A Computational Study to Identify Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) from Eucalyptus Active Compounds


 ,
,  , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Ligand Preparations
2.2. Protein Model Preparations
2.3. Molecular Docking
2.4. Molecular Dynamics (MD) Simulations and Analysis
3. Results
3.1. Molecular Docking Results
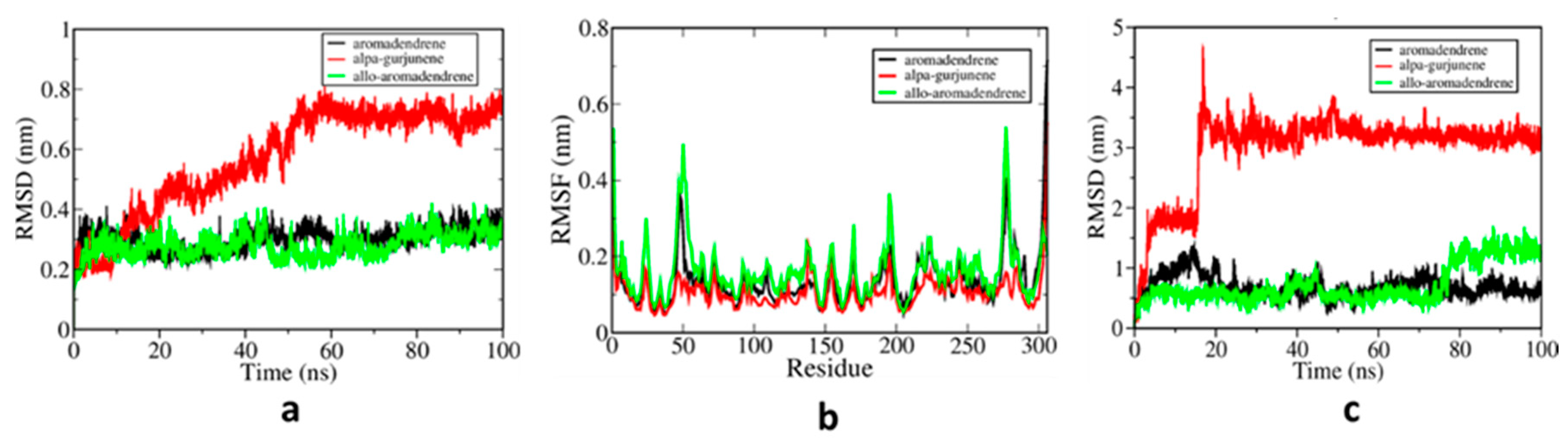
3.2. MD Simulations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020, 750, 137489. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-H.; Cai, L.; Cheng, Z.-S.; Cheng, H.; Deng, T.; Fan, Y.-P.; Fang, C.; Huang, D.; Huang, L.-Q.; Huang, Q.; et al. A rapid advice guideline for the diagnosis and treatment of 2019 novel coronavirus (2019-nCoV) infected pneumonia (standard version). Mil. Med. Res. 2020, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 20 July 2020).
- WHO. Global Nigeria WHO (COVID-19) Homepage. Available online: https://covid19.who.int/region/afro/country/ng (accessed on 20 July 2020).
- He, J.; Hu, L.; Huang, X.; Wang, C.; Zhang, Z.; Wang, Y.; Zhang, D.; Ye, W. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: Insights from structures of protease and inhibitors. Int. J. Antimicrob. Agents 2020, 56, 106055. [Google Scholar] [CrossRef]
- Delang, L.; Neyts, J. Medical treatment options for COVID-19. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 209–214. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Amanatidou, D.; Petrou, A.; Geronikaki, A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules 2020, 25, 2529. [Google Scholar] [CrossRef]
- Hall, D.C., Jr.; Ji, H.-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel. Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, D.S.N.B.K.; Murahari, M.; Chandramohan, V.; Panda, S.P.; Atmakuri, L.R.; Guntupalli, C. In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS CoV-2. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Krammer, F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Denaro, M.; Smeriglio, A.; Barreca, D.; De Francesco, C.; Occhiuto, C.; Milano, G.; Trombetta, D. Antiviral activity of plants and their isolated bioactive compounds: An update. Phytother. Res. 2020, 34, 742–768. [Google Scholar] [CrossRef]
- Covid-19: Race to eucalyptus in Maputo–Watch. Club of Mozambique, 13 April 2020; p. 1.
