Abstract
The cytochrome P450s (CYPs) play a central role in the metabolism of various endogenous and exogenous compounds including drugs. CYPs are vulnerable to inhibition and induction which can lead to adverse drug reactions. Therefore, insights into the underlying mechanism of CYP450 inhibition and the estimation of overall CYP inhibitor properties might serve as valuable tools during the early phases of drug discovery. Herein, we present a large data set of inhibitors against five major metabolic CYPs (CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) for the evaluation of important physicochemical properties and ligand efficiency metrics to define property trends across various activity levels (active, efficient and inactive). Decision tree models for CYP inhibition were developed with an accuracy >90% for both the training set and 10-folds cross validation. Overall, molecular weight (MW), hydrogen bond acceptors/donors (HBA/HBD) and lipophilicity (clogP/logPo/w) represent important physicochemical descriptors for CYP450 inhibitors. However, highly efficient CYP inhibitors show mean MW, HBA, HBD and logP values between 294.18–482.40,5.0–8.2,1–7.29 and 1.68–2.57, respectively. Our results might help in optimization of toxicological profiles associated with new chemical entities (NCEs), through a better understanding of inhibitor properties leading to CYP-mediated interactions.
Keywords:
cytochrome P450; CYP1A2; CYP2C9; CYP2C19; CYP2D6; CYP3A4; lipophilic efficiency; ligand efficiency; decision tree; drug metabolism 1. Introduction
The drug discovery and development is a grueling and lengthy process that is prone to high attrition rates throughout all phases of development [1]. However, to increase the research and development output, an improved “5R” strategy deciphering right target, right safety, right tissue, right patient and right commercial potential has been proposed by AstraZeneca [2]. Various proofs of concept examples of the application of the “5R” strategy indicates an improved success rate from candidate selection to the completion of phase III [2,3]. Thus, for high quality leads and drug candidates better insights into pharmacokinetics (PK)/pharmacodynamics (PD) along with ADMET (absorption, distribution, metabolism, excretion and toxicity) properties is highly recommended [3]. Additionally, more focused approaches towards incorporating pharmacokinetics and drug metabolism into compound design has assisted in making PK/PD and dose related predictions in humans [3]. Hitherto, drug metabolism is an influential factor in pharmacokinetics and hence modulates the behavior of a drug. Therefore, early understanding of metabolism of new chemical entities (NCE) and their affinity towards various metabolic enzymes might assist the PK/PD optimization during the drug development process [4]. Generally, amongst all metabolic enzymes, the most important are the cytochrome P450s which constitute a ubiquitous superfamily of heme proteins, playing a key role in the oxidative, peroxidative and reductive metabolism of a wide range of endogenous and exogenous compounds, including drugs [4]. In human,57 CYP isoforms have been identified with CYP1A2,2C9,2C19,2D6 and 3A4 mediating ~90% of all the phase I metabolic reactions of clinically relevant drugs [5]. The association of cytochrome P450s with toxicological events due to metabolic alterations has brought about CYP-mediated drug metabolism as the principal reason for the occurrence of several drug–drug interactions (DDIs) [6]. Moreover, the co-administration of drugs might lead to the inhibition or induction of cytochrome P450s, therefore, there is an earnest need to assess CYP mediated interaction profiles of NCEs during the drug design and development phase [7,8]. Furthermore, during the last decade, DDIs associated with the inhibition of cytochrome P450s mainly due to the broad substrate specificity of CYP family of enzymes, emerged as the most common reason for the removal of various marketed drugs [9,10,11].
Additionally, the cytochrome P450 enzymes display an inherent affinity for lipophilic substrates due to their lipophilic nature [12,13,14]. Whereas, depending on the ionization states, lipophilic compounds also show inhibition potential against the cytochrome P450s [15]. This represents lipophilicity as one of the most significant physicochemical property in the drug discovery and design programs that plays a significant role in determining the ADMET properties [16] along with selectivity, promiscuity [17] and potency [18]. Many two- and three-dimensional quantitative structure–activity relationship (2D and 3DQSAR) studies have also reported the effect of lipophilicity on the inhibition of cytochrome P450s [19,20,21,22,23,24].
Therefore, from the drug design perspective, it is anticipated that NCEs should display a suitable metabolism with negligible or no potential of CYP inhibition or induction [25]. During the recent years the availability of X-ray, crystallographic structures of various mammalian CYP isoforms and mutagenesis data has provided a better understanding of CYP structure–function relationships [26,27,28,29,30,31,32]. Most importantly, significant in silico, in vitro and experimental efforts have been made to elucidate the underlying mechanisms behind CYP inhibition [33,34,35,36,37,38,39]. Moreover, various ligand- and structure-based in silico models as well as machine learning approaches have been used for the classification of inhibitors and substrates of individual CYP isoforms [40,41,42,43,44,45,46]. Herein, we estimate a set of physicochemical parameters in combination with lipophilic efficiency (LipE) and ligand efficiency (LE) metrics to classify the most active and efficient inhibitors of the target CYPs (CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4). Additionally, we attempt to build simple and easily interpretable decision tree models for the prediction of cytochrome P450 inhibition. The identification of molecular descriptor ranges, important for CYP inhibition in general and for highly efficient binding in particular, might provide a valuable tool for the classification and prediction of CYP inhibition against the selected subtypes.
2. Materials and Methods
2.1. Database Collection
A data set of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 inhibitors with inhibitory potency (IC50) values was collated from ChEMBL database [47] using a filtering criteria of IC50 ≤ 100 µM against each CYP subtype. The dataset was further refined by removing inconsistent potency values (% age inhibition, nonabsolute) and duplicate entries. After refinement, the final data set of 6999 inhibitors of CYPs includes 612 CYP1A2,1341 CYP2C9,651 CYP2C19,1647 CYP2D6 and 2747 CYP3A4 inhibitors (Figure 1 and Tables S1–S5 in Supplementary Materials). Additionally, the inhibitor datasets of each CYP class were divided in to three activity levels including active, efficient and inactive. Generally, an activity threshold of IC50 ≤ 50 µM was used to categorize compounds as actives and remaining compounds with IC50 > 50–100 µM as inactives. Here, in this particular study, we have used the activity threshold of 50 μM to build a more generalized inhibition model for each CYP subtype as proposed by Tie et al. [5,6]. However, the active inhibitors with LipE ≥ 5, lipophilicity (clogP) values of ~1.0–3.0, IC50 ~10–150 nM and LE ≥ 0.29 (kcal/mol/heavy atom) were further classified as highly efficient (more prone to drug–drug interaction due to CYP inhibition). These include 12 CYP1A2, eight CYP2C9, five CYP2C19, eight CYP2D6 and 17 inhibitors of CYP3A4. The activity ranges and the number of actives and inactives against each CYP isoform are presented in Figure 1. Furthermore, the IC50 (µM) values were normalized by converting into pIC50 for the calculation of LipE and LE metrics. The schematic workflow used in this study for the elucidation of CYP inhibitor properties across various activity levels is shown in Figure 2.
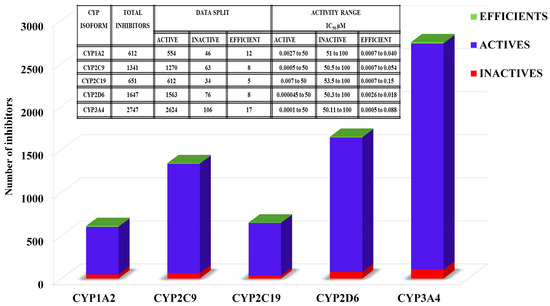
Figure 1.
The total number of cytochrome P450 (CYP) inhibitors split into “active,” “inactive” and “efficient” along with the respective potency ranges against each CYP isoform.
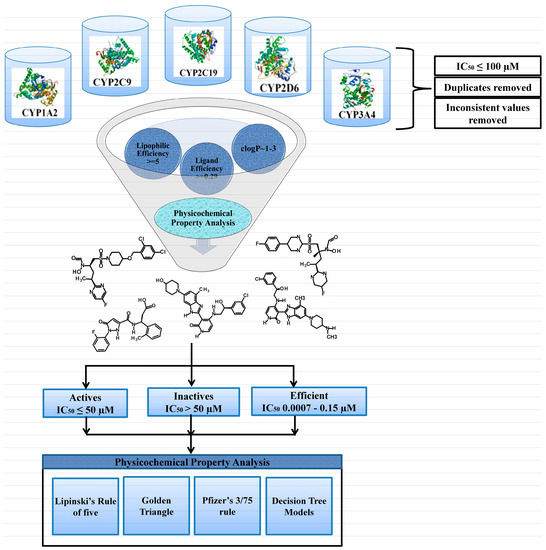
Figure 2.
The schematic work flow used in this study to probe the properties of the selected CYP inhibitors across various activity levels. IC50: Inhibitory Potency; clogP: Lipophilicity
2.2. Selection of Efficient Inhibitors of CYP Isoforms
2.2.1. Lipophilic Efficiency (LipE)
Lipophilicity contributes towards drug solubility, permeability and metabolism, thus representing an important factor in pharmacokinetics- and pharmacodynamics-mediated toxicity of a chemical entity [16]. Leeson and Springthorpe proposed the lipophilic efficiency (LipE) metric as an explicit approach to estimate drug-likeliness by providing a linkage between lipophilicity and potency [17]. However, a drug like compound may also show off-target toxicity due to its potential to interact with antitargets such as CYP450, hERG and P-glycoprotein. Herein, we apply this concept to the inhibitors of the selected CYP450 subtypes to estimate the properties of the most efficient CYP inhibitors and anticipate that avoiding these properties during lead optimization programs may reduce antitarget interaction potential of new chemical entities. The LipE profiles were generated by subtracting lipophilicity (clogP) from the negative logarithm of potency (pIC50) values against the respective CYP isoform (Equation (1)) (Tables S1–S5 in Supplementary Materials).
The clogP values were calculated through the Bio-Loom software package [48] using the SMILES of the entire data set whereas, the LipE calculations were performed using the Excel spreadsheet.
2.2.2. Ligand Efficiency (LE)
Ligand efficiency is a measure that quantifies a ligands affinity towards its target and is measured by dividing the binding free energy (ΔG) in kcal/mol to the number of heavy atoms (HA) [49,50]. The binding free energies (ΔG) were calculated using Equation (2) where, R is the ideal gas constant, T is the temperature in Kelvin and Kd is the disassociation constant. A temperature of 310 K was used to compute ligand efficiencies in kcal/mol/heavy atom. Additionally, ΔG values for CYP inhibitors were computed by substituting the dissociation constant (Kd) with pIC50 values as explicated by Hopkins et al. [49] which was also further established by the experimental findings of Kuntz et al. [51].
In order to estimate the binding quality of a compound towards the respective CYP isoforms, here ligand efficiency (LE) profiling for the entire inhibitor dataset was performed using Equation (3):
ΔG and LE values for inhibitors of each CYP subtype are shown in Supplementary Materials (Tables S1–S5 in Supplementary Materials). The Excel spreadsheet was used to perform ligand efficiency calculations.
