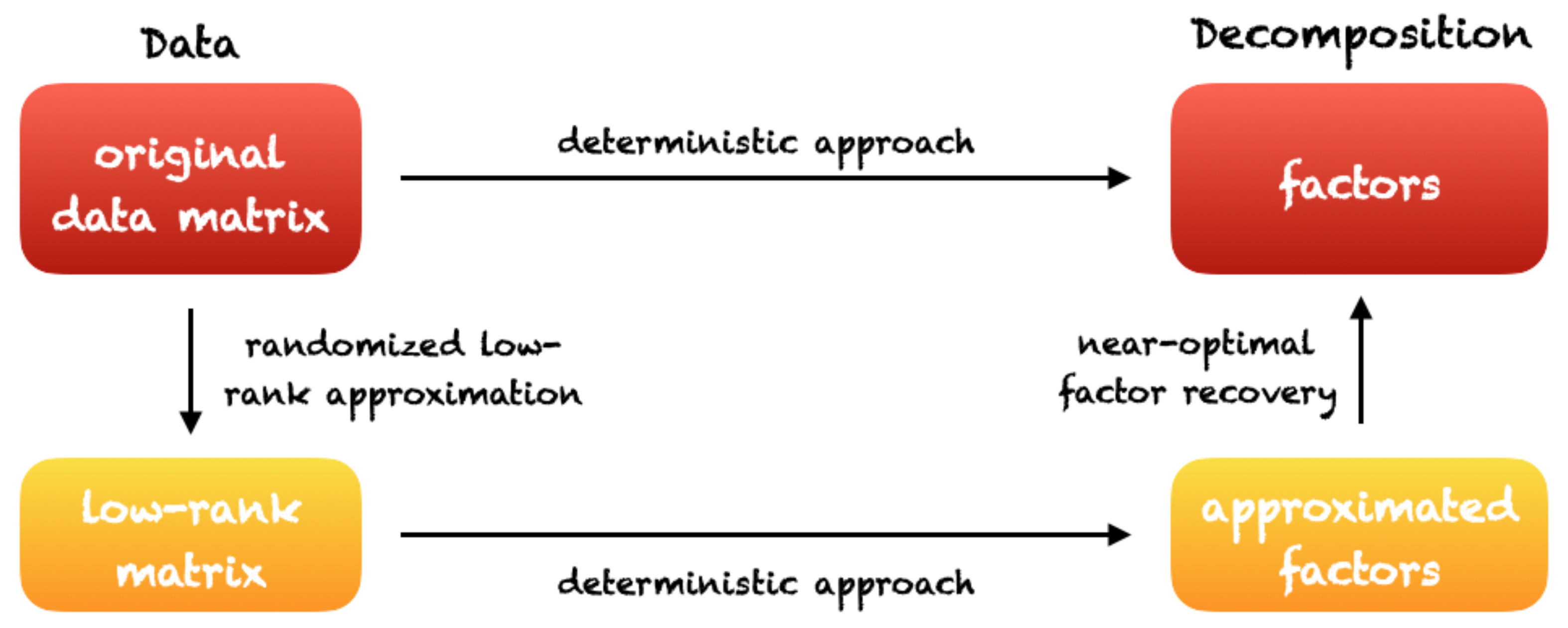
4.4. Randomized Matrix Factorizations and Low Rank Approximations with Applications to High-Dimensional Gene Clustering and Gene Regulatory Network Inference
4.4.1. Randomized Singular Value Decomposition
| Algorithm 2 Randomized Singular Value Decomposition. |
| Input: Data matrix , target rank k Output: Left and right singular vector matrices U and , singular value matrix // compute approximate basis , e.g., via Algorithm 3 // project to low-dimensional space // compute SVD of // recover left singular values // compute SVD of X |
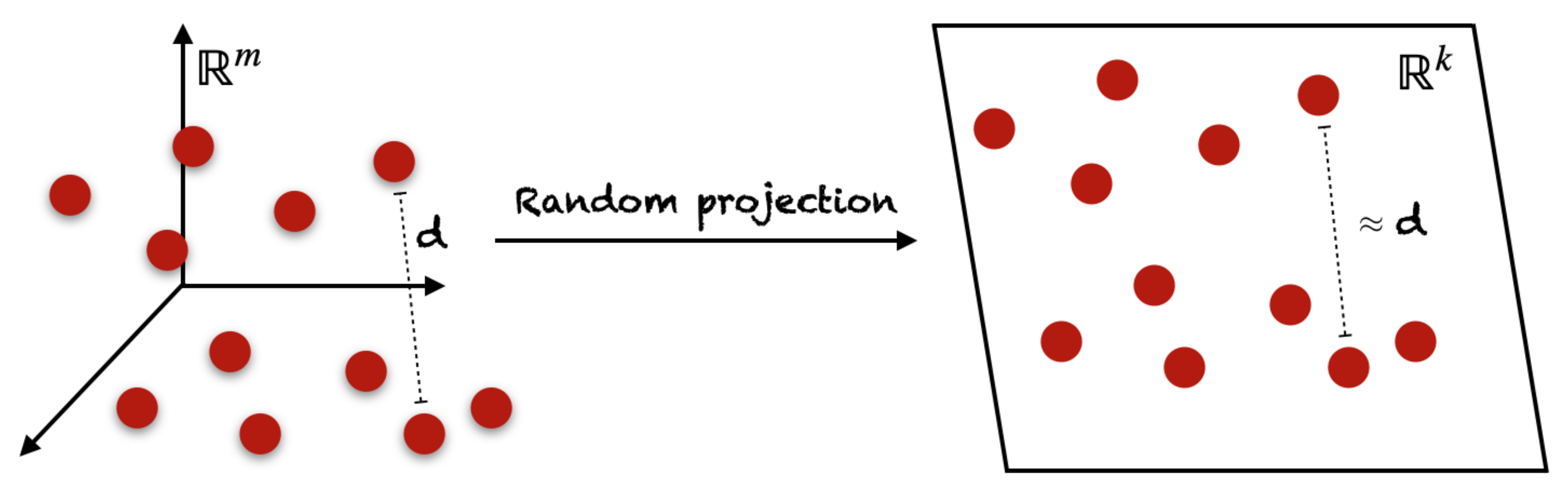
4.4.2. Random Projection
| Algorithm 3 Orthonormal basis estimation via Gaussian Random Projection. |
| Input: Data matrix , target rank k Output: Approximate basis // generate Gaussian random matrix // generate sketch // form orthonormal basis Q, e.g., using QR factorization |
4.4.3. Randomized Principal Component Analysis
| Algorithm 4 Randomized Principal Component Analysis (via SVD). |
| Input: Data matrix (centered, scaled), target rank Output: Eigenvector matrix , eigenvalue matrix , k principal component matrix // compute randomized SVD of X (Algorithm 2) // recover eigenvalues // compute k principal components |
5. A Note on Deep Learning Based Methods for Computational Biology
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bodine, D.M. Gene Regulation. In NIH Talking Glossary of Genetic Terms. Available online: https://www.genome.gov/genetics-glossary/Gene-Regulation (accessed on 20 August 2021).
