Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Approach
2.1. Quantum Calculations
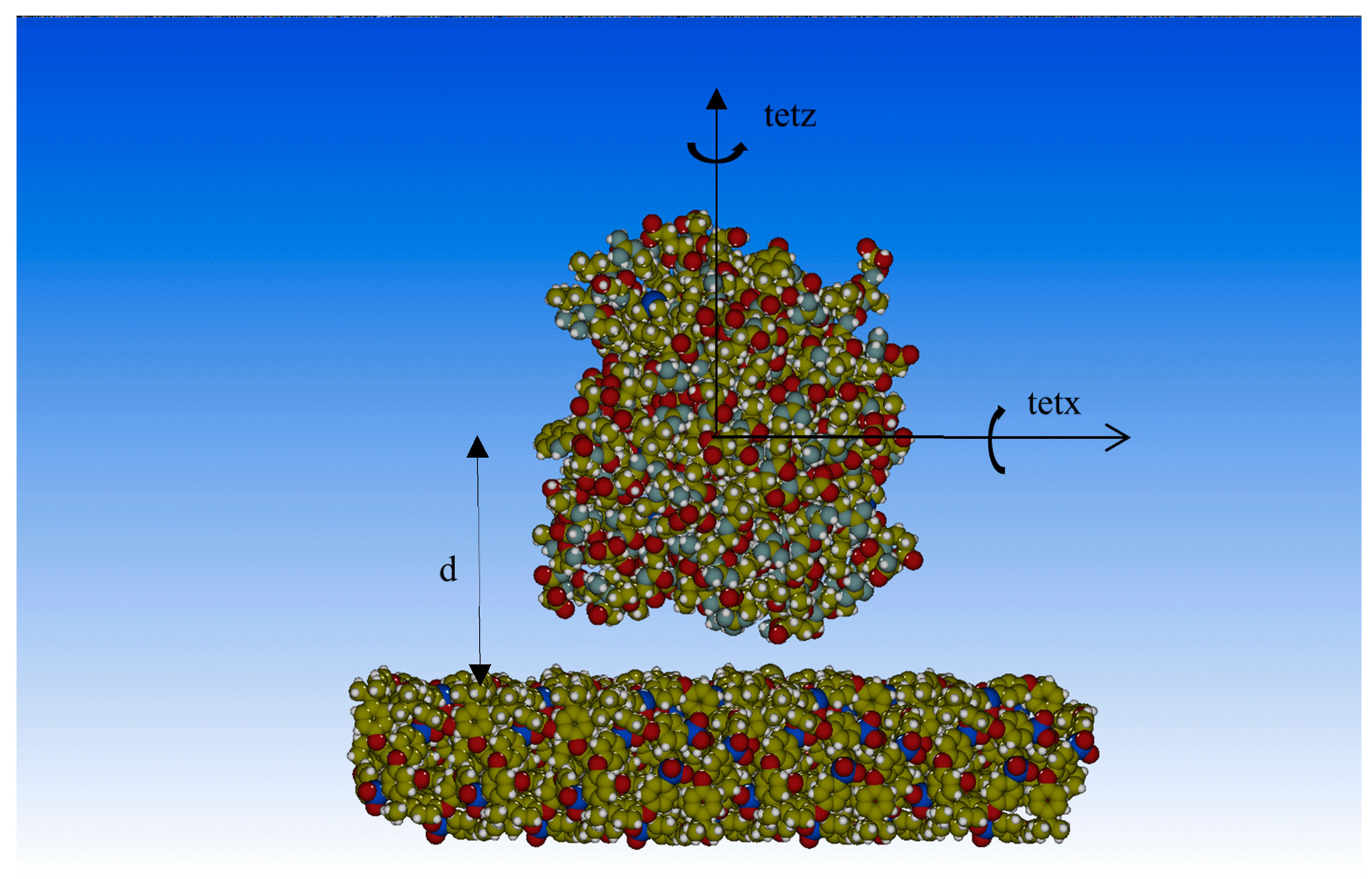
2.2. Molecular Mechanics Optimizations
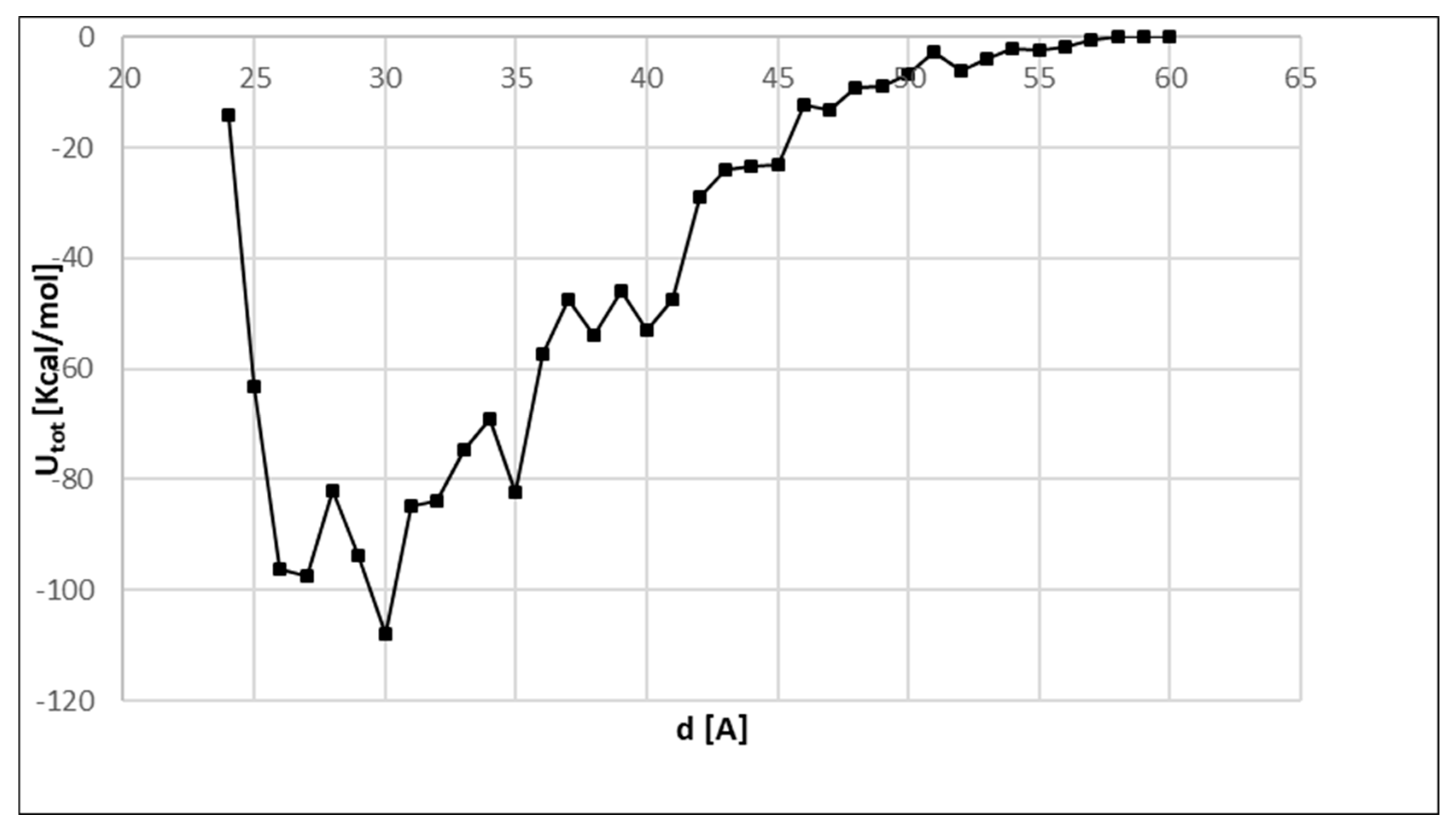
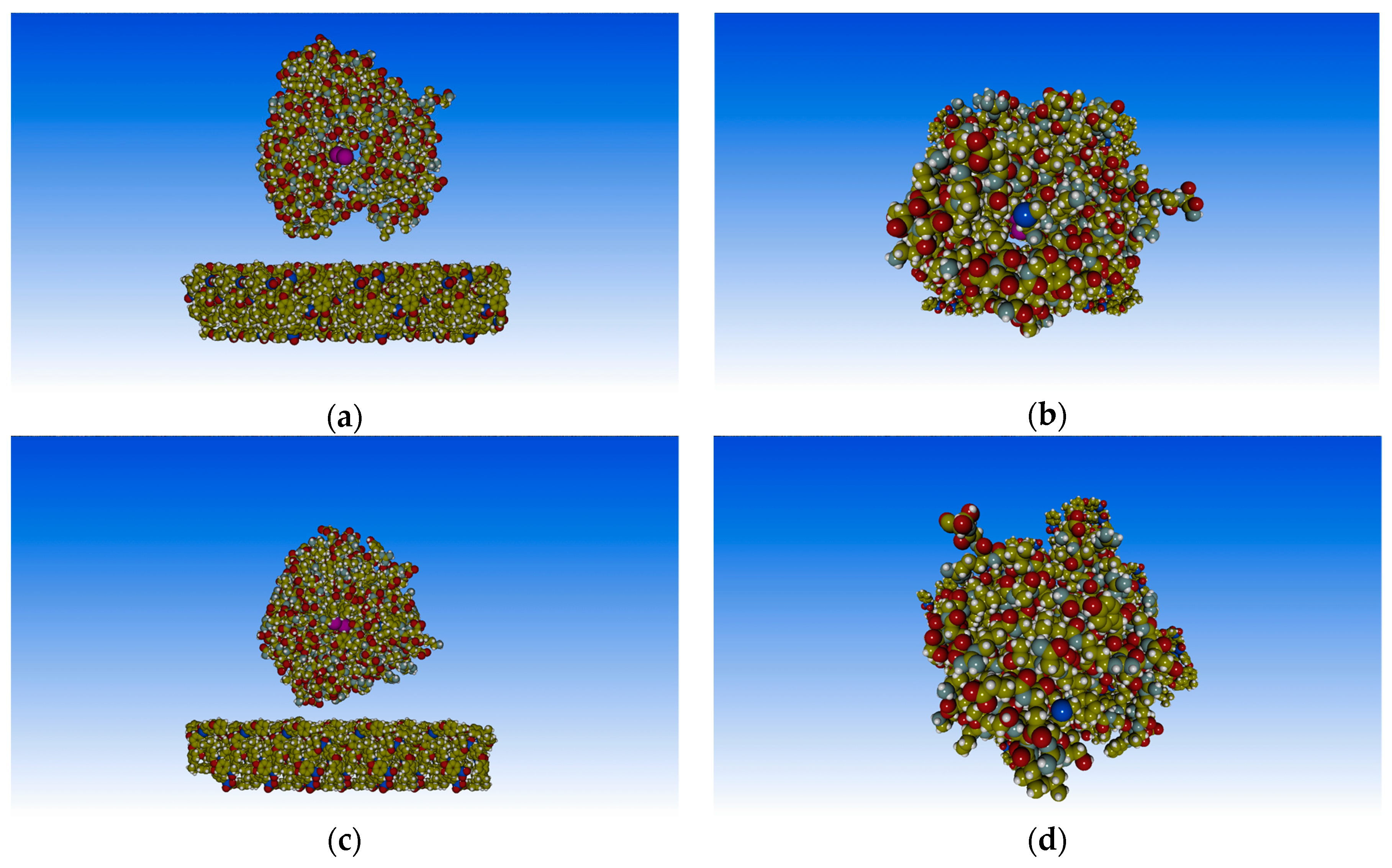
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weinan, E.; Li, X.; Vanden-Eijnden, E. Some Recent Progress in Multiscale Modeling; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Horstemeyer, M. Multiscale Modeling: A review. In Practical Aspects of Computational Chemistry; Springer: New York, NY, USA, 2009; pp. 87–135. [Google Scholar]
- Parekh, S.; Vinci, V.A.; Strobel, R.J. Improvement of microbial strains and fermentation processes. Appl. Microbiol. Biotechnol. 2000, 54, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Gurung, N.; Ray, S.; Bose, S.; Rai, V. A broader view: Microbial enzymes and their relevance in industries, medicine, and beyond. BioMed Res. Int. 2013, 2013, 329121. [Google Scholar] [CrossRef] [PubMed]
- Underkofler, L.A.; Barton, R.R.; Rennert, S.S. Production of microbial enzymes and their applications. Appl. Microbiol. 1958, 6, 212–221. [Google Scholar] [PubMed]
- Raushel, F.M.; Holden, H.M. Phosphotriesterase: An enzyme in search of its natural substrate. Adv. Enzymol. Relat. Areas Mol. Biol. 2000, 74, 51–93. [Google Scholar] [PubMed]