2.3. Computation of Physicochemical Properties
The FAF-Drugs4 [52] server was used for the estimation of physicochemical properties including molecular weight (MW), logP, logD (pH7), topological polar surface area (TPSA), rotatable bonds, hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), number of rings, stereocenters, fraction of sp3 carbons (Fsp3) and formal charges of the CYP inhibitors. Additionally, the sum of van der Waals (vdW) surface areas (Å2) of hydrogen bond acceptors (vsa_acc) and hydrogen bond donors (vsa_don) were also calculated for the inhibitors of each CYP subtype using MOE(Molecular Operating Environment) [53].
Furthermore, Lipinski’s rule of five [54], the Golden Triangle [55] and the Pfizer’s 3/75 rule [56] have also been applied using these physicochemical descriptors to probe inhibition rules for active and efficient inhibitors of the respective CYP isoform.
2.4. Decision Trees (C4.5 DT)
The decision trees for the classification of active and inactive inhibitors of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 were built using the complete set of already calculated physicochemical descriptors. WEKA software package [57] was used to train decision trees based on J48 classifier [58] using 10-fold cross-validation procedure. J48 is one of the most powerful and commonly used decision tree classifier that is an improved version of C4.5 algorithm [59]. A J48 classifier creates a binary decision tree to model the classification procedure based on the divide-and-conquer rule [60].
2.5. Model Performance Evaluation
In order to evaluate the overall performance of decision tree models, several parameters including accuracy (Equation (4)), sensitivity (Equation (5)) and specificity (Equation (6)) were calculated; where accuracy indicates the ratio of correctly categorized occurrences to the total number of entities, sensitivity and specificity correspond to the ratio of correctly classified inhibitors and correctly predicted noninhibitors, respectively [61].
Matthews correlation coefficient (MCC) metric was further used to measure the quality of classification model by taking into account the true positive (TP), true negative (TN), false positive (FP) and false negative (FN) instances (Equation (7)) [62]. The MCC values usually fall between −1 to +1, where ideally, a value of +1 for a classifier is indicative of a good agreement between predicted and experimental values of classes [62]. Another index that shows a better evaluation of the models predictive power is kappa statistic (Equation (8)), which uses by chance, the expected agreement based on the ratio between the classes (Equations (8) and (9)) [63] where 1,0 and −1 indicate perfect agreement, no agreement above that expected by chance and complete disagreement, respectively [63].
where, E is the expected agreement and calculated as follows:
Furthermore, area under the curve (AUC) based on the receiver operating characteristic (ROC) curve was also calculated to estimate the overall model performance [64]. Overall, the AUC of about 0.5 corresponds to the expected performance of random selection, whereas a value below 0.5 is indicative of inferior performance as compared to random selection [64].
3. Results
3.1. Activity and Efficiency Landscape of the Selected CYP Isoforms Inhibitors
In order to refine the CYP inhibition rules, inhibitor datasets of the selected CYP isoforms were divided in to active, efficient and inactive. The highly efficient inhibitors of CYP subtypes were selected on the basis of drug lipophilic and ligand efficiency metrics.
3.2. Lipophilic Efficiency
Previously, Leeson and Springthorpe demonstrated a clogP of ~2.5, potency in the range ~1–10 nM and LipE of ~5–7 or greater as the optimal criteria for an average oral drug against a true target [17]. In the present study, we attempt to apply this concept to a set of antitargets, the cytochrome P450 family of enzymes, to further refine the respective inhibition rules. The clogP and LipE distribution for the inhibitors of the selected CYP isoforms shows that a greater percentage of CYP inhibitors are highly lipophilic with clogP values from 2.0–7.0 and LipE values from 0.0–5.0 (Figure 3a,b).
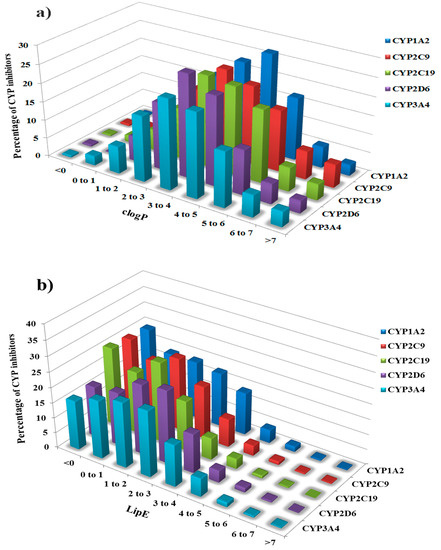
Figure 3.
(a) Distribution of clogP and (b) Lipophilic efficiency (LipE) values for each class of Cytochrome P450 inhibitors (CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4).
Further evaluation of these parameters against selected CYP450 isoforms revealed that LipE values of CYP1A2 inhibitors data set (612 compounds) vary from −5.47–7.06 with inhibitory potency (IC50) and clogP values in the range 0.0027–100 µM and −1.81–9.84, respectively, as shown in Supplementary Information Table S1. Majority of the CYP1A2 inhibitors (75.5%) displayed positive LipE values (LipE ≥ 0) while 24.67% inhibitors exhibit negative LipE values (LipE < 0) which might be due to their low IC50 or very high clogP values (Figure 4a and Table S1). Considering 13 compounds having LipE ≥5, only dehydroevidiamine showed a LipE ≥5 due to negative clogP (−1.81) and activity value of 5.63 µM. However, the remaining 12 CYP1A2 inhibitors including a neuroprotective voltage-dependent sodium channel modulator (5-(1-benzyl-1H-indazol-3-yl)-1,2,4-oxadiazol-3-yl) [65] and a photoactive agent methoxsalen [66,67] satisfy the efficiency thresholds established by Leeson and Springthorpe [17] for true therapeutic targets and therefore, represent a set of highly efficient (prone to drug–drug interaction) CYP1A2 inhibitors in terms of activity and lipophilicity as shown in Figure 4a and highlighted in Table S1.
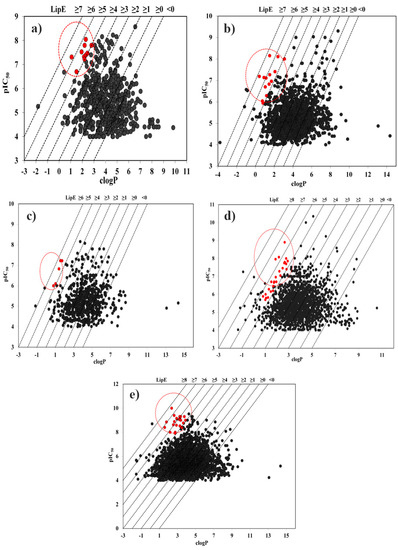
Figure 4.
LipE profiling of (a) CYP1A2, (b) CYP2C9, (c) CYP2C19, (d) CYP2D6 and (e) CYP3A4 inhibitors, where clogP is plotted against the activity (pIC50). The cytochrome P450 inhibitors fulfilling Leeson and Springthorpe criteria (LipE of ~5–7) and clogP ~1–3 are highlighted (red color) in the plot for each CYP isoform.
Similarly, LipE values of 1341 inhibitors of CYP2C9 vary from −9.5–7.87. About 25% CYP2C9 inhibitors displayed negative LipE values of <0 and 75% CYP2C9 inhibitors showed positive LipE values of ≥ 0 as shown in Table S2. Overall,20 inhibitors of CYP2C9 exhibited LipE ≥5, clogP and IC50 values in the ranges −3.77–2.4 and 0.007–79.5 µM, respectively. Six CYP2C9 inhibitors (CHEMBL179399, CHEMBL2419525, CHEMBL2419524, CHEMBL2069801, CHEMBL511410 and CHEMBL2419516) showed a LipE ≥ 5 due to negative clogP values (−3.77 to −0.15). However, only 14 CYP2C9 inhibitors displayed high LipE (≥5) due to positive clogP values (Table S2 (lilac)). These include one c-Jun N-terminal kinase (JNK) inhibitor [68] and seven other inhibitors from ChEMBL database (CHEMBL2425651, CHEMBL2419505, CHEMBL2393185, CHEMBL2419515, CHEMBL484345, CHEMBL2391567 and CHEMBL2393188) that show an overall balance of LipE (≥5), clogP (1.05–2.4) and activity values (IC50 = 0.0078–0.149 µM) against CYP2C9 (Figure 4b). These compounds are categorized as efficient inhibitors of CYP2C9 and highlighted in Figure 4b (red).
Additionally, for 651 CYP2C19 inhibitors, LipE values between −9.2–6.15, clogP in the range of −1.15–14.36 and IC50 between 0.007–100 µM were observed (Supplementary Information Table S3). Similar to CYP1A2 and CYP2C9 inhibitors, less number (26%) of CYP2C19 inhibitors were identified with negative LipE values (< 0) and a greater number (74%) of inhibitors with positive LipE values (≥0). Only two compounds (CHEMBL2069802 and CHEMBL2069801) with negative clogP (−1.15 to −0.15) and IC50 values between 1.3–9.8 µM showed LipE values ≥ 6. However, five CYP2C19 inhibitors with positive clogP values (0.8–1.73) including CHEMBL2170635, CHEMBL3110021, CHEMBL1089, CHEMBL1672632 and CHEMBL2036220 were identified with LipE ≥ 5 and IC50 values in the range 0.06–1 µM, respectively, as highlighted red in Figure 4c. Therefore, these epitomize a set of highly efficient CYP2C19 inhibitors with balanced activity and lipophilicity.
Similarly, for 1647 CYP2D6 inhibitors,83% of the data displayed positive LipE (≥ 0) and the remaining 17% displayed negative LipE (< 0) values (Supplementary Information Table S4 and Figure 4d). Thirty compounds presented a LipE ≥ 5 with clogP and IC50 values falling in the range −1.32–10.56 and 0.000045–94.3 μM, respectively. Amongst these, seven compounds with negative clogP (−1.32 to −0.15) and 23 inhibitors with positive clogP (0.04–5.11) exhibited higher LipE values (≥ 5) (Table S4 (lilac)). However, eight CYP2D6 inhibitors with LipE ≥ 5 including Yohimbine (an alpha-2 adrenergic receptor blocker) [69,70], Quinidine (class I antiarrhythmic agent) [71] and a κ opioid receptor agonist [72] derived from drug compound ICI 199441 [73] displayed a balance of clogP (1.88–2.79) and potency (0.00126–0.11 µM) values as shown in Figure 4d (red points) and thus, represent the highly efficient inhibitors of CYP2D6.
Nevertheless,2747 CYP3A4 inhibitors with LipE, clogP and IC50 values in the range −9.17–8.5, −1.55–14.36 and 0.00001–100 µM, respectively, were observed as shown in Supplementary Information Table S5. Amongst these,2304 (84%) compounds were identified with positive LipE values however, remaining 443 (16%) presented negative LipE values (Table S4 and Figure 4e). Overall, a total of 53 compounds showed LipE values ≥ 5 including 43 inhibitors with positive clogP (0.2–4.25) and ten showing negative clogP values (−1.55 to −0.12). Out of these 43 inhibitors of CYP3A4, only 17 compounds including five N-hydroxyformamide inhibitors used for treating osteoarthritis [74], four insulin-like growth factor 1 kinase (IGF-IR) inhibitors [75] and one antibacterial benzothiazole that inhibits Staphylococcus aureus cell division [72] fulfilled the efficiency criteria (LipE ≥ 5) exhibiting clogP and potency values within 1.24–2.93 and 0.0001–0.088 µM, respectively (Figure 4e). The mean values and ranges of IC50, clogP and LipE for all inhibitors and highly efficient inhibitors against each CYP subtype are presented in Table 1.