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Krouk, G.; Lingeman, J.; Marshall-Colon, A.; Coruzzi, G.; Shasha, D. Gene regulatory networks in plants: Learning causality from time and perturbation. Genome Biol. 2013, 14, 123. [Google Scholar] [CrossRef]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Iwase, A.; Matsui, K.; Ohme-Takagi, M. Manipulation of plant metabolic pathways by transcription factors. Plant Biotechnol. 2009, 26, 29–38. [Google Scholar] [CrossRef]
- Muhammad, D.; Schmittling, S.; Williams, C.; Long, T. More than meets the eye: Emergent properties of transcription factors networks in Arabidopsis. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1860, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Maetschke, S.; Madhamshettiwar, P.; Davis, M.; Ragan, M. Supervised, semi-supervised and unsupervised inference of gene regulatory networks. Briefings Bioinform. 2013, 15, 195–211. [Google Scholar] [CrossRef]
- Marbach, D.; Costello, J.; Küffner, R.; Vega, N.; Prill, R.; Camacho, D.; Allison, K.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef]
- Banf, M.; Rhee, S. Computational inference of gene regulatory networks: Approaches, limitations and opportunities. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1860. [Google Scholar] [CrossRef]
- MacQuarrie, K.; Fong, A.; Morse, R.; Tapscott, S. Genome-wide transcription factor binding: Beyond direct target regulation. Trends Genet. TIG 2011, 27, 141–148. [Google Scholar] [CrossRef]
- Marbach, D.; Roy, S.; Ay, F.; Meyer, P.; Candeias, R.; Kahveci, T.; Bristow, C.; Kellis, M. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012, 22, 1334–1349. [Google Scholar] [CrossRef]
- Banf, M.; Rhee, S. Enhancing gene regulatory network inference through data integration with markov random fields. Sci. Rep. 2017, 7, 41174. [Google Scholar] [CrossRef]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Verleyen, W.; Ballouz, S.; Gillis, J. Measuring the wisdom of the crowds in network-based gene function inference. Bioinformatics 2014, 31, 745–752. [Google Scholar] [CrossRef]
- Lee, T.; Yang, S.; Kim, E.; Ko, Y.; Hwang, S.; Shin, J.; Shim, J.; Shim, H.; Kim, H.; Kim, C.; et al. AraNet v2: An improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucleic Acids Res. 2014, 43. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Yang, S.; Kim, E.; Kim, C.; Shim, H.; Cho, A.; Kim, H.; Hwang, S.; Shim, J.; Lee, I. FlyNet: A versatile network prioritization server for the Drosophila community. Nucleic Acids Res. 2015, 43, W91–W97. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Nanjundiah, V. Barbara McClintock and the discovery of jumping genes. Resonance 1996, 1, 56–62. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–0356. [Google Scholar] [CrossRef]
- Martín-Arevalillo, R.; Nanao, M.; Larrieu, A.; Vinos-Poyo, T.; Mast, D.; Galvan-Ampudia, C.; Brunoud, G.; Vernoux, T.; Dumas, R.; Parcy, F. Structure of the Arabidopsis TOPLESS corepressor provides insight into the evolution of transcriptional repression. Proc. Natl. Acad. Sci. USA 2017, 114, 201703054. [Google Scholar] [CrossRef] [PubMed]
- Park, P. ChIP-Seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Furey, T. ChIP-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, L.; Wu, D.; Chen, G. Advances in bulk and single-cell multi-omics approaches for systems biology and precision medicine. Briefings Bioinform. 2021, 22, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Ma, K. Transcription Factors. Wikipedia. 2021. Available online: https://commons.wikimedia.org/wiki/File:Transcription_Factors.svg (accessed on 20 August 2021).
