
In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds

 ,
, 
 , , , and
, , , and 
Abstract
:1. Introduction
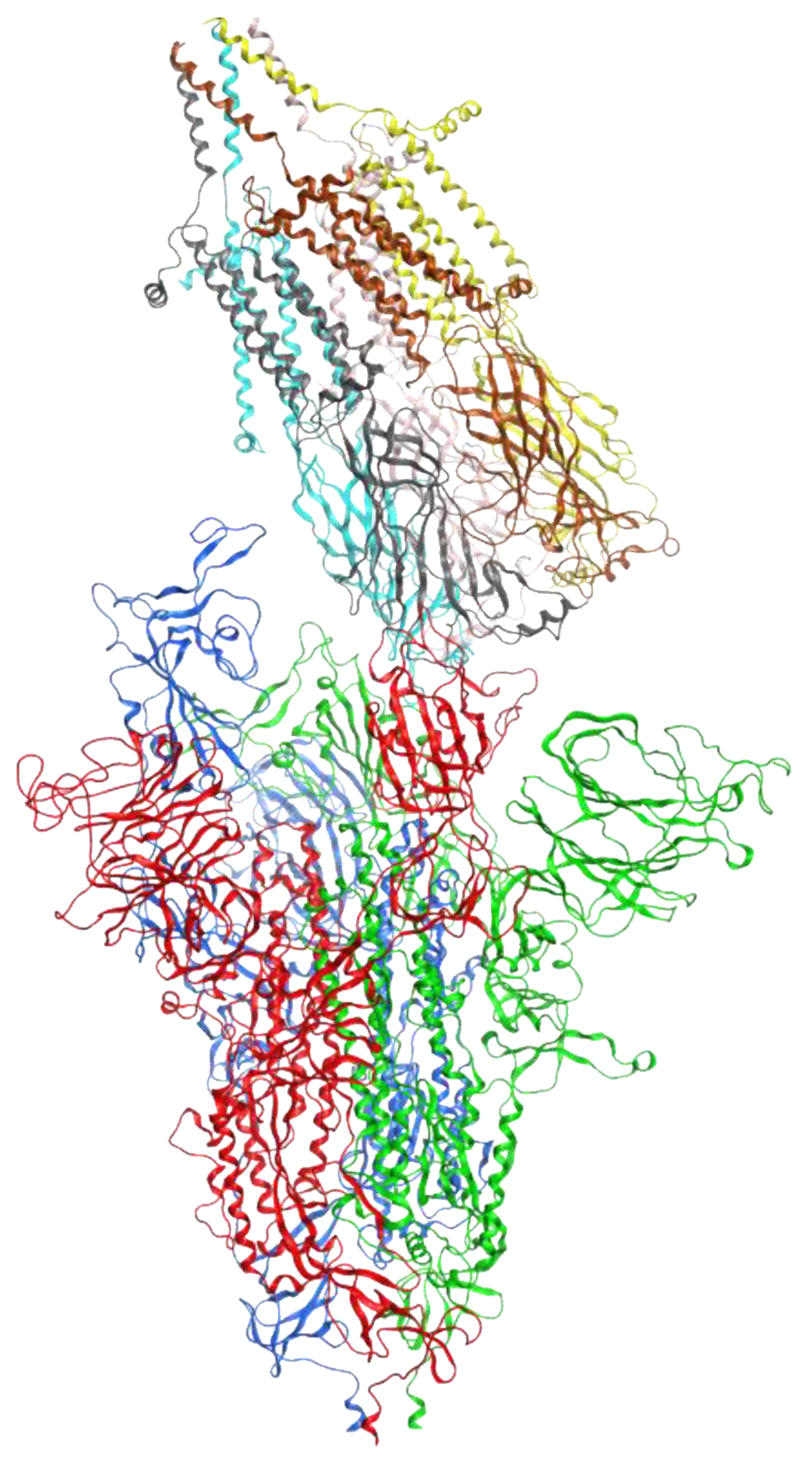
1.1. Binding of SARS-CoV-2 Spike Protein to Host Cell SA and CD147 Surface Molecules
1.2. The Role of CD147 in the Inflammatory Response
1.3. Competitive Binding of Ivermectin to SARS-CoV-2 Spike Protein Binding Sites
1.4. Nicotinic Acetylcholine Receptors: Anti-Inflammatory Modulation and Blockage of Viral Bindings
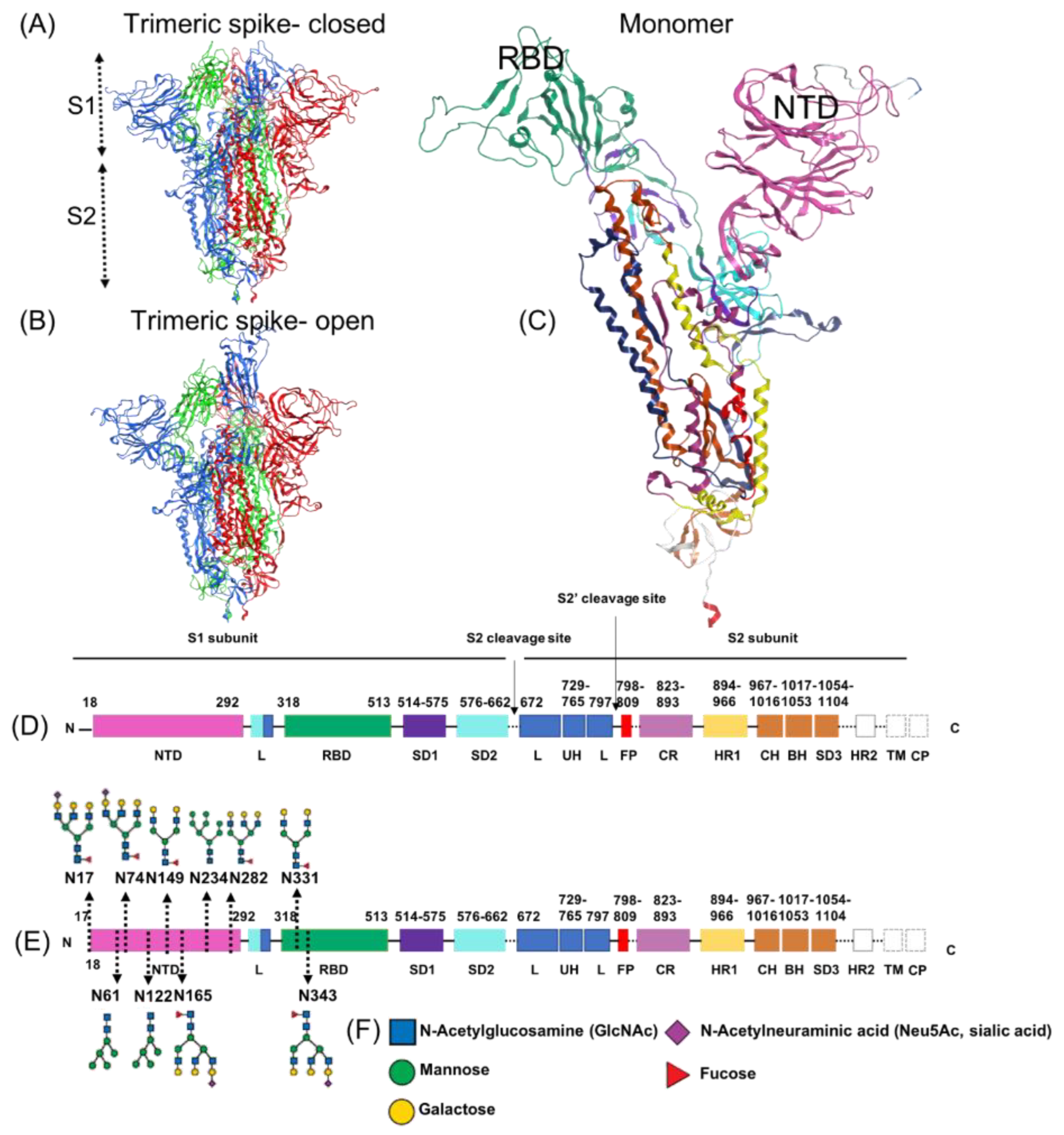
1.5. Subdomains of the SARS-CoV-2 Spike Protein, S1 Region
2. Materials and Methods
2.1. Ligand Database Preparation
2.2. Protein Preparation
2.3. Binding Sites
2.4. Molecular Docking Simulations
2.5. Molecular Dynamics (MD) Simulations
2.6. Ligand Interaction Fingerprint
3. Results
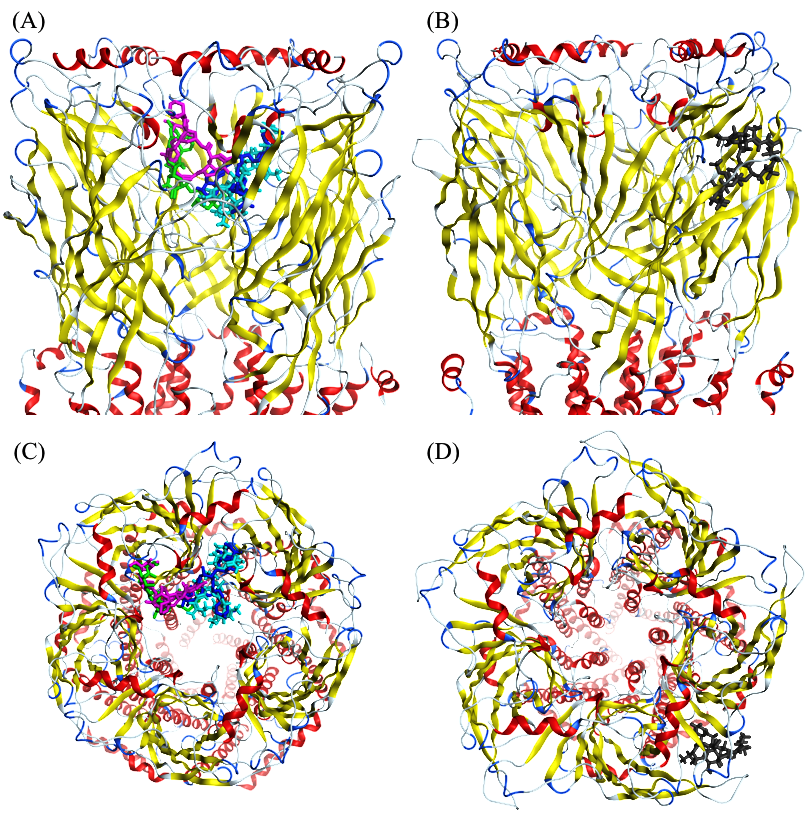