Table 1.
The summary of IC50, clogP, LipE, heavy atom (HA) and ligand efficiency (LE) ranges for all inhibitors and highly efficient inhibitors of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 that fulfill the efficiency criteria of clogP ~1.0–3.0, LipE ≥ 5 and LE ≥ 0.29.
3.3. Ligand Efficiency
To gain insight into the highly efficient inhibitors of the selected CYP450 isoforms in terms of binding free energy with the respective enzyme, we computed the ligand efficiency (LE) metric for the entire inhibitor dataset as outlined in the Materials and Method section. For our dataset of CYP inhibitors, a greater percentage (97.9%) of inhibitors displayed heavy atom count (HA) from 10–50 with LE values from 0.1–0.5 (kcal/mol/heavy atom) as shown by distribution plots in Figure 5a,b. Generally, LE values for the entire CYP inhibitor data set vary from 0.016–1.07 kcal/mol/heavy atom (Table 1 and Tables S1–S5). It is evident that out of the total data, about 365 CYP1A2,361 CYP2C9,236 CYP2C19,739 CYP2D6 and 675 CYP3A4 inhibitors showed LE within the range of already established threshold (≥ 0.29 kcal/mol/heavy atom) for optimal binding with true therapeutic target, which may reflect an optimal fit inside the respective binding site [49]. Therefore, in the present study, compounds having LE ≥ 0.29 kcal/mol/heavy atom along with LipE ≥ 5 and clogP ~1.0–3.0 were classified as the highly efficient inhibitors of the respective CYP subtype. The overall ranges and mean values of LE and HA for the entire set of inhibitors as well as for the most efficient inhibitors against each CYP subtype are shown in Table 1. However, the absolute LE, HA count and ΔG values of the inhibitors of CYP1A2,2C9,2C19,2D6 and 3A4 are presented in Tables S1–S5 respectively.
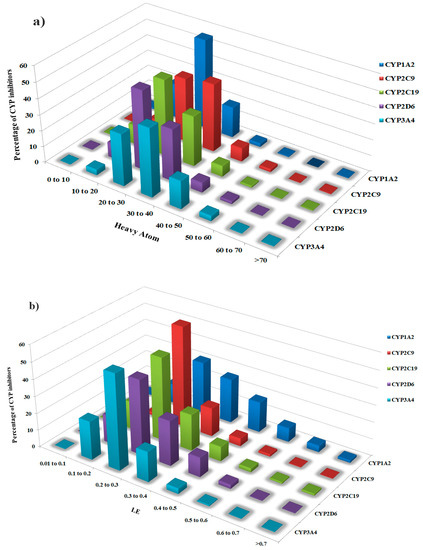
Figure 5.
(a) Distribution of heavy atom count and (b) LE values for each class of cytochrome P450 inhibitors including CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4.
3.4. Physicochemical Properties
The physicochemical properties associated with chemical compounds might influence the overall efficacy, metabolism and safety profiles. Therefore, various studies elucidating the relationships between potency, ADME and physicochemical properties of chemical entities have been reported in literature [17,76,77,78,79]. For various classes of compounds, a better understanding of the physicochemical properties might assist in differentiating target families and ultimately avoiding the undesirable binding to off-targets. Additionally, it may also contribute towards the design of compounds capable of binding to multiple biological targets which might prove beneficial for the treatment of complex disease conditions [80]. Here, physicochemical properties including MW, logP, logD, TPSA, rotatable bond, HBDs and HBAs, vsa_acc, vsa_don, rings, number of stereocenters, fraction of sp3 carbons (Fsp3) and the formal charges have been computed to probe the general and specific properties of CYP inhibitors across various classes and activity levels.
Additionally, the two most important applications of physicochemical parameters to assess drug-likeness are the well-known Lipinski’s rule of five (RO5) [54] and the Golden Triangle rule [55] that were originally proposed by taking into account the properties of successful drug compounds of that time. The application of these rules to true therapeutic targets has been extensively reported in literature [81,82,83,84,85]. However, here we have monitored the RO5 and Golden Triangle violations for the inhibitors of antitargets, the cytochrome P450 family of enzymes.
The physicochemical properties of oral drugs reaching clinical phase II were estimated by Lipinski et al. to frame the well-known rule of five, indicating that a logP ≤ 5, MW ≤ 500, HBAs (O + N atom count) ≤ 10 and HBDs (OH + NH count) ≤ 5 is necessary for absorption or permeation [54]. Considering the trends of these important descriptors across the family of CYP inhibitors it is notable that the CYP3A4 inhibitors show the highest mean (466.29) and median (455.63) molecular weights with 95% percentile of 677.69 (Table 2) which is well explicated by the fact that CYP3A4 accommodates large and structurally diverse compounds due its promiscuous binding site [86]. Similarly, the highly efficient CYP3A4 inhibitors with optimal LipE and LE values show the highest mean and median MW (M: 482.46, Mdn: 493.99) in comparison to the highly efficient inhibitors of the remaining CYP isoforms in the data set. Overall, CYP1A2 inhibitors including those fulfilling the efficiency criteria display the lowest mean and median MW (all inhibitors M: 345.14, Mdn: 330.37, efficient inhibitors M: 294.18, Mdn: 288.3) as compared to other CYP isoforms which expounds the fact that molecular planarity with a small volume to surface ratio may favor CYP1A2 inhibition [11]. For the analysis of molecular weight property for other CYP isoforms, refer to Table 2 and Table S6. The 95% confidence intervals (CI) for the difference between calculated property means were also computed for all datasets (Table S6).
Table 2.
The range (R), mean (M), standard error of mean (SEM), median (Mdn) and 95% percentile (P) values for physicochemical parameters of all inhibitors and highly efficient inhibitors of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. TPSA: Topological polar surface area; HBAs: Hydrogen bond acceptors; HBDs: Hydrogen bond donors; Fsp3: Fraction of sp3 carbons; Vsa_acc: Sum of van der Waals (vdW) surface areas (Å2) of Hydrogen bond acceptors; Vsa_don: Sum of van der Waals (vdW) surface areas (Å2) of Hydrogen bond donors; MW: Molecular weight.
An important component of the RO5 is lipophilicity, which is a major contributing factor in several ADMET parameters as well as potency. A higher lipophilicity-associated with the chemical entities might lead to unsuitable metabolism and solubility, whereas reduced permeability might be an outcome of lower lipophilicity [87]. Specifically, for the CYP family of enzymes, lipophilicity is crucial for determining binding affinity of a compound and selectivity towards the specific CYP isoforms [88]. Previously, various in silico models based on logP/logD, hydrogen bonding potential or polar surface area for the prediction of ADME and efficiency have been reported in literature [89,90,91]. Herein, the lipophilicity values characterized by computed logP and logD were compiled for each CYP subtype, showing that overall CYP inhibitors of the selected subtypes are highly lipophilic. This clearly indicates the inherent affinity of cytochrome P450 family of enzymes for lipophilic compounds [12,13,14]. From our datasets the highest mean/median logP values are shown by CYP2C9 (M: 4.03/Mdn: 3.9) and CYP2C19 (M: 3.9/Mdn: 3.88) inhibitors. Whereas, the highly efficient inhibitors of each CYP subtype show mean/median logP values of ~2.5 (Table 1). Moreover, in comparison to all other CYP inhibitor datasets, the lowest mean/median logD values were displayed by CYP2D6 inhibitors (M: 2.24/Mdn: 2.135) as shown in Table 2. Briefly, these are basic compounds with positive charge on nitrogen [36]. Thus, CYP2D6 inhibitors might show lower logD values due to lower partitioning of the protonated amines at pH 7.4 into the organic phase [92]. Similarly, the lowest mean/median logD (M: 0.57/Mdn: 0.405) values were shown by highly efficient CYP2D6 inhibitors.
CYP-family-based property analysis of inhibitors, in terms of hydrogen bonding potential (HBA and HBD), was also performed. It is well explicated that the overall shape and flexibility along with molecular size, compound lipophilicity and hydrogen bonding potential are of extreme importance for the estimation of permeability of chemical entities [93]. Therefore, all these parameters were also assessed for the selected CYP inhibitor datasets (Table 2, Table S6). We observed mean hydrogen bond donor (HBD) values within 1 to 2 for all inhibitors and 1 to 3 for the highly efficient inhibitors of each CYP subtype (Table 2). The highest mean/median hydrogen bond acceptor (HBA) values were shown by all CYP3A4 inhibitors (M: 6.7/Mdn: 7), including highly efficient inhibitors of CYP3A4 (M: 8/Mdn: 9), which can be explained by the fact that CYP3A4 inhibitors exhibit high molecular weight that ultimately increases the atom count, thus subsequently increasing the hydrogen bonding potential. In contrast, the lowest mean/median values of HBA count was shown by all CYP1A2 inhibitors (M: 4/Mdn: 4) mainly due to planar aromatic compounds with small volume to surface ratio [94]. A similar trend in HBA count was observed for the highly efficient inhibitors of CYP1A2 (M: 5/Mdn: 5) (Table 2).
Figure 6 represents the overall Lipinski’s violations for each CYP inhibitor dataset. Overall, greater number of CYP1A2 inhibitors (81.37%) do not violate the RO5 followed by inhibitors of CYP2D6 (74.32%), CYP2C19 (70.20%), CYP2C9 inhibitors (64.50%) with only 57.26% CYP3A4 inhibitors as shown in Figure 6. Various studies elucidating the relationships between CYP enzymes and calculated properties have been reported extensively in literature, however, the CYP inhibition is of utmost concern in terms of RO5 violations [15,55,95,96]. The RO5 guidelines are based on molecular properties and, therefore, do not take into account the affinity of a ligand towards its particular target [97]. Thus, RO5 is a simplistic criteria solely based on molecular properties that does not consider a ligands affinity towards its target [97] and safety profiling. Therefore, majority of the CYP inhibitors from our dataset showing drug-like properties (no RO5 violations) also show greater chances of toxicological outcomes due to the inhibition of CYP isoforms.
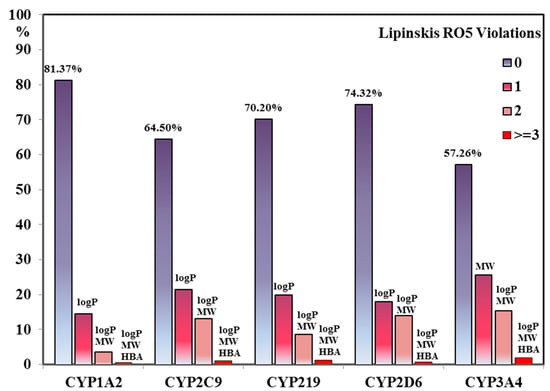
Figure 6.
The bar chart distribution showing the number of Lipinski’s violations (RO5) against CYP inhibitor datasets. The most commonly violated RO5 descriptor pairs for each CYP subtype including logP, MW and HBA have also been labelled. For each data set bars are color-coded according to the number of RO5 violations, where 0 indicates no violation and 1,2 and ≥ 3 show one, two and three or more RO5 violations, respectively.