- Herz, H.M.; Hu, D.; Shilatifard, A. Enhancer Malfunction in Cancer. Mol. Cell 2014, 53, 859–866. [Google Scholar] [CrossRef]
- Herz, H.M. Enhancer deregulation in cancer and other diseases. BioEssays 2016, 38. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Denker, A.; Laat, W. The second decade of 3C technologies: Detailed insights into nuclear organization. Genes Dev. 2016, 30, 1357–1382. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Berkum, N.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.; Sabo, P.; Dorschner, M.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.; Rubin, A.; Flynn, R.; Dai, C.; Khavari, P.; Greenleaf, W.; Chang, H. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Fullwood, M.; Ruan, Y. ChIP-Based Methods for the Identification of Long-Range Chromatin Interactions. J. Cell. Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef]
- Casamassimi, A.; Ciccodicola, A. Transcriptional Regulation: Molecules, Involved Mechanisms, and Misregulation. Int. J. Mol. Sci. 2019, 20, 1281. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, S.; Ng, N.; Dong, S.; Porter, R.; Morterud, C.; Williams, C.; Asman, C.; Switzenberg, J.; Boyle, A. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics 2019, 36, 364–372. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Brandi, N.; Hata, A. MicroRNA in Cancer The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer 2010, 1, 1100–1114. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.; Beech, J.; Kulshreshtha, R.; Abdelmohsen, K.; Weinstock, D.; Gorospe, M.; et al. MiR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef]
- Schep, A.N.; Buenrostro, J.D.; Denny, S.K.; Schwartz, K.; Sherlock, G.; Greenleaf, W.J. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015, 25, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Lamparter, D.; Marbach, D.; Rueedi, R.; Bergmann, S.; Kutalik, Z. Genome-Wide Association between Transcription Factor Expression and Chromatin Accessibility Reveals Regulators of Chromatin Accessibility. PLoS Comput. Biol. 2017, 13, e1005311. [Google Scholar] [CrossRef]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.; Garcia, S.; Clement, K.; Andrade, M.; Buenrostro, J.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.; Kidner, C.; Hall, I.; Teng, G.; Grewal, S.; Martienssen, R. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef]
- Bannister, A.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Guertin, M.; Lis, J. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr. Opin. Genet. Dev. 2012, 23, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G. DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5384. [Google Scholar] [CrossRef]
- Buenrostro, J.; Giresi, P.; Zaba, L.; Chang, H.; Greenleaf, W. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Savadel, S.; Hartwig, T.; Turpin, Z.; Vera, D.; Lung, P.Y.; Sui, X.; Blank, M.; Frommer, W.; Dennis, J.; Zhang, J.; et al. The native cistrome and sequence motif families of the maize ear. PLoS Genet. 2021, 17, e1009689. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2014, 16, 258–264. [Google Scholar] [CrossRef]
- Niederhuth, C.; Schmitz, R. Putting DNA methylation in context: From genomes to gene expression in plants. Biochim. Biophys. Acta 2016, 1860, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M.; Zhenyuan, L.; Kendall, J.; Donoghue, M.; Reinders, J.; Llaca, V.; Deschamps, S.; Smith, A.; Levy, D.; Mccombie, W.; et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013, 23, 1651–1662. [Google Scholar] [CrossRef]
- Yong Syuan, C.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef]
- Harris, R.; Wang, T.; Coarfa, C.; Nagarajan, R.; Hong, C.; Downey, S.; Johnson, B.; Fouse, S.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Lander, E.; Altshuler, D.; Daly, M.; Grossman, S.; Jaffe, D.; Korn, J. A map of human genome variation from population-scale sequencing. Nature 2012, 457, 1061. [Google Scholar]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-Specific Effects of Genetic and Epigenetic Variation on Gene Regulation and Splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Lazar, M. Shining light on dark matter in the genome. Proc. Natl. Acad. Sci. USA 2019, 116, 201918894. [Google Scholar] [CrossRef]
- Broekema, R.; Bakker, O.; Jonkers, I. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020, 10, 190221. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Kim, S.; Zhi, D.; Cui, X. Predicting gene expression using DNA methylation in three human populations. PeerJ 2019, 7, e6757. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Banf, M.; Prietsch, G.; Engelhorn, J.; Yang, J.; Wang, Z.Y. Hybrid allele-specific ChIP-Seq analysis links variation in transcription factor binding to traits in maize. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Zarayeneh, N.; Ko, E.; Oh, J.H.; Suh, S.; Liu, C.; Gao, J.; Kim, D.; Kang, M. Integration of multi-omics data for integrative gene regulatory network inference. Int. J. Data Min. Bioinform. 2017, 18, 223. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.P.; Bodein, A.; Périn, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021, 19. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Rehani, P.; Ying, M.; Huang, J.; Liu, S.; Roussos, P.; Wang, D. scGRNom: A computational pipeline of integrative multi-omics analyses for predicting cell-type disease genes and regulatory networks. Genome Med. 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Graw, S.; Chappell, K.; Washam, C.; Gies, A.; Bird, J.; Robeson, M.; Byrum, S. Multi-omics data integration considerations and study design for biological systems and disease. Mol. Omics 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayanan, A.; Gupta, R.; Thompson, E.; Nyholt, D.; Bauer, D.; Nagaraj, S. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Briefings Bioinform. 2019, 21, 1920–1936. [Google Scholar] [CrossRef]