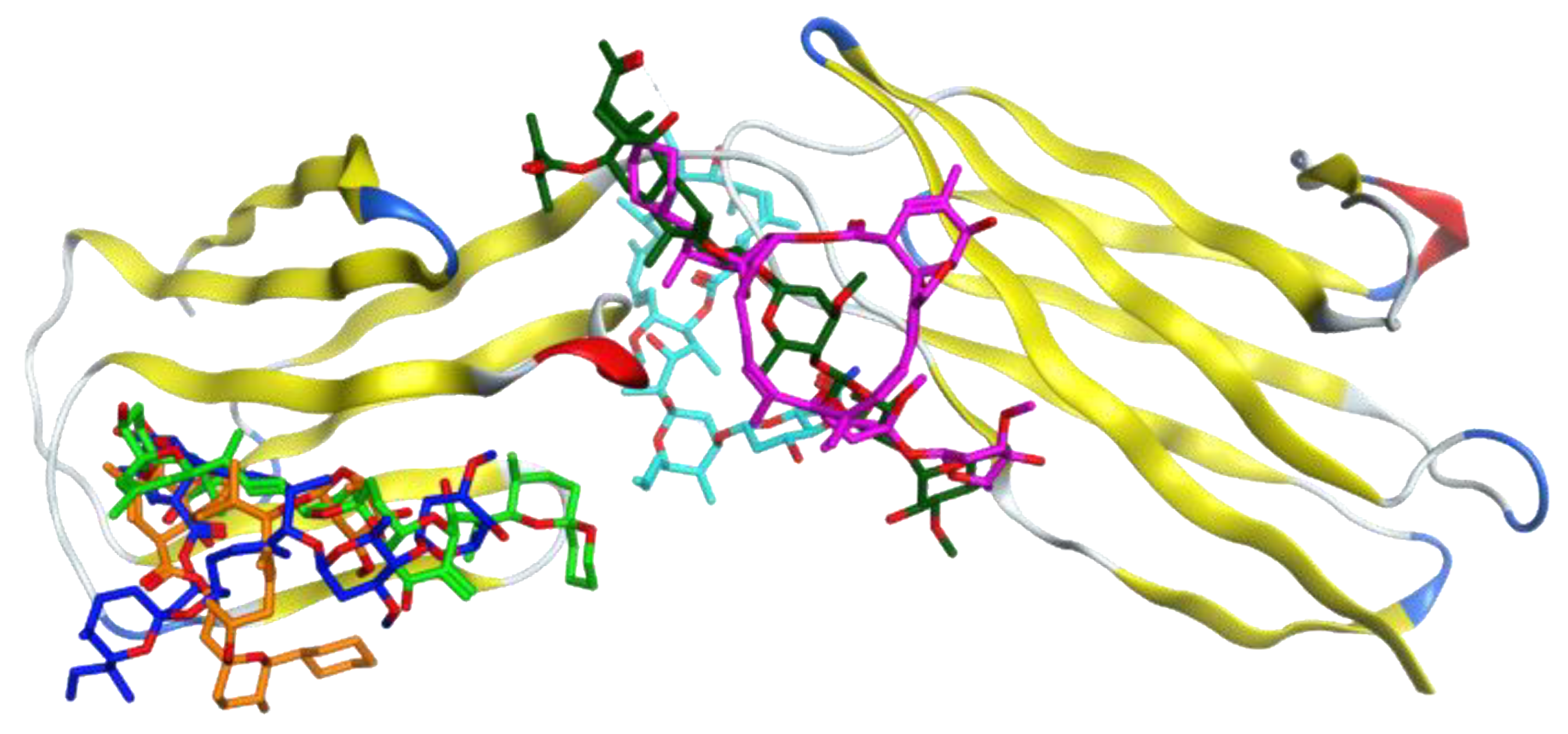
3.1. Molecular Docking Analysis
3.2. Selection of the Most Promising Compounds
3.3. Molecular Dynamics Simulations and RMSD Analysis
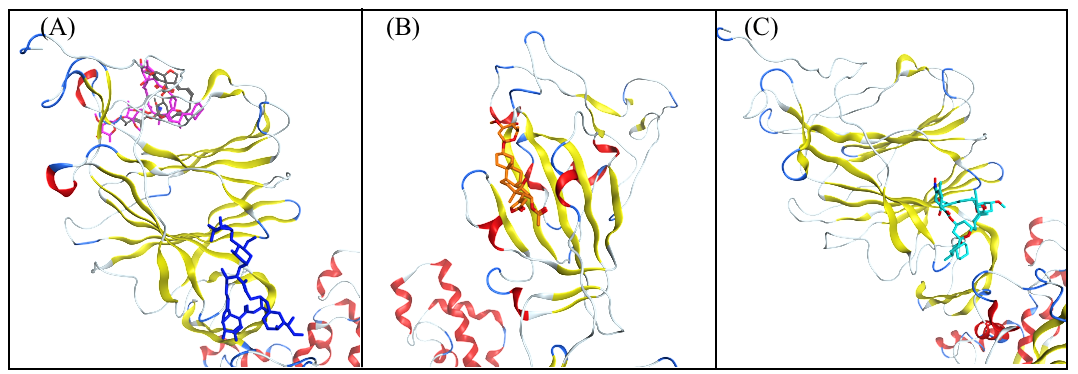
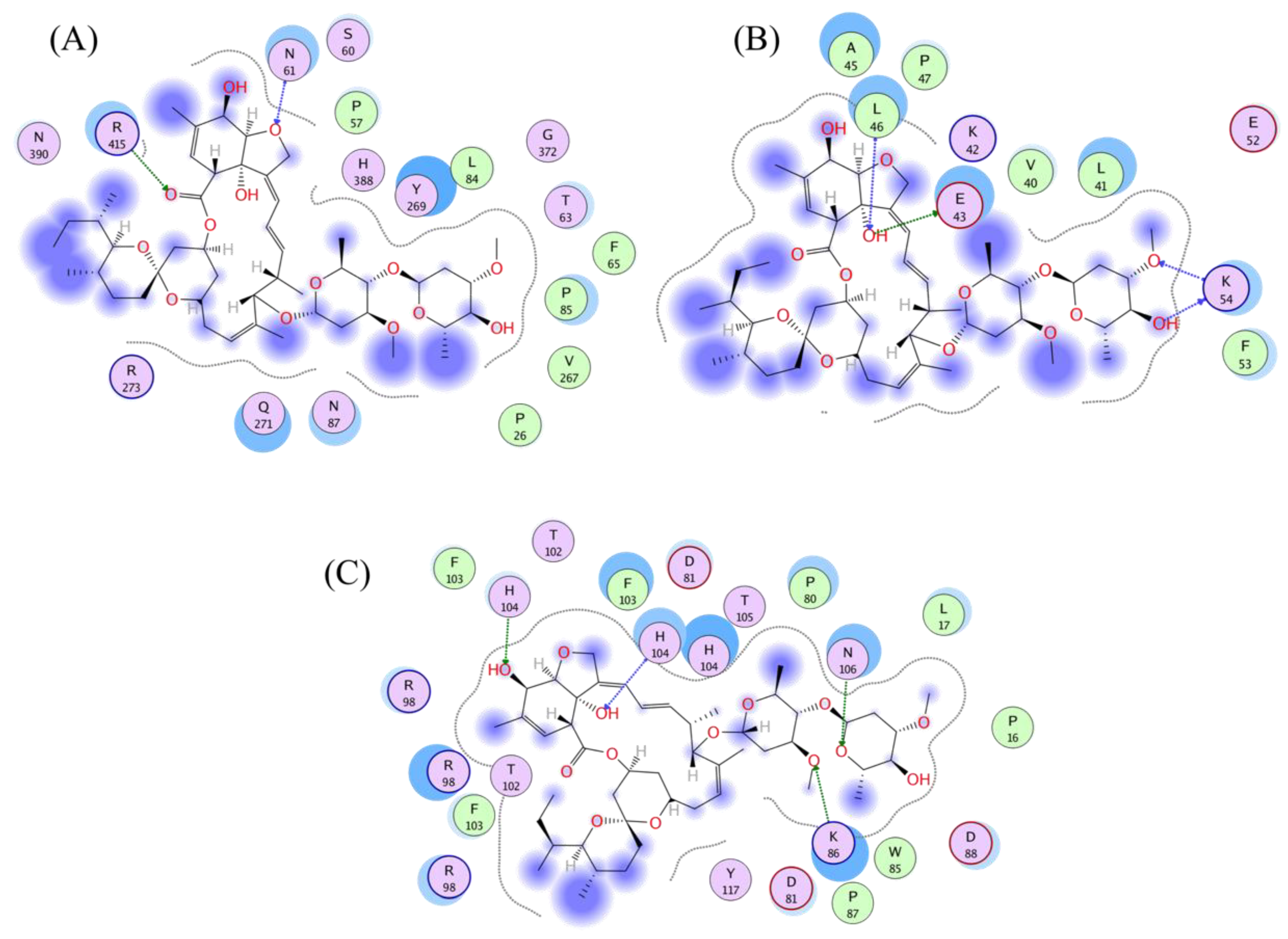
3.4. Analysis of the Protein–Ligand Interactions
3.5. Bioactivity of the Test Agents with Greatest Binding Strength
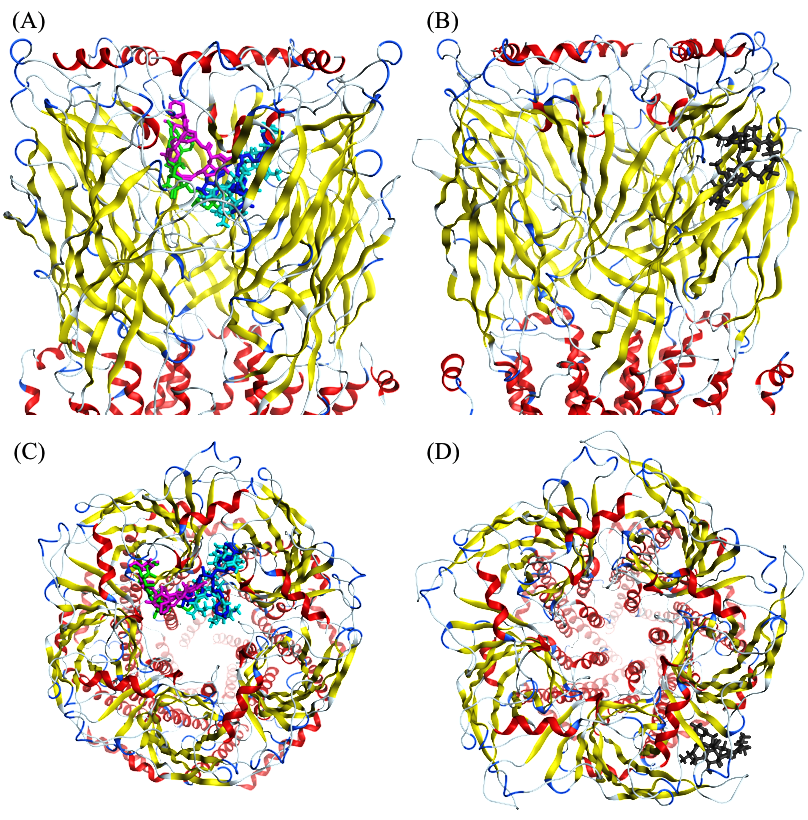
3.6. Protein-Protein Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α7nAChr | alpha-7 nicotinic acetylcholine receptor |
| ACE2 | angiotensin converting enzyme 2 |
| ACh | acetylcholine |
| BCov | bovine coronavirus |
| CD147 | cluster of differentiation 147 protein, encoded by the BSG gene |
| Co-IP | co-immunoprecipitation |
| COVID-19 | coronavirus disease 2019 |
| ELISA | enzyme-linked immunosorbent assay |
| GPU | graphics processing unit |