Interestingly, the greatest numbers of RO5 violations were observed for logP and MW descriptors which can be explained by the fact that CYP inhibitors are larger and highly lipophilic in nature. Generally, for the two most important RO5 descriptors (logP and MW) it is shown that an increased lipophilicity (logP) is associated to target promiscuity and toxicity, whereas an increased MW leads to decreased promiscuity [17,96,98,99]. It is also well explicated that highly lipophilic compounds show a greater potential for hERG and CYP inhibition which clearly explains the trend observed for highest RO5 logP violations in our dataset [15,87]. However, CYP3A4 inhibition has also been correlated to increased MW and lipophilicity with decreased Fsp3 which might lead to potential drug–drug interactions and clearance issues [15,55,95,96]. This is also depicted by the greatest RO5 MW and logP violations for CYP3A4 inhibitors from our data set (Figure 6).
Moreover, the Golden Triangle hypothesis was originally proposed by Johnson et al. that aids the selection of molecules with better permeability, metabolic stability and improved potency by simultaneously optimizing the overall absorption and clearance of chemical entities. Principally, in vitro permeability (Caco-2 cells: 16,227 compounds) and metabolic data (human liver microsomes (HLM): 47,018 compounds) were used for analysis with physicochemical properties including MW and logD, where a positive correlation was observed between logD and permeability at a given MW. However, for metabolic clearance, a negative correlation was observed with logD and MW. Therefore, the combination of permeability and HLM data were used to define favorable thresholds with baseline logD ranging from −2.0–5.0 at MW of 200 Da and an apex at logD 1.0–2.0 and MW of 450 Da for compounds with better permeability and metabolic stability properties [55]. Since the logD and MW parameters are also closely related to LipE, LE and lipophilic metabolic efficiency (LipMetE) parameters, therefore, the Golden Triangle can be effectively used by designing leads against true therapeutic targets with optimal LipE, LE and LipMetE into the center of Golden Triangle to provide better potency, absorption/permeability, metabolic stability and suitable clearance properties for new chemical entities [55].
Herein, the logD and MW properties have been calculated for our antitarget inhibitor datasets (selected CYP450 isoforms). The highly lipophilic CYP inhibitors lying outside the Golden Triangle fail to display better permeability and show low in vitro clearance and, thus, represent poor pharmacokinetics. The CYP inhibitors from our datasets with high MW and lower logD values do not lie with the Golden Triangle mainly due to low permeability, whereas a greater number of highly lipophilic CYP inhibitors with high MW lie outside this region due to higher in vitro clearance. Ideally, while screening against an antitarget (toxicity prediction), compounds within the Golden Triangle represent safer chemical entities while the ones lying outside this region represent more notorious compounds due to poor absorption and permeability properties. However, for our datasets, the highest number of CYP1A2 (70.75%) and the lowest number of CYP3A4 (44.70%) inhibitors were observed within the Golden Triangle region mainly due to high MW and highly lipophilicity. Moreover, a better prevalence of inhibitors within the Golden Triangle has also been observed for CYP2D6 (66.48%), CYP2C19 (62.52%) and CYP2C9 (52.57%). It is also observed that the majority of the highly efficient inhibitors of each CYP subtype also lie within this window mainly due to the fulfillment of the efficiency criteria (clogP ~1.0–3.0, LipE ≥ 5, LE ≥ 0.29, MW ≤ 500) (Figure 7a–e). Overall, the presence of most efficient, as well as highly active, inhibitors of CYP isoforms (toxic) within the Golden Triangle indicates that majority of the CYP inhibitors show properties of safer compounds but still they are notorious and show a greater degree of CYP inhibition potential.
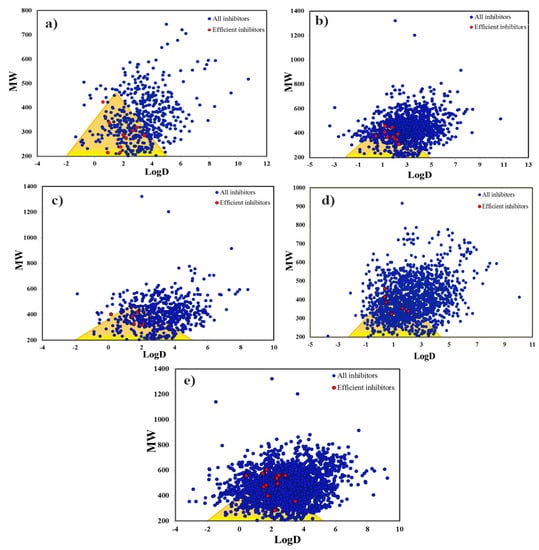
Figure 7.
Golden Triangle rule positioning of (a) CYP1A2 inhibitors, (b) CYP2C9 inhibitors, (c) CYP2C19 inhibitors, (d) CYP2D6 inhibitors and (e) CYP3A4 inhibitors. Here, all compounds are denoted by blue points, whereas highly efficient inhibitors are shown by red points. The compounds located in the Golden Triangle show a greater likelihood of an optimal permeability, low clearance and a better metabolic stability.
The 3/75 rule is yet another important rule introduced by Pfizer that takes in account physicochemical properties and is mainly based on the observation that at a plasma concentration < 10 µM (Cmax), a logP > 3 and TPSA < 75Å2 leads to a greater possibility of adverse and toxicological outcomes [56]. TPSA is an important physicochemical parameter related to hydrogen bonding that shows the sum of surfaces of all polar atoms (mainly oxygen and nitrogen) and is frequently used for the assessment of oral bioavailability and permeability [96,100]. Moreover, an increasing trend in TPSA values is indicative of reduced permeability and the overall bioavailability [100]. The trends of TPSA for our datasets were also monitored across various classes and activity levels and are shown in Table 2.
Additionally, the Pfizer 3/75 rule was also applied to all inhibitor datasets and the greatest number of CYP1A2 inhibitors (54.24%) followed by 46.1% CYP2D6,44.4% CYP2C19,34.5% CYP2C9 and 29% CYP3A4 inhibitors were observed in the pink region, indicating a greater likelihood to cause toxicity and experimental promiscuity (Figure 8a–e). A compound with clogP > 3 and TPSA < 75 is observed within the unacceptable region indicated by pink whereas, a chemical entity with clogP < 3 and TPSA > 75 defines the acceptable region of safety (green region). Similar to the Golden Triangle results, a substantial number of CYP inhibitors, including the highly efficient inhibitors, reside outside the pink region but still they are capable of causing toxicological outcomes mediated by CYP inhibition. Therefore, there is an earnest need to assess the physicochemical property trends of CYP inhibitors and noninhibitors at different classification levels and to construct highly accurate predictive models for the safety profiling.
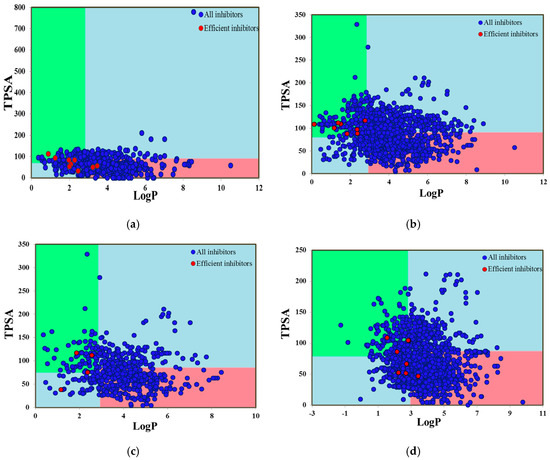
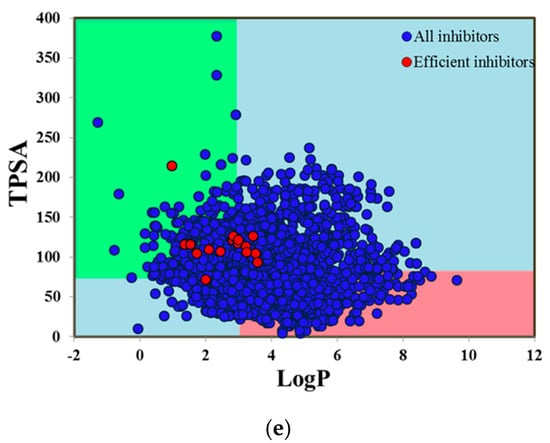
Figure 8.
Pfizer 3/75 rule positioning of (a) CYP1A2 inhibitors, (b) CYP2C9 inhibitors, (c) CYP2C19 inhibitors, (d) CYP2D6 inhibitors and (e) CYP3A4 inhibitors. Here, all compounds are denoted by blue points and highly efficient inhibitors of CYP isoforms are shown by red points. The compounds located in the pink square show a greater likelihood to cause toxicity and experimental promiscuity.
Other important descriptors encoding molecular flexibility of CYP inhibitors were also assessed by the number of rings, rotatable bonds, stereocenters and the fraction of sp3 hybridized carbons (Fsp3) since it plays an influential role in determining the overall permeability, bioavailability and promiscuity against a particular target [95,101]. Overall, an increased risk of hERG toxicity and CYP inhibition has also been associated with an aromatic ring count greater than three [101,102]. Therefore, higher ring counts (between 0 to 7) were shown by all inhibitors and highly efficient inhibitors of CYPs and no significant variations were observed in the mean/median ranges as shown in Table 2. Additionally, the greatest molecular flexibility in terms of rotatable bonds was shown by all CYP3A4 inhibitors, including highly efficient CYP3A4 inhibitors and the lowest being shown by CYP1A2 inhibitors which correlates with the molecular planarity of CYP1A2 inhibitors [36]. For a detailed analysis of the calculated properties of all classes of inhibitors (all/highly efficient) against each CYP isoform, refer to Table 2.
4. Decision Trees
In the current study, predictive decision tree models were obtained with HBD_HBA count, sCenters, number of rings, HBAs, HBDs, total charge, molecular weight, logD, logP and vsa_acc. The values of selected descriptors for active, efficient and inactive compounds against each CYP subtype are summarized in Table 3. The statistical parameters of each model are shown in Table 4.
Table 3.
The relevant set of descriptors for each CYP inhibition decision tree classifier along with the description, average values of selected descriptors for active, efficient and inactive inhibitors against each CYP isoform are presented.
Table 4.
The model evaluation parameters for the inhibitor-based decision tree classifiers of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. MCC: Matthews correlation coefficient; AUC: Area under the curve.
A decision tree model for the classification of CYP1A2 inhibition was built using a training set of 612 inhibitors with data split into 566 active compounds (including 12 efficient) with IC50 values ≤ 50 µM and 46 inactives with IC50 > 50 µM. Most prominently, for CYP1A2 inhibitors the HBD and HBA count (HBA_HBD) was identified as an important classification descriptor. Other discerning descriptors for this class include stereocenters (sCenters), hydrogen bond acceptors (HBA) and molecular weight (Figure 9a). Additionally, for active inhibitors a HBA count of ≤6 is shown by the decision tree classifier for CYP1A2 inhibition. A similar range of HBA count for CYP1A2 inhibitors has been reported by Vasanthanathan et al. [103]. Overall, it is shown by the decision tree classifier that the active inhibitors of CYP1A2 show lower HBA_HBD counts (≤ 11), sCenters, HBA (≤ 6) and molecular weights (≤ 507) in comparison to inactive CYP1A2 inhibitors. However, the highly efficient CYP1A2 inhibitors display lower mean molecular weights and sCenters along with higher HBA in comparison to actives (Mean: 294.18/0.11/5) and inactives (Mean: 422.65/0.80/5.78) (Table 3).