- Blencowe, M.; Arneson, D.; Ding, J.; Chen, Y.W.; Saleem, Z.; Yang, X. Network modeling of single-cell omics data: Challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019, 3, ETLS20180176. [Google Scholar] [CrossRef]
- Hasin-Brumshtein, Y.; Seldin, M.; Lusis, A. Multi-omics Approaches to Disease. Genome Biol. 2017, 18, 1–5. [Google Scholar] [CrossRef]
- Suravajhala, P.; Kogelman, L.; Kadarmideen, H. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications In animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, J.; Zhang, Y.; Wang, J. Discover novel disease-associated genes based on regulatory networks of long-range chromatin interactions. Methods 2020, 189, 22–33. [Google Scholar] [CrossRef]
- Laplace, P.S. A Philosophical Essay on Probabilities, 1st ed.; John Wiley & Sons: New York, NY, USA, 1902. [Google Scholar]
- Gleick, J. Chaos: Making a New Science; Viking: New York, NY, USA, 1987; p. 352. [Google Scholar]
- ’t Hooft, G. Entangled quantum states in a local deterministic theory. arXiv 2009, arXiv:0908.3408. [Google Scholar]
- Einstein, A.; Born, M. Briefwechsel 1916–1955; Rowohlt: Hamburg, Germany, 1972. [Google Scholar]
- Born, M. Zur Quantenmechanik der Stoßvorgänge. Zeitschrift Physik 1926, 37, 863–867. [Google Scholar] [CrossRef]
- Bera, M.; Acín, A.; Kuś, M.; Mitchell, M.; Lewenstein, M. Randomness in Quantum Mechanics: Philosophy, Physics and Technology. Rep. Prog. Phys. 2016, 80, 124001. [Google Scholar] [CrossRef]
- Landsman, K. Randomness? What Randomness? Found. Phys. 2020, 50, 61–104. [Google Scholar] [CrossRef]
- Osborne, M.; Rubinstein, A. A Course in Game Theory; MIT Press: Cambridge, UK, 1994; Volume 63. [Google Scholar] [CrossRef]
- Moreh, J. Randomness, game theory and free will. Erkenntnis 1994, 41, 49–64. [Google Scholar] [CrossRef]
- Heams, T. Randomness in Biology. Math. Struct. Comp. Sci. Spec. Issue 2014, 24. [Google Scholar] [CrossRef]
- Kaplan, R.W. Th. Dobzhansky, F. J. Ayala, G. L. Stebbins, and J. W. Valentine. Evolution. 572 S., 123 Zeichnungen. Schemata und Kurven. San Francisco 1977. H. W. Freeman & Co. Ltd. £ 18.60. J. Basic Microbiol. 1979, 19, 228–229. [Google Scholar] [CrossRef]
- Mayo, O. A Century of Hardy–Weinberg Equilibrium. Twin Res. Hum. Genet. Off. J. Int. Soc. Twin Stud. 2008, 11, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Chown, M. The Omega Man. New Scientist Magazine, 10 March 2001. [Google Scholar]
- Terwijn, S.A. The Mathematical Foundations of Randomness; Springer International Publishing: Cham, Switzerland, 2016; pp. 49–66. [Google Scholar] [CrossRef]
- Mises, R. Grundlagen der Wahrscheinlichkeitsrechnung. Math. Z. 1919, 5, 52–99. [Google Scholar] [CrossRef]
- Wald, A. Die Widerspruchsfreiheit des Kollektivbegriffes. Actualités Sci. Indust. 1938, 735. [Google Scholar]
- Church, A. On the Concept of a Random Sequence. Bull. Am. Math. Soc. 1940, 46, 130–135. [Google Scholar] [CrossRef]
- Plato, J. AN Kolmogorov, Grundbegriffe der wahrscheinlichkeitsrechnung (1933). In Landmark Writings in Western Mathematics 1640–1940; Elsevier Science: Amsterdam, The Netherlands, 2005; pp. 960–969. [Google Scholar] [CrossRef]
- Martin-Löf, P. The Definition of Random Sequences. Inf. Control. 1966, 9, 602–619. [Google Scholar] [CrossRef]
- Downey, R.; Hirschfeldt, D. Algorithmic randomness. Commun. ACM 2019, 62, 70–80. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Gödel, K. Über formal unentscheidbare Sätze der Principia Mathematica und Verwandter Systeme I. Monatshefte Math. Und Phys. 1931, 38, 173–198. [Google Scholar] [CrossRef]
- Turing, A. On Computable Numbers, with an Application to the Entscheidungsproblem. Proc. Lond. Math. Soc. 1938, 43. [Google Scholar] [CrossRef]
- Li, M.; Vitányi, P. An Introduction to Kolmogorov Complexity and Its Applications; Springer: Berlin, Germany, 2019. [Google Scholar] [CrossRef]
- Motwani, R.; Raghavan, P. Randomized Algorithms. ACM Comput. Surv. (CSUR) 1995, 28. [Google Scholar] [CrossRef]
- Metropolis, N. The beginning of the Monte Carlo method. Los Alamos Sci. 1987, 125–130. [Google Scholar]
- Cipra, B. The best of the 20th century: Editors name Top 10 Algorithms. SIAM News 2000, 33, 1–2. [Google Scholar]
- List, B.; Maucher, M.; Schöning, U.; Schuler, R. Randomized QuickSort and the Entropy of the Random Source. Lect. Notes Comput. Sci. 2005, 3595, 450–460. [Google Scholar] [CrossRef]
- Karger, D.; Stein, C. A New Approach to the Minimum Cut Problem. J. ACM 1996, 43, 601–640. [Google Scholar] [CrossRef]
- Karp, R. An introduction to randomized algorithms. Discret. Appl. Math. 1991, 34, 165–201. [Google Scholar] [CrossRef]
- Sharma, K.; Garg, D. Randomized Algorithms: Methods and Techniques. Int. J. Comput. Appl. 2011, 28. [Google Scholar] [CrossRef]