- Vecchio, M.G.; Loganes, C.; Minto, C. Beneficial and Healthy Properties of Eucalyptus Plants: A Great Potential Use. Open Agric. J. 2016, 10, 52–57. [Google Scholar] [CrossRef]
- Salehi, B.; Sharifi-Rad, J.; Quispe, C.; Llaique, H.; Villalobos, M.; Smeriglio, A.; Trombetta, D.; Ezzat, S.M.; Salem, M.A.; Zayed, A.; et al. Insights into Eucalyptus genus chemical constituents, biological activities and health-promoting effects. Trends Food Sci. Technol. 2019, 91, 609–624. [Google Scholar] [CrossRef]
- The Jakarta Post. Available online: https://www.thejakartapost.com/news/2020/05/09/agriculture-ministry-claims-to-have-developed-eucalyptus-based-covid-19-treatment.html (accessed on 21 July 2020).
- Tempo Misleading: COVID-19 Can Be Cured with Eucalyptus Oil. Available online: https://www.poynter.org/?ifcn_misinformation=covid-19-can-be-cured-with-eucalyptus-oil (accessed on 17 July 2020).
- Adeniyi, B.A.; Ayepola, O.O.; Adu, F.D. The antiviral activity of leaves of Eucalyptus camaldulensis (Dehn) and Eucalyptus torelliana (R. Muell). Pak. J. Pharm. Sci. 2015, 28, 1773–1776. [Google Scholar]
- Usachev, E.V.; Pyankov, O.V.; Usacheva, O.V.; Agranovski, I.E. Antiviral activity of tea tree and eucalyptus oil aerosol and vapour. J. Aerosol Sci. 2013, 59, 22–30. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Khunrae, P.; Sutthibutpong, T. Effects of oligolignol sizes and binding modes on a GH11 xylanase inhibition revealed by molecular modeling techniques. J. Mol. Model. 2020, 26, 124. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular docking in modern drug discovery: Principles and recent applications. In Drug Discovery and Development-New Advances; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Das, P.; Majumder, R.; Mandal, M.; Basak, P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Mpiana, P.T.; Ngbolua, K.-T.-N.; Tshibangu, D.S.T.; Kilembe, J.T.; Gbolo, B.Z.; Mwanangombo, D.T.; Inkoto, C.L.; Lengbiye, E.M.; Mbadiko, C.M.; Matondo, A.; et al. Identification of potential inhibitors of SARS-CoV-2 main protease from Aloe vera compounds: A molecular docking study. Chem. Phys. Lett. 2020, 754, 137751. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020, 255, 117831. [Google Scholar] [CrossRef]
- Gajula, M.N.V.P.P.; Kumar, A.; Ijaq, J. Protocol for Molecular Dynamics Simulations of Proteins. Bio-Protocol 2016, 6, e2051. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, Y.; Yuan, T.; Li, Q.; Tian, J.; Guan, W.; Liu, B.; Zhang, W.; Xu, X.; Zhang, Y. Enhanced Thermostability of Glucose Oxidase through Computer-Aided Molecular Design. Int. J. Mol. Sci. 2018, 19, 425. [Google Scholar] [CrossRef]
- English, A.C.; Groom, C.R.; Hubbard, R.E. Experimental and computational mapping of the binding surface of a crystalline protein. Protein Eng. Des. Sel. 2001, 14, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.; Tian, S.-H.; Meng, Z.-H.; Yang, L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783. [Google Scholar] [CrossRef]
- Al-Khafaji, K.; Al-Duhaidahawi, D.; Taskin Tok, T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dash, J.J.; Purohit, P.; Muya, J.T.; Meher, B.R. Drug Repurposing of Allophenylnorstatine Containing HIV-Protease Inhibitors Against SARS-CoV-2 Mpro: Insights from Molecular Dynamics Simulations and Binding Free Energy Estimations. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020, 1–13. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eucalyptol Compounds | PubChem ID | Molecular Weight (g/mol) |
|---|---|---|
| 1,8-cineole (eucalyptol) | 2758 | 154.25 |