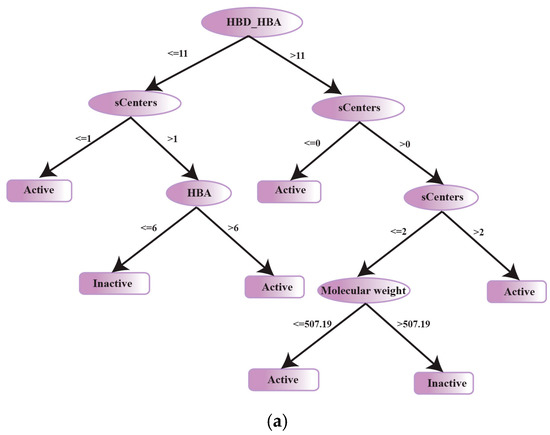
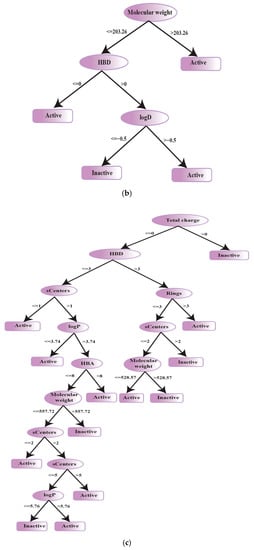
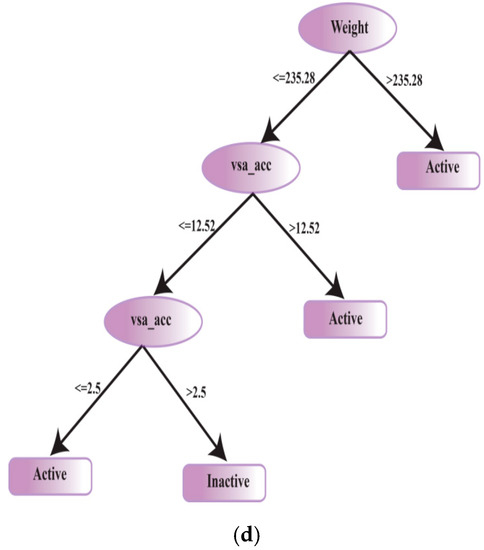
Figure 9.
(a) A J-48-pruned decision tree for CYP1A2 inhibitors based on HBD_HBA, sCenters, HBA and molecular weight. (b) A J-48-pruned decision tree for CYP2C9 inhibitors based on molecular weight, HBD and logD. (c) A J-48-pruned decision tree for CYP2D6 inhibitors based on total charge, stereocenters, HBD, HBA, logP, rings and molecular weight descriptors. (d) A J-48-pruned decision tree for CYP3A4 inhibitors based molecular weight and vsa_acc from the set of selected descriptors.
For the CYP2C9 pruned decision tree model, molecular weight appeared to be the branching descriptor. Other discriminating descriptors for CYP2C9 inhibitors include HBD and logD as shown in Figure 9b. Previously, Jónsdóttir et al. proposed that, in comparison to CYP2C9 substrates, the CYP2C9 inhibitors exhibit larger mean molecular weight and polar surface area which further strengthens the selection of our descriptor set [104]. In contrast, Ekins et al. have delineated, through different inhibitor data sets, that CYP2C9 inhibitor binding is controlled by multiple factors within the binding site, such as hydrophobic, hydrogen bond acceptor and donor interactions which reflects the significance of hydrogen bonding potential as described by descriptors (HBD, logD) in our model [105]. Largely, the trends of MW, logD and HBD descriptors selected by our CYP2C9 inhibition classification model have already been discussed across various activity levels in the physicochemical property analysis section (Table 3).
For CYP2D6 inhibition classification shape, atomic polarizability, electrostatic, hydrophobic, lipophilicity and acid base features have already been reported in literature [106,107]. Herein, a CYP2D6 decision tree classifier was built using 1647 compounds and a set of seven descriptors as explained in Table 3. Total charge descriptor appeared as the branching node and the other selected descriptors include HBD, HBA, rings, sCenters, logP and molecular weight (Figure 9c). Two extremely important descriptors including logP(o/w) and molecular weight were selected by our CYP2D6 inhibitor classification showing mean logP(o/w) and MW of 400.19/3.69 and 395.50/3.61 for actives and inactives, respectively. Considering the selected descriptors for the three categories of active, efficient and inactive CYP2D6 inhibitors, it is evident that efficient inhibitors represent the highest mean HBD, HBA, total charge and stereocenters with lower molecular weights, logD values and ring counts (Table 3).
Finally, CYP3A4 inhibition-based decision tree was built using a set of 2641 active including 43 efficient and 106 inactive compounds (Figure 1). Briefly, size and hydrophobicity of a chemical entity are the molecular properties that have an influential role in determining CYP3A4 inhibition [82]. Choi et al. have built recursive partitioning trees for CYP3A4 inhibitor and noninhibitor classification using a set of 2D descriptors indicating molecular weight as the most conclusive feature which is also shown by the physicochemical property analysis and decision tree of CYP3A4 inhibitors. For the decision tree, the other discerning descriptor was vsa_acc (Figure 9d). The chemical entities with a molecular weight >235.28 were classified as active CYP3A4 inhibitors. These also include 17 highly efficient inhibitors with LE and LipE values in the range of already discussed thresholds. It is obvious that the highest mean values of molecular weight and vsa_acc were shown by highly efficient CYP3A4 inhibitors followed by active and inactive compounds (Table 3).
Additionally, for our decision tree classifiers, the model evaluation was performed using specificity, sensitivity, accuracy, MCC and kappa statistics. A large number of independent studies based on classification models to probe inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 with variable data sets (109–17,143 compounds), overall accuracies, MCC and kappa statistics in the ranges 61.9–97%, 0.287–1 and 0.38–0.65 have been reported in literature until now [36,103,106,107,108,109,110,111,112,113,114,115]. The details of statistical parameters to evaluate the model performance have been provided in Table 4. It is notable that all four CYP inhibition-based decision tree models in this study show an overall accuracy and sensitivity above 90% for both the training set and 10-folds cross validation. Moreover, based on the model evaluation parameters the best performance was shown by the decision tree classifier for CYP1A2 followed by CYP2D6 inhibition (Table 4).
5. Discussion
The discovery of small-molecule drugs is a challenging endeavor that relies on parallel optimization of several parameters including efficacy, pharmacokinetics and safety [116]. Therefore, ADMET properties, in addition to the pharmacological parameters, are of extreme importance to achieve clinical success of a drug candidate [117]. Amongst these, drug metabolism plays a major role in determining the therapeutic fate of drugs where high metabolic liability can ultimately lead to high clearance and loss of pharmacological activity. However, poor metabolic turnover might lead to toxicity and adverse drug reactions due to the accumulation of drugs or active metabolites. Additionally, the inhibition or induction of drug metabolism due to co-administration of drugs might also lead to potential drug–drug interactions. Furthermore, during the recent years, high attrition rates during preclinical and later stages of clinical drug development have been associated with drug safety problems [118,119,120,121]. For small-molecule drug candidates, toxicology at preclinical phase and drug safety concerns at clinical trial remain the major reasons for higher attrition rates and clinical failures accounting for 25% phase I and 14% phase II failures during 2013 to 2015 [122,123]. Therefore, for the elucidation of drug metabolism at molecular level, a better understanding of metabolic properties and a revised research and development (R&D) strategy might assist in the optimization of the metabolic stability and safety properties of NCEs, eventually leading to an efficacious drug discovery and development process [124]. Thus, for the improvement of R&D productivity, recently a 5R framework based on right target, right tissue, right safety, right patient and right commercial potential has been applied that increased the success rates from 4% (2005–2010) to 19% (2012–2016) from candidate selection to the successful completion of phase III [3].
Furthermore, probing toxicological profiles of new chemical entities remains an important cornerstone of the drug development process which is experimentally expensive and the translation of animal model results to humans are also challenging. Therefore, a number of in silico models based on machine learning techniques using different combinations of data types have been developed for toxicity prediction of thousands of NCEs/drugs yet with their own strengths and weaknesses [125]. Herein, we presented a data set of 6999 inhibitors against five CYP isoforms mainly CYP1A2,2C9,2C19,2D6, and 3A4 with known activity values. A combination of ligand efficiency metrics (LipE and LE), physicochemical parameters and decision tree models have been used to discern the important property trends across various activity levels (actives- > highly efficient- > inactive) to probe CYP inhibition using large datasets of CYP inhibitors.
Table 1 summarizes clogP, LipE, LE and heavy atom ranges across the activity levels of CYP inhibitors which might provide valuable ranges and mean values generally for all CYP inhibitors and particularly for most efficient CYP inhibitors. It is important to note that a CYP inhibitor might not necessarily be an efficient inhibitor. Therefore, the estimation of these parameter ranges might prove useful for the differentiation between general inhibitors and efficient/potent inhibitors against the selected CYP450s. Additionally, during the recent years, many studies probing the properties of fragments, HTS High Throughput Screening) hits, corresponding leads, clinical candidates and marketed drugs for the investigation and identification of successful trends in physicochemical parameters leading to the formulation of several rules for future drug designing programs have been reported [49,54,126,127,128,129]. The physicochemical properties of oral drugs reaching clinical phase II were estimated by Lipinski et al. to frame the well-known rule of five indicating that a logP ≤ 5, MW ≤ 500, HBAs ≤ 10 and HBDs ≤ 5 is necessary for absorption or permeation [54]. For fragments and drug-like compounds, these parameters have already been estimated (see Table 5) but the current study is aimed at identifying the ranges of RO5 parameters, TPSA, rotatable bonds, LipE and LE generally for all CYP inhibitors and more specifically, for active and highly efficient inhibitors against the selected CYP isoforms.
Table 5.
The ranges and estimated average values of rule of five parameters and ligand efficiency metrics for fragments, drug-like compounds and CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 active and highly efficient inhibitors.
Amongst the RO5 physicochemical properties, MW of a chemical entity is one of the most important parameter in the drug discovery programs that can influence absorption, elimination, blood brain barrier penetration and interactions with on-targets and off-targets [15,76]. For fragment-like and drug-like compounds, MW < 300 and < 500 have been reported [54,127,130] (Table 5), whereas from the analysis of MW trends in our dataset, mean values between 340 to 470.8 and 294 to 482 have been observed for active and highly efficient CYP inhibitors, respectively. Additionally, an increasing order of MW across CYP1A2- > 2D6- > 2C19- > 2C9- > 3A4 and CYP1A2- > 2C19- > 2C9- > 2D6- > 3A4 has been observed for the active and efficient CYP inhibitors. Overall, amongst all subtypes the lowest molecular weight was observed for highly efficient (MW: 195.22–423.46) and active inhibitors (MW: 108.14–742.95) of CYP1A2, whereas the highest molecular weight is shown by highly efficient (MW: 284.34–604.14) and active inhibitors (MW: 108.14–1202.61) of CYP3A4; thus, indicating the potential of lower MW (195–423) entities to inhibit CYP1A2 and higher MW (284.34–604.14) compounds to inhibit CYP3A4.