- Sipser, M. Introduction to the Theory of Computation; Cengage Learning: Boston, MA, USA, 1997. [Google Scholar]
- Aitken, S.; Akman, O. Nested sampling for parameter inference in systems biology: Application to an exemplar circadian model. BMC Syst. Biol. 2013, 7, 72. [Google Scholar] [CrossRef]
- Aalto, A.; Viitasaari, L.; Ilmonen, P.; Mombaerts, L.; Goncalves, J. Gene regulatory network inference from sparsely sampled noisy data. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Bernardi, R.; Cardoso dos Reis Melo, M.; Schulten, K. Enhanced Sampling Techniques in Molecular Dynamics Simulations of Biological Systems. Biochim. Biophys. Acta 2014, 1850, 872–877. [Google Scholar] [CrossRef]
- Johnson, R.; Kirk, P.; Stumpf, M. SYSBIONS: Nested sampling for systems biology. Bioinformatics 2014, 31, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Ye, Y.; Wu, Q.; Huang, J.; Ng, M.; Li, X. Stratified sampling for feature subspace selection in random forests for high dimensional data. Pattern Recognit. 2013, 46, 769–787. [Google Scholar] [CrossRef]
- Halko, N.; Martinsson, P.G.; Tropp, J. Finding Structure with Randomness: Probabilistic Algorithms for Constructing Approximate Matrix Decompositions. SIAM Rev. 2011, 53, 217–288. [Google Scholar] [CrossRef]
- Yau, C.; Campbell, K. Bayesian statistical learning for big data biology. Biophys. Rev. 2019, 11, 95–102. [Google Scholar] [CrossRef]
- Ram, R.; Chetty, M. MCMC Based Bayesian Inference for Modeling Gene Networks. In Pattern Recognition in Bioinformatics; Kadirkamanathan, V., Sanguinetti, G., Girolami, M.A., Niranjan, M., Noirel, J., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5780, pp. 293–306. [Google Scholar]
- Lee, J.; Sung, W.; Choi, J.-H. Metamodel for Efficient Estimation of Capacity-Fade Uncertainty in Li-Ion Batteries for Electric Vehicles. Energies 2015, 6, 5538–5554. [Google Scholar] [CrossRef]
- Ko, Y.; Kim, J.; Rodriguez-Zas, S. Markov chain Monte Carlo simulation of a Bayesian mixture model for gene network inference. Genes Genom. 2019, 41, 547–555. [Google Scholar] [CrossRef]
- Agostinho, N.; Machado, K.; Werhli, A. Inference of regulatory networks with a convergence improved MCMC sampler. BMC Bioinform. 2015, 16, 306. [Google Scholar] [CrossRef] [PubMed]
- Low, S.; Mohamad, M.; Omatu, S.; Chai, L.E.; Bin Deris, S.; Yoshioka, M. Inferring gene regulatory networks from perturbed gene expression data using a dynamic Bayesian network with a Markov Chain Monte Carlo algorithm. In Proceedings of the 2014 IEEE International Conference on Granular Computing, GrC, Noboribetsu, Japan, 22–24 October 2014; pp. 179–184. [Google Scholar] [CrossRef]
- Buhler, J.; Tompa, M. Finding Motifs Using Random Projections. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2002, 9, 225–242. [Google Scholar] [CrossRef]
- Wang, L.; Dong, L. Randomized algorithms for motif detection. J. Bioinform. Comput. Biol. 2005, 3, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.S.; Ju, H.; Jung, H.J. Adaptive Markov chain Monte Carlo algorithms for Bayesian inference: Recent advances and comparative study. Struct. Infrastruct. Eng. 2019, 15, 1548–1565. [Google Scholar] [CrossRef]
- Werhli, A.; Husmeier, D. Gene Regulatory Network Reconstruction by Bayesian Integration of Prior Knowledge and/or Different Experimental Conditions. J. Bioinform. Comput. Biol. 2008, 6, 543–572. [Google Scholar] [CrossRef]
- Barreto, N.M.; dos Santos Machado, K.; Werhli, A.V. Inference of regulatory networks with MCMC sampler guided by mutual information. Proc. Symp. Appl. Comput. 2017, 18–23. [Google Scholar]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef]
- Qi, Y. Random Forest for Bioinformatics; Springer: Boston, MA, USA, 2012; pp. 307–323. [Google Scholar] [CrossRef]
- Pratapa, A.; Jalihal, A.; Law, J.; Bharadwaj, A.; Murali, T. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 2020, 17, 1–8. [Google Scholar] [CrossRef]
- Stephan, J.; Stegle, O.; Beyer, A. A random forest approach to capture genetic effects in the presence of population structure. Nat. Commun. 2015, 6, 7432. [Google Scholar] [CrossRef]
- Svetlichnyy, D.; Imrichova, H.; Fiers, M.; Kalender Atak, Z.; Aerts, S. Identification of High-Impact cis-Regulatory Mutations Using Transcription Factor Specific Random Forest Models. PLoS Comput. Biol. 2015, 11, e1004590. [Google Scholar] [CrossRef] [PubMed]
- Choobdar, S.; Ahsen, M.; Crawford, J.; Tomasoni, M.; Fang, T.; Lamparter, D.; Lin, J.; Hescott, B.; Hu, X.; Mercer, J.; et al. Assessment of network module identification across complex diseases. Nat. Methods 2018. [Google Scholar] [CrossRef]
- Satuluri, V.; Parthasarathy, S.; Ucar, D. Markov Clustering of Protein Interaction Networks with Improved Balance and Scalability. In Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology, Niagara Falls, NY, USA, 2–4 August 2010; pp. 247–256. [Google Scholar] [CrossRef]
- Enright, A.; Dongen, S.; Ouzounis, C. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Drineas, P.; Mahoney, M.W. RandNLA: Randomized Numerical Linear Algebra. Commun. ACM 2016, 59, 80–90. [Google Scholar] [CrossRef]
- Mahoney, M.; Drineas, P. Structural Properties Underlying High-Quality Randomized Numerical Linear Algebra Algorithms. In Handbook of Big Data; Chapman and Hall/CRC: London, UK, 2016; pp. 137–154. [Google Scholar]