| HE | hemagglutinin esterase |
| HIV | human immunodeficiency virus |
| IL-1 | interleukin 1 |
| IL-6 | interleukin 6 |
| MD | molecular dynamics |
| MERS | Middle East respiratory syndrome |
| MHV-4 | mouse hepatitis virus 4, JHM strain |
| MOE | Molecular Operating Environment |
| NAG | N-acetyl-D-glucosamine |
| NTD | N-terminal domain |
| PDB | Protein Data Bank |
| PLB | propensity for ligand binding |
| RBC | red blood cell |
| RBD | receptor binding domain |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| RCT | randomized clinical trial |
| RMSD | root mean square deviation |
| SA | sialic acid |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SPR | surface plasmon resonance |
| TNF | tumor necrosis factor |
References
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Aminpour, M.; Cannariato, M.; Zucco, A.; Di Gregorio, E.; Israel, S.; Perioli, A.; Tucci, D.; Rossi, F.; Pionato, S.; Marino, S.; et al. Computational Study of Potential Galectin-3 Inhibitors in the Treatment of COVID-19. Biomedicines 2021, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- Scheim, D.E. A Deadly Embrace: Hemagglutination Mediated by SARS-CoV-2 Spike Protein at its 22 N-Glycosylation Sites, Red Blood Cell Surface Sialoglycoproteins, and Antibody. Int. J. Mol. Sci. 2022, 23, 2558. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Amoura, Z.; Rey, F.A.; Miyara, M. A nicotinic hypothesis for COVID-19 with preventive and therapeutic implications. Comptes Rendus Biol. 2020, 343, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lagoumintzis, G.; Chasapis, C.T.; Alexandris, N.; Kouretas, D.; Tzartos, S.; Eliopoulos, E.; Farsalinos, K.; Poulas, K. Nicotinic cholinergic