Additionally, lipophilicity parameter is an important mediator of the overall ADMET properties, where a high lipophilicity might hamper compound solubility and metabolism, whereas lower lipophilicity might ultimately lead to decreased permeability [87]. It is also well explicated that high lipophilicity associated with a chemical entity might lead to target promiscuity and toxicity issues arising from hERG inhibition, phospholipidosis or cytochrome P450 (CYP) inhibitions [15,87]. Generally, for fragments and drug-like compounds a clogP of < 3 and < 5 has been reported [54,127,130], but for our dataset of CYP inhibitors, a mean clogP of ~4 has been observed for active inhibitors of all CYP subtypes, whereas for highly efficient inhibitors, mean clogP values between 1.56–2.36 have been observed (Table 5). Moreover, lipophilicity of active compounds increased from CYP2D6- > 3A4- > 1A2- > 2C9- > 2C19, whereas an inverse order was observed for highly efficient CYP inhibitors. Thus, new chemical entities displaying clogP values of 1.56 to ~4 might show greater CYP450 inhibition potential.
However, as far as the hydrogen bonding potential is considered, fragment- and drug-like compounds generally follow the rule of three and RO5, whereas from our datasets, the mean HBAs and HBDs counts for active and efficient CYP inhibitors were observed between 3 to 5/3 to 6 and 1 to 2/1 to 3, respectively. For active compounds, the increase in both the number of HBA and HBD was observed in the order 1A2- > 2C9- > 2C19- > 2D6- > 3A4. However, with the exception of CYP1A2 and CYP2D6 inhibitors, all highly efficient inhibitors displayed a greater number of HBA/HBD as shown in Table 5. Thus, indicating that new compounds containing a lower number of HBA/HBD (Mean: 2–3/1–2) screened against the studied parameters might display greater inhibition potential against CYP1A2 and CYP2D6, respectively. Another descriptor associated to hydrogen bonding is the topological surface area (TPSA) [131,132] that was monitored for the CYP inhibitor datasets. A previous QSPR(Quantitative Structure Property Relationship) study associated an increase in the TPSA and the number of rotatable bonds to decreased oral bioavailability, and proposed that TPSA can be used effectively with number of rotatable bonds to reveal flexibility of molecules [133]. Moreover, thresholds of rotatable bonds (≤ 10) and TPSA (≤ 140 Å2) were defined to obtain direct correlation with oral bioavailability in the rat [133]. A TPSA of ≤ 60 Å2 and ≤ 140 Å2 has been described in literature for fragments and drugs [54,127,130,133] in humans, whereas for our CYP inhibitor data sets, increasing trend of TPSA was observed while moving from actives to efficient inhibitors with CYP3A4 showing the highest TPSA values of > 83.46 Å2 and CYP1A2 showing the lowest TPSA values (> 57.25 Å2), as shown in Table 5. For CYP1A2 and CYP2C9, an increase in number of rotatable bonds was observed while moving from efficient to active inhibitors, whereas for CYP2C19, CYP2D6 and CYP3A4, no difference was observed between active and efficient inhibitors in terms of rotatable bonds. Overall, our results show that new chemical entities displaying mean TPSA values between 67.6–105 and mean rotatable bonds between 2 to 8 might show better bioavailability ultimately leading to a greater inhibition potential against the major metabolic CYP isoforms during first pass metabolism. Specifically, new chemical entities with mean TPSA values and number of rotatable bonds falling within 56.25–67.61/2–5,81.3–93.75/6–7,71.9–98.81/6,62.58–71.67/7 and 83.46–105/8 might show greater inhibition potential against CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 due to increased bioavailability.
Various classification models to probe the inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 with variable data sets, overall accuracies, MCC and kappa statistics have been reported in literature [35,71,72,73,74,75,76,78,79,95,96,97]. However, a direct comparison with already reported instances in literature is difficult since variable datasets, descriptor selection and description methods have been used. Thus, a comparison of model evaluation parameters of studies reporting CYP inhibition classification models would be reasonable. Overall, an accuracy and sensitivity above 90% is shown by our CYP inhibition-based decision tree models using training sets and 10-folds cross validation (Table 4). The MCC, kappa statistics and AUC values for the CYP inhibition-based decision tree models were observed in the ranges 0.299–0.50,0.233–0.43 and 0.597–0.78, respectively, with CYP3A4 classifier showing the lowest MCC, kappa and AUC values. Based on the model evaluation parameters, the best performance was shown by the decision tree classifier for CYP1A2 inhibition (accuracy = 94.43, sensitivity = 94.6, specificity = 87.50, MCC = 0.50, kappa = 0.43) followed by CYP2D6 (accuracy = 96.49, sensitivity = 96.63, specificity = 86.36, MCC = 0.46, kappa = 0.39) and CYP2C9 (accuracy = 96.01, sensitivity = 95.98, specificity = 100.0, MCC = 0.37, kappa = 0.25) classifiers, respectively (Table 4).
Generally, for the prediction of drug–drug interactions associated to CYP inhibition, various in silico approaches and web based computational tools have been reported in literature [134,135,136,137,138,139]. These include WhichCyp [134], vNN Web Server [136], admetSAR [140] and yet other freely available tools based on classification models for the prediction of CYP inhibition potential associated with new chemical entities. So far, other more sophisticated methods, based on dynamic mechanistic model taking into account the simultaneous influence of reversible and irreversible CYP inhibition and DDI module of GastroPlusTM for the prediction of time dependent CYP inhibition, have also been reported in the literature [137,139]. However, in this particular study, the trends of ligand efficiency metrics and physicochemical properties for cytochrome P450 enzymes using large datasets of CYP inhibitors have been calculated as a simple step towards a better understanding of cytochrome P450 inhibition by estimating activity thresholds across various classes and activity levels (while considering active, efficient and finally the inactive inhibitors) which might assist in the optimization of overall properties of new chemical entities during the drug discovery phases.
6. Conclusions
Here, we estimated LipE and LE profiles along with the physicochemical properties and decision tree models for CYP1A2,2C9,2C19,2D6 and 3A4 inhibitor classification to effectively distinguish active inhibitors from inactive and highly efficient inhibitors. Moreover, the features important for inhibition against each CYP isoform were encoded by the relevant set of descriptors including molecular weight, lipophilicity, number of hydrogen bond acceptor and donors, total charges, stereocenters and ring counts. Additionally, the clogP, LipE, heavy atom count and LE trends were analyzed for CYP1A2,2C9,2C19,2D6 and 3A4 inhibitor data sets to provide the thresholds of these parameters for active (IC50 ≤ 50 µM), highly potent (clogP ~1.0–3.0, LipE ≥ 5, LE ≥ 0.29) and inactive (IC50 > 50–100 µM) inhibitors against each CYP subtype. Generally, amongst the entire data set of CYP inhibitors, the highly efficient inhibitors show mean MW, HBA, HBD and logP values between 294.18–482.40,5.0–8.2,1–7.29 and 1.68–2.57, respectively. Overall, our results could aid the early prediction of CYP inhibition against the major players of drug metabolism (CYP1A2,2C9,2C19,2D6 and 3A4) during the drug discovery phases.
Supplementary Materials
The following are available online at https://www.mdpi.com/2079-3197/7/2/26/s1, Table S1: LipE and LE profiling results of CYP1A2 inhibitors dataset, Table S2: LipE and LE profiling results of CYP2C9 inhibitors dataset, Table S3: LipE and LE profiling results of CYP2C19 inhibitors dataset, Table S4: LipE and LE profiling results of CYP2D6 inhibitors dataset and Table S5: LipE and LE profiling results of CYP3A4 inhibitors dataset.
Author Contributions
Y.S.K. and I.J. conceived and designed the project. Y.S.K. and I.J. carried out all the work, analyzed the results and wrote the paper.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Meanwell, N.A. Improving drug design: An update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 2016, 29, 564–616. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamrén, B.; Johnson, A.; March, R.E. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 2018, 17, 167–181. [Google Scholar] [PubMed]
- Taavitsainen, P.; Honkakoski, P.; Juvonen, R.; Pelkonen, O.; Raunio, H. Role of xenobiotic metabolism in drug discovery and development. Biotechnology-Volume XII: Fundamentals in Biotechnology, 2009; 11, 108. [Google Scholar]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Bertz, R.J.; Granneman, G.R. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin. Pharmacokinet. 1997, 32, 210–258. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E. Clinically important pharmacokinetic drug–drug interactions: Role of cytochrome P450 enzymes. J. Clin. Pharm. Ther. 1998, 23, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Caldwell, G.W. Metabolism profiling, and cytochrome P450 inhibition & induction in drug discovery. Curr. Top. Med. Chem. 2001, 1, 403–425. [Google Scholar] [PubMed]
- Friedman, M.A.; Woodcock, J.; Lumpkin, M.M.; Shuren, J.E.; Hass, A.E.; Thompson, L.J. The safety of newly approved medicines: Do recent market removals mean there is a problem? JAMA 1999, 281, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Lasser, K.E.; Allen, P.D.; Woolhandler, S.J.; Himmelstein, D.U.; Wolfe, S.M.; Bor, D.H. Timing of new black box warnings and withdrawals for prescription medications. JAMA 2002, 287, 2215–2220. [Google Scholar] [CrossRef]
- Hasler, J.A.; Estabrook, R.; Murray, M.; Pikuleva, I.; Waterman, M.; Capdevila, J.; Holla, V.; Helvig, C.; Falck, J.R.; Farrell, G. Human cytochromes P450. Mol. Asp. Med. 1999, 20, 1–137. [Google Scholar] [CrossRef]
- Nassar, A.-E.F.; Kamel, A.M.; Clarimont, C. Improvingthe decision-making process in structural modification of drug candidates: Reducing toxicity. Drug Discov. Today 2004, 9, 1055–1064. [Google Scholar] [CrossRef]