- Wan, S.; Kim, J.; Won, K. SHARP: Hyper-fast and accurate processing of single-cell RNA-seq data via ensemble random projection. Genome Res. 2020, 30, gr.254557.119. [Google Scholar] [CrossRef] [PubMed]
- Anjing, F.; Wang, H.; Xiang, H.; Zou, X. Inferring Large-Scale Gene Regulatory Networks Using a Randomized Algorithm Based on Singular Value Decomposition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 1997–2008. [Google Scholar] [CrossRef]
- Brooks, S.; Gelman, A.; Jones, G.; Meng, X.L. Handbook of Markov Chain Monte Carlo; Chapman and Hall/CRC: London, UK, 2011; pp. 1–592. [Google Scholar]
- Hastings, W. Monte Carlo Sampling Methods Using Markov Chains and Their Application. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Chib, S. Understanding the Metropolis-Hastings Algorithm. Am. Stat. 1995, 49, 327–335. [Google Scholar] [CrossRef]
- Betancourt, M. A Conceptual Introduction to Hamiltonian Monte Carlo. arXiv 2017, arXiv:1701.02434. [Google Scholar]
- Blei, D.; Kucukelbir, A.; McAuliffe, J. Variational Inference: A Review for Statisticians. J. Am. Stat. Assoc. 2016, 112, 859–877. [Google Scholar] [CrossRef]
- Efroymson, M. Multiple Regression Analysis; John Wiley: New York, NY, USA, 1960; pp. 192–203. [Google Scholar]
- Hoerl, A.; Kennard, R. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 2012, 12, 55–67. [Google Scholar] [CrossRef]
- Efron, B.; Hastie, T.; Johnstone, L.; Tibshirani, R. Least Angle Regression. Ann. Stat. 2002, 32, 407–499. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B (Methodological) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Geurts, P. Unsupervised Gene Network Inference with Decision Trees and Random Forests: Methods and Protocols. Methods Mol. Biol. 2019, 195–215. [Google Scholar] [CrossRef]
- Maduranga, D.A.K.; Zheng, J.; Mundra, P.A.; Rajapakse, J.C. Inferring Gene Regulatory Networks from Time-Series Expressions Using Random Forests Ensemble. In Pattern Recognition in Bioinformatics; Ngom, A., Formenti, E., Hao, J.K., Zhao, X.M., van Laarhoven, T., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2013; Volume 7986, pp. 13–22. [Google Scholar]
- Huynh-Thu, V.A.; Sanguinetti, G. Combining tree-based and dynamical systems for the inference of gene regulatory networks. Bioinformatics 2015, 31, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Petralia, F.; Wang, P.; Yang, J.; Tu, Z. Integrative random forest for gene regulatory network inference. Bioinformatics 2015, 31, i197–i205. [Google Scholar] [CrossRef] [PubMed]
- Cliff, A.; Romero, J.; Kainer, D.; Walker, A.M.; Furches, A.; Jacobson, D.A. A High-Performance Computing Implementation of Iterative Random Forest for the Creation of Predictive Expression Networks. Genes 2019, 10, 996. [Google Scholar] [CrossRef]
- Dai, H. Perfect sampling methods for random forests. Adv. Appl. Probab. 2008, 40, 897–917. [Google Scholar] [CrossRef][Green Version]
- Huynh-Thu, V.A.; Geurts, P. DynGENIE3: Dynamical GENIE3 for the inference of gene networks from time series expression data. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Awek, J.; Arodz, T. ENNET: Inferring large gene regulatory networks from expression data using gradient boosting. BMC Syst. Biol. 2013, 7, 106. [Google Scholar] [CrossRef]
- Aibar, S.; Bravo González-Blas, C.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.; Shin, W.; Han, S.; Jeon, M.; Jang, H.; Jang, I.S.; Kang, J. BTNET: Boosted tree based gene regulatory network inference algorithm using time-course measurement data. BMC Syst. Biol. 2018, 12, 20. [Google Scholar] [CrossRef]
- Zheng, R.; Li, M.; Chen, X.; Wu, F.X.; Pan, Y.; Wang, J. BiXGBoost: A scalable, flexible boosting based method for reconstructing gene regulatory networks. Bioinformatics 2018, 35, 1893–1900. [Google Scholar] [CrossRef]
- Dimitrakopoulos, G. XGRN: Reconstruction of Biological Networks Based on Boosted Trees Regression. Computation 2021, 9, 48. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R. A Short Introduction to Boosting. J. Jpn. Soc. Artif. Intell. 1999, 14, 771–780. [Google Scholar]
- Roy, S.; Lagree, S.; Hou, Z.; Thomson, J.; Stewart, R.; Gasch, A. Integrated Module and Gene-Specific Regulatory Inference Implicates Upstream Signaling Networks. PLoS Comput. Biol. 2013, 9, e1003252. [Google Scholar] [CrossRef] [PubMed]
- Reiss, D.; Plaisier, C.; Wu, W.; Baliga, N. cMonkey2: Automated, systematic, integrated detection of co-regulated gene modules for any organism. Nucleic Acids Res. 2015, 43, e87. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Pavlopoulos, G.; Ouzounis, C.; Kyrpides, N.; Buluç, A. HipMCL: A high-performance parallel implementation of the Markov clustering algorithm for large-scale networks. Nucleic Acids Res. 2018, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rosvall, M.; Bergstrom, C. Maps of Random Walks on Complex Networks Reveal Community Structure. Proc. Natl. Acad. Sci. USA 2008, 105, 1118–1123. [Google Scholar] [CrossRef]
- Ramesh, A.; Trevino, R.; Von Hoff, D.; Kim, S. Clustering context-specific gene regulatory networks. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 2010, 444–455. [Google Scholar] [CrossRef]
- Ginanjar, R.; Bustamam, A.; Tasman, H. Implementation of regularized Markov clustering algorithm on protein interaction networks of schizophrenia’s risk factor candidate genes. In Proceedings of the 2016 International Conference on Advanced Computer Science and Information Systems (ICACSIS), Malang, Indonesia, 15–16 October 2016; pp. 297–302. [Google Scholar]