| α-pinene | 440968 | 136.23 |
| β-pinene | 440967 | 136.23 |
| Terpinen-4-ol | 11230 | 154.25 |
| Piperitone | 92998 | 168.23 |
| Allo-aromadendrene | 42608158 | 204.35 |
| Aromadendrene | 91354 | 204.35 |
| α-gurjunene | 15560276 | 204.35 |
| Eucalyptol Compounds | Binding Energy (kJ/mol) | Inhibition Constant (Ki) (µM) | Mpro Residues Interacting with Natural Compounds |
|---|---|---|---|
| 1,8-cineole (eucalyptol) | −26.90 | 19.5 | His41, Met49, Tyr54, His164, Met165, Asp187, Arg188, and Gln189 |
| α-pinene | −26.23 | 25.55 | His41, Met49, His164, Met165, Asp187, Arg188, and Gln189 |
| β-pinene | −26.57 | 22.34 | His41, Met49, Tyr54, His164, Met165, Asp187, Arg188, and Gln189 |
| Terpinen-4-ol | −23.89 | 65.5 | His41, Met49, Pro52, Tyr54, His164, Arg188, and Gln189 |
| Piperitone | −25.52 | 33.95 | His41, Met49, Tyr54, Cys145, His164, Met165, Glu166, Asp187, and Arg188 |
| Allo-aromadendrene | −29.99 | 5.54 | His41, Met49, Tyr54, Cys145, His164, Met165, Glu166, Asp187, Arg188, and Gln189 |
| Aromadendrene | −30.25 | 5.06 | His41, Met49, Tyr54, Cys145, His164, Met165, Asp187, Arg188, and Gln189 |
| α-gurjunene | −30.71 | 4.15 | His41, Met49, Tyr54, Cys145, His164, Met165, Glu166, Asp187, Arg188, and Gln189 |
| Complex Structures | Van der Waals Energy (±SD) (kJ/mol) | Electrostatic Energy (±SD) (kJ/mol) | Polar Solvation Energy (±SD) (kJ/mol) | Apolar Energy (±SD) (kJ/mol) | Total Binding Energy (±SD) (kJ/mol) |
|---|---|---|---|---|---|
| Mpro-allo-aromadendrene | −90.61 (±9.73) | −2.06 (±6.57) | 27.31 (±5.34) | −9.59 (±1.23) | −74.95 (±8.88) |
| Mpro-aromadendrene | −88.82 (±12.08) | −3.42 (±4.77) | 21.89 (±4.92) | −9.11 (±1.25) | −79.45 (±12.62) |
| Mpro-α-gurjunene | −99.00 (±8.53) | −0.33 (±1.91) | 25.55 (±6.93) | −11.42 (±1.06) | −85.21 (±9.44) |
| Ligands | Estimated Binding Energy (kJ/mol) | Reference | |
|---|---|---|---|
| N3 | −51.07 | −44.46 | [48] |
| Indinavir | −72.11 | −23.42 | [48] |
| Darunavir | −95.53 | - | [48] |
| Favipiravir | −36.07 | −59.46 | [49] |
| Fosfomycin | −60.63 | −34.90 | [49] |
| Aspirin | −78.37 | −17.16 | [49] |
| Allo-aromadendrene | −74.95 | −20.58 | This study |
| Aromadendrene | −79.45 | −16.08 | This study |
| α-gurjunene | −85.21 | −10.32 | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, I.A.; Muangchoo, K.; Muhammad, A.; Ajingi, Y.S.; Muhammad, I.Y.; Umar, I.D.; Muhammad, A.B. A Computational Study to Identify Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) from Eucalyptus Active Compounds. Computation 2020, 8, 79. https://doi.org/10.3390/computation8030079
Muhammad IA, Muangchoo K, Muhammad A, Ajingi YS, Muhammad IY, Umar ID, Muhammad AB. A Computational Study to Identify Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) from Eucalyptus Active Compounds. Computation. 2020; 8(3):79. https://doi.org/10.3390/computation8030079
Chicago/Turabian StyleMuhammad, Ibrahim Ahmad, Kanikar Muangchoo, Auwal Muhammad, Ya’u Sabo Ajingi, Ibrahim Yahaya Muhammad, Ibrahim Dauda Umar, and Abubakar Bakoji Muhammad. 2020. "A Computational Study to Identify Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) from Eucalyptus Active Compounds" Computation 8, no. 3: 79. https://doi.org/10.3390/computation8030079
APA StyleMuhammad, I. A., Muangchoo, K., Muhammad, A., Ajingi, Y. S., Muhammad, I. Y., Umar, I. D., & Muhammad, A. B. (2020). A Computational Study to Identify Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) from Eucalyptus Active Compounds. Computation, 8(3), 79. https://doi.org/10.3390/computation8030079