- Lewis, D.F.; Dickins, M. Substrate SARs in human P450s. Drug Discov. Today 2002, 7, 918–925. [Google Scholar] [CrossRef]
- Lewis, D.F.; Dickins, M. Baseline lipophilicity relationships in human cytochromes P450 associated with drug metabolism. Drug Metab. Rev. 2003, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Davis, A.M.; Chohan, K.K.; Paine, S.W.; Boyer, S.; Gavaghan, C.L.; Arnby, C.H.; Kankkonen, C.; Albertson, N. Generation of in-silico cytochrome P450 1A2,2C9,2C19,2D6, and 3A4 inhibition QSAR models. J. Comput. Aided Mol. Des. 2007, 21, 559–573. [Google Scholar] [CrossRef]
- Lewis, D.F.; Lake, B.G.; Ito, Y.; Dickins, M. Lipophilicity relationships in inhibitors of CYP2C9 and CYP2C19 enzymes. J. Enzym. Inhib. Med. Chem. 2006, 21, 385–389. [Google Scholar] [CrossRef]
- Lewis, D.F.; Lake, B.G.; Dickins, M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: The importance of compound lipophilicity. J. Enzym. Inhib. Med. Chem. 2007, 22, 1–6. [Google Scholar] [CrossRef]
- Ai, C.; Li, Y.; Wang, Y.; Chen, Y.; Yang, L. Insight into the effects of chiral isomers quinidine and quinine on CYP2D6 inhibition. Bioorg. Med. Chem. Lett. 2009, 19, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Didziapetris, R.; Dapkunas, J.; Sazonovas, A.; Japertas, P. Trainable structure–activity relationship model for virtual screening of CYP3A4 inhibition. J. Comput. Aided Mol. Design 2010, 24, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Roy, P.P. Comparative chemometric modeling of cytochrome 3A4 inhibitory activity of structurally diverse compounds using stepwise MLR, FA-MLR, PLS, GFA, G/PLS and ANN techniques. Eur. J. Med. Chem. 2009, 44, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Yano, J.K.; Wester, M.R.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structure of human microsomal cytochrome P450 2C8 evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004, 279, 9497–9503. [Google Scholar] [CrossRef]
- Szklarz, G.D.; Halpert, J.R. Molecular basis of P450 inhibition and activation: Implications for drug development and drug therapy. Drug Metab. Dispos. 1998, 26, 1179–1184. [Google Scholar] [PubMed]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Vinković, D.M.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinković, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Wester, M.R.; Yano, J.K.; Schoch, G.A.; Yang, C.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-Å resolution. J. Biol. Chem. 2004, 279, 35630–35637. [Google Scholar] [CrossRef]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Wester, M.R.; Johnson, E.F.; Marques-Soares, C.; Dansette, P.M.; Mansuy, D.; Stout, C.D. Structure of a substrate complex of mammalian cytochrome P450 2C5 at 2.3 Å resolution: Evidence for multiple substrate binding modes. Biochemistry 2003, 42, 6370–6379. [Google Scholar] [CrossRef]
- Fowler, S.; Zhang, H. In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: Current status on methodologies and their utility for predicting drug–drug interactions. AAPS J. 2008, 10, 410–424. [Google Scholar] [CrossRef]
- Nembri, S.; Grisoni, F.; Consonni, V.; Todeschini, R. In silico prediction of cytochrome p450-drug interaction: Qsars for cyp3a4 and cyp2c9. Int. J. Mol. Sci. 2016, 17, 914. [Google Scholar] [CrossRef]
- Dimelow, R.J.; Metcalfe, P.D.; Thomas, S. In Silico Models of Drug Metabolism and Drug Interactions. In Encyclopedia of Drug Metabolism and Interactions; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Refsgaard, H.H.; Jensen, B.F.; Christensen, I.T.; Hagen, N.; Brockhoff, P.B. In silico prediction of cytochrome P450 inhibitors. Drug Dev. Res. 2006, 67, 417–429. [Google Scholar] [CrossRef]
- Marechal, J.-D.; Yu, J.; Brown, S.; Kapelioukh, I.; Rankin, E.M.; Wolf, C.R.; Roberts, G.C.; Paine, M.J.; Sutcliffe, M.J. In silico and in vitro screening for inhibition of cytochrome P450 CYP3A4 by comedications commonly used by patients with cancer. Drug Metab. Dispos. 2006, 34, 534–538. [Google Scholar] [CrossRef]
- Ashour, M.L.; Youssef, F.S.; Gad, H.A.; Wink, M. Inhibition of cytochrome P450 (CYP3A4) activity by extracts from 57 plants used in traditional chinese medicine (TCM). Pharmacogn. Mag. 2017, 13, 300. [Google Scholar]
- Nookala, A.R.; Li, J.; Ande, A.; Wang, L.; Vaidya, N.K.; Li, W.; Kumar, S.; Kumar, A. Effect of methamphetamine on spectral binding, ligand docking and metabolism of anti-hiv drugs with CYP3A4. PLoS ONE 2016, 11, e0146529. [Google Scholar] [CrossRef]
- Hochleitner, J.; Akram, M.; Ueberall, M.; Davis, R.A.; Waltenberger, B.; Stuppner, H.; Sturm, S.; Ueberall, F.; Gostner, J.M.; Schuster, D. A combinatorial approach for the discovery of cytochrome P450 2D6 inhibitors from nature. Sci. Rep. 2017, 7, 8071. [Google Scholar] [CrossRef]
- Su, B.-H.; Tu, Y.-s.; Lin, C.; Shao, C.-Y.; Lin, O.A.; Tseng, Y.J. Rule-based prediction models of cytochrome P450 inhibition. J. Chem. Inf. Model. 2015, 55, 1426–1434. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Carbonell, P.; Chevillard, F.; Moroy, G.; Nicot, A.B.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Integrated structure-and ligand-based in silico approach to predict inhibition of cytochrome P450 2D6. Bioinformatics 2015, 31, 3930–3937. [Google Scholar]
- Kaur, P.; Chamberlin, A.R.; Poulos, T.L.; Sevrioukova, I.F. Structure-based inhibitor design for evaluation of a CYP3A4 pharmacophore model. J. Med. Chem. 2015, 59, 4210–4220. [Google Scholar] [CrossRef]
- Baudry, J.; Li, W.; Pan, L.; Berenbaum, M.R.; Schuler, M.A. Molecular docking of substrates and inhibitors in the catalytic site of CYP6B1, an insect cytochrome P450 monooxygenase. Protein Eng. 2003, 16, 577–587. [Google Scholar] [CrossRef][Green Version]
- Mukhtar, S.; Kiani, Y.S.; Jabeen, I. Molecular docking simulations and GRID-independent molecular descriptor (GRIND) analysis to probe stereoselective interactions of CYP3A4 inhibitors. Med. Chem. Res. 2017, 26, 2322–2335. [Google Scholar] [CrossRef]
- Jensen, B.F.; Vind, C.; Padkjær, S.B.; Brockhoff, P.B.; Refsgaard, H.H. In silico prediction of cytochrome P450 2D6 and 3A4 inhibition using Gaussian kernel weighted k-nearest neighbor and extended connectivity fingerprints, including structural fragment analysis of inhibitors versus noninhibitors. J. Med. Chem. 2007, 50, 501–511. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Bio-Loom Program, T.V. BioByte Co. Available online: http://biobyte.com/bb/prod/bioloom.html (accessed on 23 May 2019).
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.r.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of Ligand Efficiency Metrics; ACS Publications: Washington, DC, USA, 2014. [Google Scholar]
- Kuntz, I.; Chen, K.; Sharp, K.; Kollman, P. The maximal affinity of ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef]
- Moe. Molecular Operating Environment; Chemical Computing Group Inc.: Montreal, QC, Canada, 2013; Volume 8, H3A 2R7,2015.49. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef]
- Jantan, H.; Hamdan, A.R.; Othman, Z.A. Human talent prediction in HRM using C4. 5 classification algorithm. Int. J. Comput. Sci. Eng. 2010, 2, 2526–2534. [Google Scholar]
- Patil, T.R.; Sherekar, S. Performance analysis of Naive Bayes and J48 classification algorithm for data classification. Int. J. Comput. Sci. Appl. 2013, 6, 256–261. [Google Scholar]
- Baratloo, A.; Hosseini, M.; Negida, A.; El Ashal, G. Part 1: Simple definition and calculation of accuracy, sensitivity and specificity. Emergency 2015, 3, 48–49. [Google Scholar]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta BBA Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Cohen, J. A coefficient of agreement for nominal scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Clutterbuck, L.A.; Posada, C.G.; Visintin, C.; Riddall, D.R.; Lancaster, B.; Gane, P.J.; Garthwaite, J.; Selwood, D.L. Oxadiazolylindazole sodium channel modulators are neuroprotective toward hippocampal neurones. J. Med. Chem. 2009, 52, 2694–2707. [Google Scholar] [CrossRef]
- Fulton, J.E., Jr.; Leyden, J.; Papa, C. TReatment of vitiligo with topical methoxsalen and blacklite. Arch. Dermatol. 1969, 100, 224–229. [Google Scholar] [CrossRef]
- Parrish, J.A.; Fitzpatrick, T.B.; Tanenbaum, L.; Pathak, M.A. Photochemotherapy of Psoriasis with Oral Methoxsalen and Longwave Ultraviolet Light. N. Engl. J. Med. 1974, 291, 1207–1211. [Google Scholar] [CrossRef]
- Palmer, W.S.; Alam, M.; Arzeno, H.B.; Chang, K.-C.; Dunn, J.P.; Goldstein, D.M.; Gong, L.; Goyal, B.; Hermann, J.C.; Hogg, J.H. Development of amino-pyrimidine inhibitors of c-Jun N-terminal kinase (JNK): Kinase profiling guided optimization of a 1,2,3-benzotriazole lead. Bioorg. Med. Chem. Lett. 2013, 23, 1486–1492. [Google Scholar] [CrossRef]
- Mustafa, S.M.; Bavadekar, S.A.; Ma, G.; Moore, B.M.; Feller, D.R.; Miller, D.D. Synthesis and biological studies of yohimbine derivatives on human α 2C-adrenergic receptors. Bioorg. Med. Chem. Lett. 2005, 15, 2758–2760. [Google Scholar] [CrossRef]
- Saint-Ruf, G.; Chanh, P.H.; Hoi, B. Yohimbine Derivatives, Process for Their Preparation and Their Applications. Google Patents US3940387A, 1976. [Google Scholar]
- Hii, J.T.; Wyse, D.G.; Gillis, A.M.; Duff, H.J.; Solylo, M.A.; Mitchell, L.B. Precordial QT interval dispersion as a marker of torsade de pointes. Disparate effects of class Ia antiarrhythmic drugs and amiodarone. Circulation 1992, 86, 1376–1382. [Google Scholar] [CrossRef]
- Haydon, D.J.; Bennett, J.M.; Brown, D.; Collins, I.; Galbraith, G.; Lancett, P.; Macdonald, R.; Stokes, N.R.; Chauhan, P.K.; Sutariya, J.K. Creating an antibacterial with in vivo efficacy: Synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 2010, 53, 3927–3936. [Google Scholar] [CrossRef]
- Gallagher, E.P.; Kunze, K.L.; Stapleton, P.L.; Eaton, D.L. The Kinetics of Aflatoxin B1Oxidation by Human cDNA-Expressed and Human Liver Microsomal Cytochromes P450 1A2 and 3A4. Toxicol. Appl. Pharmacol. 1996, 141, 595–606. [Google Scholar] [CrossRef]
- De Savi, C.; Pape, A.; Cumming, J.G.; Ting, A.; Smith, P.D.; Burrows, J.N.; Mills, M.; Davies, C.; Lamont, S.; Milne, D. The design and synthesis of novel N-hydroxyformamide inhibitors of ADAM-TS4 for the treatment of osteoarthritis. Bioorg. Med. Chem. Lett. 2011, 21, 1376–1381. [Google Scholar] [CrossRef]