- Shih, Y.K.; Parthasarathy, S. Identifying functional modules in interaction networks through overlapping Markov clustering. Bioinformatics 2012, 28, i473–i479. [Google Scholar] [CrossRef]
- Valdeolivas, A.; Tichit, L.; Navarro, C.; Perrin, S.; Odelin, G.; Lévy, N.; Cau, P.; Remy, E.; Baudot, A. Random Walk with Restart on Multiplex and Heterogeneous Biological Networks. Bioinformatics 2018, 35, 497–505. [Google Scholar] [CrossRef]
- Liu, W.; Sun, X.; Peng, L.; Zhou, L.; Lin, H.; Jiang, Y. RWRNET: A Gene Regulatory Network Inference Algorithm Using Random Walk With Restart. Front. Genet. 2020, 11, 591461. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yan, G. Drug-target interaction prediction by random walk on the heterogeneous network. Mol. bioSyst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef]
- Chen, M.; Liao, B.; Li, Z. Global Similarity Method Based on a Two-tier Random Walk for the Prediction of microRNA–Disease Association. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Liu, L.; Hawkins, D.; Ghosh, S.; Young, S. Robust Singular Value Decomposition Analysis of Microarray Data. Proc. Natl. Acad. Sci. USA 2003, 100, 13167–13172. [Google Scholar] [CrossRef]
- Wall, M.; Rechtsteiner, A.; Rocha, L. Singular Value Decomposition and Principal Component Analysis. Pract. Approach Microarray Data Anal. 2002, 5, 91–109. [Google Scholar] [CrossRef]
- Brunet, J.P.; Tamayo, P.; Golub, T.; Mesirov, J. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef]
- Devarajan, K. Nonnegative Matrix Factorization: An Analytical and Interpretive Tool in Computational Biology. PLoS Comput. Biol. 2008, 4, e1000029. [Google Scholar] [CrossRef]
- Frigyesi, A.; Höglund, M. Non-Negative Matrix Factorization for the Analysis of Complex Gene Expression Data: Identification of Clinically Relevant Tumor Subtypes. Cancer Informat. 2008, 6, 275–292. [Google Scholar] [CrossRef]
- Liao, J.; Boscolo, R.; Yang, Y.L.; Tran, L.; Sabatti, C.; Roychowdhury, V. Network component analysis: Reconstruction of regulatory signals in biological systems. Proc. Natl. Acad. Sci. USA 2004, 100, 15522–15527. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.J.; Galbraith, S.; Liao, J.; Eskin, E. Using Network Component Analysis to Dissect Regulatory Networks Mediated by Transcription Factors in Yeast. PLoS Comput. Biol. 2009, 5, e1000311. [Google Scholar] [CrossRef]
- Siqi, W.; Joseph, A.; Hammonds, A.; Celniker, S.; Yu, B.; Frise, E. Stability-driven nonnegative matrix factorization to interpret Spatial gene expression and build local gene networks. Proc. Natl. Acad. Sci. USA 2016, 113, 201521171. [Google Scholar] [CrossRef]
- Ochs, M.; Fertig, E. Matrix Factorization for Transcriptional Regulatory Network Inference. In Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA, USA, 9–12 May 2012; pp. 387–396. [Google Scholar] [CrossRef]
- Wani, N.; Raza, K. iMTF-GRN: Integrative Matrix Tri-Factorization for Inference of Gene Regulatory Networks. IEEE Access 2019, 7, 126154–126163. [Google Scholar] [CrossRef]
- Baiyi, A.; Wei, S. A novel gene regulatory network construction method based on singular value decomposition. In Proceedings of the 2016 IEEE International Conference on Big Data Analysis (ICBDA), Hangzhou, China, 12–14 March 2016; pp. 1–4. [Google Scholar]
- He, Y.; Chhetri, S.; Arvanitis, M.; Srinivasan, K.; Aguet, F.; Ardlie, K.; Barbeira, A.; Bonazzola, R.; Im, H.; Brown, C.; et al. Sn-spMF: Matrix factorization informs tissue-specific genetic regulation of gene expression. Genome Biol. 2020, 21, 235. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational Drug Repositioning using Low-Rank Matrix Approximation and Randomized Algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef]
- Chen, M.; Zeleznik, O.; Thallinger, G.; Kuster, B.; Moghaddas Gholami, A.; Culhane, A. Dimension reduction techniques for the integrative analysis of multi-omics data. Briefings Bioinform. 2016, 17, bbv108. [Google Scholar] [CrossRef]
- Stein-O’Brien, G.; Arora, R.; Culhane, A.; Favorov, A.; Garmire, L.; Greene, C.; Goff, L.; Li, Y.; Ngom, A.; Ochs, M.; et al. Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends Genet. 2018, 34, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Drineas, P.; Kannan, R.; Mahoney, M. Fast Monte Carlo Algorithms for Matrices II: Computing a Low-Rank Approximation to a Matrix. SIAM J. Comput. 2004, 36, 158–183. [Google Scholar] [CrossRef]
- Liberty, E.; Woolfe, F.; Martinsson, P.G.; Rokhlin, V.; Tygert, M. Randomized algorithms for the low-rank approximation of matrices. Proc. Natl. Acad. Sci. USA 2008, 104, 20167–20172. [Google Scholar] [CrossRef] [PubMed]
- Erichson, N.; Voronin, S.; Brunton, S.; Kutz, J. Randomized Matrix Decompositions Using R. J. Stat. Softw. 2019, 89. [Google Scholar] [CrossRef]
- Eckart, C.; Young, G. The Approximation of One Matrix by Another of Lower Rank. Psychometrika 1936, 1, 211–218. [Google Scholar] [CrossRef]
- Kumar, N.; Schneider, J. Literature survey on low rank approximation of matrices. Linear Multilinear Algebra 2016, 65. [Google Scholar] [CrossRef]
- Bingham, E.; Mannila, H. Random projection in dimensionality reduction: Applications to image and text data. In Proceedings of the Seventh ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 26–29 August 2001. [Google Scholar] [CrossRef]