- Nguyen, H.; Kim, M. An overview of techniques in enzyme immobilization. Appl. Sci. Converg. Technol. 2017, 26, 157–163. [Google Scholar]
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, P.; Renka, R.J. A Levenberg–Marquardt method based on Sobolev gradients. Nonlinear Anal. 2012, 16, 6170–6179. [Google Scholar] [CrossRef]
- Han, L.; Neumann, M. Effect of dimensionality on the Nelder–Mead simplex method. Optim. Methods Softw. 2006, 21, 1–16. [Google Scholar] [CrossRef]
- Curcio, S.; Petrosino, F.; Morrone, M.; De Luca, G. Interactions between proteins and the membrane surface in multiscale modeling of organic fouling. J. Chem. Inf. Model. 2018, 58, 1815–1827. [Google Scholar] [CrossRef]
- De Luca, G.; Bisignano, F.; Paone, F.; Curcio, S. Multi-scale modeling of protein fouling in ultrafiltration process. J. Membr. Sci. 2014, 452, 400–414. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Liu, X.; Zhou, W.; Rao, Z. The crystal structure of the phosphotriesterase from M.tuberculosis, another member of phosphotriesterase-like lactonase family. Biochem. Biophys. Res. Commun. 2019, 510, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Malki, Z.E.; Bouzzine, S.M.; Bejjit, L.; Haddad, M.; Hamidi, M.; Bouachrine, M. Density functional theory [B3LYP/6-311G(d,p)] study of a new copolymer based on carbazole and (3,4-Ethylenedioxythiophene) in their aromatic and polaronic states. J. Appl. Polym. Sci. 2011, 122, 3351–3360. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Lund, M.; Jönsson, B. A mesoscopic model for Protein-Protein interactions in solution. Biophys. J. 2003, 85, 2940–2947. [Google Scholar] [CrossRef]
- Zhou, A.Q.; O’Hern, C.S.; Regan, L. The power of hard-sphere models: Explaining side-chain dihedral angle distributions of thr and val. Biophys. J. 2012, 102, 2345–2352. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput.Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Van der Waals Potential Energy. Available online: http://www.csb.yale.edu/userguides/datamanip/autodock/html/Using_AutoDock_305.a.html (accessed on 17 July 2019).
- Attaway, S. Matlab: A Practical Introduction to Programming and Problem Solving; Butterworth-Heinemann: Oxford, UK, 2009; ISBN 978-0-08-094325-1. [Google Scholar]
- Lu, D.R.; Lee, S.J.; Park, K. Calculation of solvation interaction energies for protein adsorption on polymer surfaces. J. Biomater. Sci. Polym. Ed. 1992, 3, 127–147. [Google Scholar] [CrossRef]
- Jeyachandran, Y.L.; Mielczarski, E.; Rai, B.; Mielczarski, J.A. Quantitative and qualitative evaluation of adsorption/desorption of bovine serum albumin on hydrophilic and hydrophobic surfaces. Langmuir 2009, 25, 11614–11620. [Google Scholar] [CrossRef]
- Salgın, S.; Takaç, S.; Özdamar, T.H. Adsorption of bovine serum albumin on polyether sulfone ultrafiltration membranes: Determination of interfacial interaction energy and effective diffusion coefficient. J. Membr. Sci. 2006, 278, 251–260. [Google Scholar] [CrossRef]
- May, S.; Harries, D.; Ben-Shaul, A. Lipid Demixing and Protein-Protein interactions in the adsorption of charged proteins on mixed membranes. Biophys. J. 2000, 79, 1747–1760. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrosino, F.; Curcio, S.; Chakraborty, S.; De Luca, G. Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study. Computation 2019, 7, 56. https://doi.org/10.3390/computation7040056
Petrosino F, Curcio S, Chakraborty S, De Luca G. Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study. Computation. 2019; 7(4):56. https://doi.org/10.3390/computation7040056
Chicago/Turabian StylePetrosino, Francesco, Stefano Curcio, Sudip Chakraborty, and Giorgio De Luca. 2019. "Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study" Computation 7, no. 4: 56. https://doi.org/10.3390/computation7040056
APA StylePetrosino, F., Curcio, S., Chakraborty, S., & De Luca, G. (2019). Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study. Computation, 7(4), 56. https://doi.org/10.3390/computation7040056