- Zimmermann, K.; Wittman, M.D.; Saulnier, M.G.; Velaparthi, U.; Langley, D.R.; Sang, X.; Frennesson, D.; Carboni, J.; Li, A.; Greer, A. Balancing oral exposure with Cyp3A4 inhibition in benzimidazole-based IGF-IR inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4075–4080. [Google Scholar] [CrossRef]
- Paul Gleeson, M.; Hersey, A.; Hannongbua, S. In-silico ADME models: A general assessment of their utility in drug discovery applications. Curr. Top. Med. Chem. 2011, 11, 358–381. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- van de Waterbeemd, H. Improving compound quality through in vitro and in silico physicochemical profiling. Chem. Biodivers. 2009, 6, 1760–1766. [Google Scholar] [CrossRef]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef]
- Mugumbate, G.; Overington, J.P. The relationship between target-class and the physicochemical properties of antibacterial drugs. Bioorg. Med. Chem. 2015, 23, 5218–5224. [Google Scholar] [CrossRef]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Alsherbiny, M.A.; Aly, M.H.; Ghabbour, H.A.; Abdel-Aziz, H.A. Synthesis and cytotoxic activity of biphenylurea derivatives containing indolin-2-one moieties. Molecules 2016, 21, 762. [Google Scholar] [CrossRef]
- Elekofehinti, O.O.; Ejelonu, O.C.; Kamdem, J.P.; Akinlosotu, O.B.; Famuti, A.; Adebowale, D.D.; Iwaloye, O.; Bulu, Y.I.; Kade, I.J.; Rocha, J.B.T. Discovery of potential visfatin activators using in silico docking and ADME predictions as therapy for type 2 diabetes. Beni Suef Univ. J. Basic Appl. Sci. 2018, 7, 241–249. [Google Scholar] [CrossRef]
- Hobson, A.D.; Harris, C.M.; van der Kam, E.L.; Turner, S.C.; Abibi, A.; Aguirre, A.L.; Bousquet, P.; Kebede, T.; Konopacki, D.B.; Gintant, G. Discovery of A-971432, an orally bioavailable selective sphingosine-1-phosphate receptor 5 (S1P5) agonist for the potential treatment of neurodegenerative disorders. J. Med. Chem. 2015, 58, 9154–9170. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opinion Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Lewis, D.F.; Jacobs, M.N.; Dickins, M. Compound lipophilicity for substrate binding to human P450s in drug metabolism. Drug Discov. Today 2004, 9, 530–537. [Google Scholar] [CrossRef]
- Stenberg, P.; Norinder, U.; Luthman, K.; Artursson, P. Experimental and computational screening models for the prediction of intestinal drug absorption. J. Med. Chem. 2001, 44, 1927–1937. [Google Scholar] [CrossRef]
- Cummins, C.L.; Jacobsen, W.; Benet, L.Z. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002, 300, 1036–1045. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure–activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef]
- Upthagrove, A.L.; Nelson, W.L. Importance of Amine pK a and distribution coefficient in the metabolism of fluorinated propranolol derivatives. preparation, identification of metabolite regioisomers, and metabolism by CYP2D6. Drug Metab. Dispos. 2001, 29, 1377–1388. [Google Scholar]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Lee, H.; Yeom, H.; Kim, Y.G.; Yoon, C.N.; Jin, C.; Choi, J.S.; Kim, B.-R.; Kim, D.-H. Structure-related inhibition of human hepatic caffeine N3-demethylation by naturally occurring flavonoids. Biochem. Pharmacol. 1998, 55, 1369–1375. [Google Scholar] [CrossRef]
- Lovering, F. Escape from flatland 2: Complexity and promiscuity. MedChemComm 2013, 4, 515–519. [Google Scholar] [CrossRef]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand Efficiency Indices for Drug Discovery: Towards An Atlas-Guided Paradigm; Academic Press: Waltham, MA, USA, 2013. [Google Scholar]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef]
- Price, D.A.; Blagg, J.; Jones, L.; Greene, N.; Wager, T. Physicochemical drug properties associated with in vivo toxicological outcomes: A review. Expert Opin. Drug Metab. Toxicol. 2009, 5, 921–931. [Google Scholar]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J. The impact of aromatic ring count on compound developability–are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef]
- Vasanthanathan, P.; Taboureau, O.; Oostenbrink, C.; Vermeulen, N.P.; Olsen, L.; Jorgensen, F.S. Classification of cytochrome P450 1A2 inhibitors and non-inhibitors by machine learning techniques. Drug Metab. Dispos. 2008, 37, 658–664. [Google Scholar] [CrossRef]
- Jónsdóttir, S.Ó.; Ringsted, T.; Nikolov, N.G.; Dybdahl, M.; Wedebye, E.B.; Niemelä, J.R. Identification of cytochrome P450 2D6 and 2C9 substrates and inhibitors by QSAR analysis. Bioorg. Med. Chem. 2012, 20, 2042–2053. [Google Scholar] [CrossRef]
- Ekins, S.; Bravi, G.; Binkley, S.; Gillespie, J.S.; Ring, B.J.; Wikel, J.H.; Wrighton, S.A. Three-and four-dimensional-quantitative structure activity relationship (3D/4D-QSAR) analyses of CYP2C9 inhibitors. Drug Metab. Dispos. 2000, 28, 994–1002. [Google Scholar]
- Yap, C.W.; Chen, Y.Z. Prediction of cytochrome P450 3A4,2D6, and 2C9 inhibitors and substrates by using support vector machines. J. Chem. Inf. Model. 2005, 45, 982–992. [Google Scholar] [CrossRef]
- Burton, J.; Ijjaali, I.; Barberan, O.; Petitet, F.; Vercauteren, D.P.; Michel, A. Recursive partitioning for the prediction of cytochromes P450 2D6 and 1A2 inhibition: Importance of the quality of the dataset. J. Med. Chem. 2006, 49, 6231–6240. [Google Scholar] [CrossRef]
- Kriegl, J.M.; Arnhold, T.; Beck, B.; Fox, T. A support vector machine approach to classify human cytochrome P450 3A4 inhibitors. J. Comput. Aided Mol. Des. 2005, 19, 189–201. [Google Scholar] [CrossRef]
- Chohan, K.K.; Paine, S.W.; Mistry, J.; Barton, P.; Davis, A.M. A rapid computational filter for cytochrome P450 1A2 inhibition potential of compound libraries. J. Med. Chem. 2005, 48, 5154–5161. [Google Scholar] [CrossRef]
- Arimoto, R.; Prasad, M.-A.; Gifford, E.M. Development of CYP3A4 inhibition models: Comparisons of machine-learning techniques and molecular descriptors. J. Biomol. Screen. 2005, 10, 197–205. [Google Scholar] [CrossRef]
- Burton, J.; Danloy, E.; Vercauteren, D. Fragment-based prediction of cytochromes P450 2D6 and 1A2 inhibition by recursive partitioning. SAR QSAR Environ. Res. 2009, 20, 185–205. [Google Scholar] [CrossRef]
- Choi, I.; Kim, S.Y.; Kim, H.; Kang, N.S.; Bae, M.A.; Yoo, S.-E.; Jung, J.; No, K.T. Classification models for CYP450 3A4 inhibitors and non-inhibitors. Eur. J. Med. Chem. 2009, 44, 2354–2360. [Google Scholar] [CrossRef]
- Cheng, F.; Yu, Y.; Shen, J.; Yang, L.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Inf. Model. 2011, 51, 996–1011. [Google Scholar] [CrossRef]
- Hammann, F.; Gutmann, H.; Baumann, U.; Helma, C.; Drewe, J. Classification of cytochrome P450 activities using machine learning methods. Mol. Pharm. 2009, 6, 1920–1926. [Google Scholar] [CrossRef]
- Lee, J.; Basith, S.; Cui, M.; Kim, B.; Choi, S. In silico prediction of multiple-category classification model for cytochrome P450 inhibitors and non-inhibitors using machine-learning method. SAR QSAR Environ. Res. 2017, 28, 863–874. [Google Scholar] [CrossRef]
- Schneider, G. Automating drug discovery. Nat. Rev. Drug Discov. 2017, 17, 97. [Google Scholar] [CrossRef]
- Li, A.P. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Krauss, R.M.; Roden, D.M.; Eichelbaum, M.; Hayden, M.R.; Nakamura, Y. When good drugs go bad. Nature 2007, 446, 975. [Google Scholar] [CrossRef]
- Arrowsmith, J. Phase III and submission failures: 2007–2010. Nature Rev. Drug Discov. 2011, 10, 1–2. [Google Scholar] [CrossRef]
- Arrowsmith, J. Trial Watch: Phase II Failures: 2008–2010; Nature Publishing Group: London, UK, 2011. [Google Scholar]
- Arrowsmith, J.; Miller, P. Trial Watch: Phase II and Phase III Attrition Rates 2011–2012; Nature Publishing Group: London, UK, 2013. [Google Scholar]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475. [Google Scholar] [CrossRef]
- Harrison, R.K. Phase II and Phase III Failures: 2013–2015; Nature Publishing Group: London, UK, 2016. [Google Scholar]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387. [Google Scholar] [CrossRef]
- Huang, R.; Xia, M.; Sakamuru, S.; Zhao, J.; Lynch, C.; Zhao, T.; Zhu, H.; Austin, C.P.; Simeonov, A. Expanding biological space coverage enhances the prediction of drug adverse effects in human using in vitro activity profiles. Sci. Rep. 2018, 8, 3783. [Google Scholar] [CrossRef]
- Orita, M.; Ohno, K.; Niimi, T. Two ‘Golden Ratio’indices in fragment-based drug discovery. Drug Discov. Today 2009, 14, 321–328. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar]
- Perola, E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J. Med. Chem. 2010, 53, 2986–2997. [Google Scholar] [CrossRef]
- Tarcsay, A.k.; Nyíri, K.; Keserü, G.r.M. Impact of lipophilic efficiency on compound quality. J. Med. Chem. 2012, 55, 1252–1260. [Google Scholar] [CrossRef]
- Schultes, S.; de Graaf, C.; Haaksma, E.E.; de Esch, I.J.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, e157–e162. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Leeson, P.D. Molecular inflation, attrition and the rule of five. Adv. Drug Deliv. Rev. 2016, 101, 22–33. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Rostkowski, M.; Spjuth, O.; Rydberg, P. WhichCyp: Prediction of cytochromes P450 inhibition. Bioinformatics 2013, 29, 2051–2052. [Google Scholar] [CrossRef]
- Raunio, H.; Kuusisto, M.; Juvonen, R.O.; Pentikäinen, O.T. Modeling of interactions between xenobiotics and cytochrome P450 (CYP) enzymes. Front. Pharmacol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN web server for ADMET predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef]
- Grime, K.; D Ferguson, D.; J Riley, R. The use of HepaRG and human hepatocyte data in predicting CYP induction drug-drug interactions via static equation and dynamic mechanistic modelling approaches. Curr. Drug Metab. 2010, 11, 870–885. [Google Scholar] [CrossRef]
- Lu, Y.; Figler, B.; Huang, H.; Tu, Y.-C.; Wang, J.; Cheng, F. Characterization of the mechanism of drug-drug interactions from PubMed using MeSH terms. PLoS ONE 2017, 12, e0173548. [Google Scholar] [CrossRef]
- Sohlenius-Sternbeck, A.-K.; Meyerson, G.; Hagbjörk, A.-L.; Juric, S.; Terelius, Y. A strategy for early-risk predictions of clinical drug–drug interactions involving the GastroPlusTM DDI module for time-dependent CYP inhibitors. Xenobiotica 2018, 48, 348–356. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties; ACS Publications: Washington, DC, USA, 2012. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).