- Johnson, W.; Lindenstrauss, J. Extensions of Lipschitz mappings into a Hilbert space. Conf. Mod. Anal. Probab. 1982, 26, 189–206. [Google Scholar]
- Xie, H.; Li, J.; Qiaosheng, Z.; Wang, Y. Comparison among dimensionality reduction techniques based on Random Projection for cancer classification. Comput. Biol. Chem. 2016, 65, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T. Principal Component Analysis. In Springer Series in Statistics; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Saad, Y. Numerical Methods for Large Eigenvalue Problems; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 2011. [Google Scholar]
- Agrawal, A.; Chiu, A.; Le, M.; Halperin, E.; Sankararaman, S. Scalable probabilistic PCA for large-scale genetic variation data. PLoS Genet. 2020, 16, e1008773. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, K.; Bhatia, G.; Loh, P.R.; Georgiev, S.; Mukherjee, S.; Patterson, N.; Price, A. Fast Principal-Component Analysis Reveals Convergent Evolution of ADH1B in Europe and East Asia. Am. J. Hum. Genet. 2016, 98, 456–472. [Google Scholar] [CrossRef]
- Abraham, G.; Inouye, M. Fast Principal Component Analysis of Large-Scale Genome-Wide Data. PLoS ONE 2014, 9, e93766. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 1–4. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.; Frey, B. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Zeng, H.; Edwards, M.; Liu, G.; Gifford, D. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef]
- Long-Chen, S.; Liu, Y.; Song, J.; Yu, D.J. SAResNet: Self-attention residual network for predicting DNA-protein binding. Briefings Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Yuan, Y.; Bar-Joseph, Z. GCNG: Graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020, 21, 1–6. [Google Scholar] [CrossRef]
- Knauer-Arloth, J.; Eraslan, G.; Andlauer, T.; Martins, J.; Iurato, S.; Kühnel, B.; Waldenberger, M.; Frank, J.; Gold, R.; Hemmer, B.; et al. DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLoS Comput. Biol. 2020, 16, e1007616. [Google Scholar] [CrossRef]
- Qin, Q.; Feng, J. Imputation for transcription factor binding predictions based on deep learning. PLoS Comput. Biol. 2017, 13, e1005403. [Google Scholar] [CrossRef]
- Wang, X.; Dizaji, K.; Huang, H. Conditional generative adversarial network for gene expression inference. Bioinformatics 2018, 34, i603–i611. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V.; Gayoso, A.; Yosef, N.; Pachter, L. Interpretable factor models of single-cell RNA-seq via variational autoencoders. Bioinformatics 2020, 36, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Simidjievski, N.; Bodnar, C.; Tariq, I.; Scherer, P.; Andres Terre, H.; Shams, Z.; Jamnik, M.; Lio, P. Variational Autoencoders for Cancer Data Integration: Design Principles and Computational Practice. Front. Genet. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Timotheou, S. A novel weight initialization method for the random neural network. Neurocomputing 2009, 73, 160–168. [Google Scholar] [CrossRef]
- Hao, Y.; Mu, T.; Hong, R.; Wang, M.; Liu, X.; Goulermas, J. Cross-Domain Sentiment Encoding through Stochastic Word Embedding. IEEE Trans. Knowl. Data Eng. 2020, 32, 1909–1922. [Google Scholar] [CrossRef]
- Poernomo, A.; Kang, D. Biased Dropout and Crossmap Dropout: Learning towards effective Dropout regularization in convolutional neural network. Neural Networks 2018, 104, 60–67. [Google Scholar] [CrossRef]
- Welling, M.; Teh, Y. Bayesian Learning via Stochastic Gradient Langevin Dynamics. In Proceedings of the 28th International Conference on International Conference on Machine Learning, Bellevue, WA, USA, 28 June 2011–2 July 2011; Volume 21, pp. 1–6. [Google Scholar]
- Xie, Z.; Sato, I.; Sugiyama, M. A Diffusion Theory for Deep Learning Dynamics: Stochastic Gradient Descent Escapes From Sharp Minima Exponentially Fast. In Proceedings of the International Conference on Learning Representations, Vienna, Austria, 4 May 2021. [Google Scholar]
- Koumakis, L. Deep learning models in genomics; are we there yet? Comput. Struct. Biotechnol. J. 2020, 18, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Pan, Z.; Yin, K.; Khateeb, A. Recent Advances of Deep Learning in Bioinformatics and Computational Biology. Front. Genet. 2019, 10, 214. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banf, M.; Hartwig, T. The Reasonable Effectiveness of Randomness in Scalable and Integrative Gene Regulatory Network Inference and Beyond. Computation 2021, 9, 146. https://doi.org/10.3390/computation9120146
Banf M, Hartwig T. The Reasonable Effectiveness of Randomness in Scalable and Integrative Gene Regulatory Network Inference and Beyond. Computation. 2021; 9(12):146. https://doi.org/10.3390/computation9120146
Chicago/Turabian StyleBanf, Michael, and Thomas Hartwig. 2021. "The Reasonable Effectiveness of Randomness in Scalable and Integrative Gene Regulatory Network Inference and Beyond" Computation 9, no. 12: 146. https://doi.org/10.3390/computation9120146
APA StyleBanf, M., & Hartwig, T. (2021). The Reasonable Effectiveness of Randomness in Scalable and Integrative Gene Regulatory Network Inference and Beyond. Computation, 9(12), 146. https://doi.org/10.3390/computation9120146