system and COVID-19: In Silico identification of interactions between alpha7 nicotinic acetylcholine receptor and the cryptic epitopes of SARS-Co-V and SARS-CoV-2 Spike glycoproteins. Food Chem. Toxicol. 2021, 149, 112009. [Google Scholar] [CrossRef]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef]
- Chen, W.; Hui, Z.; Ren, X.; Luo, Y.; Shu, J.; Yu, H.; Li, Z. The N-glycosylation sites and Glycan-binding ability of S-protein in SARS-CoV-2 Coronavirus. bioRxiv 2020. [Google Scholar] [CrossRef]
- Choi, Y.K.; Cao, Y.; Frank, M.; Woo, H.; Park, S.-J.; Yeom, M.S.; Croll, T.I.; Seok, C.; Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. J. Chem. Theory Comput. 2021, 17, 2479–2487. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.N.; Richards, S.-J.; Guy, C.S.; Congdon, T.R.; Hasan, M.; Zwetsloot, A.J.; Gallo, A.; Lewandowski, J.R.; Stansfeld, P.J.; Straube, A.; et al. The SARS-CoV-2 Spike Protein Binds Sialic Acids and Enables Rapid Detection in a Lateral Flow Point of Care Diagnostic Device. ACS Cent. Sci. 2020, 6, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Bharara, R.; Singh, S.; Pattnaik, P.; Chitnis, C.E.; Sharma, A. Structural analogs of sialic acid interfere with the binding of erythrocyte binding antigen-175 to glycophorin A, an interaction crucial for erythrocyte invasion by Plasmodium falciparum. Mol. Biochem. Parasitol. 2004, 138, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus–sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Levine, S.; Levine, M.; Sharp, K.A.; Brooks, D.E. Theory of the electrokinetic behavior of human erythrocytes. Biophys. J. 1983, 42, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Odièvre, M.-H.; Bony, V.; Benkerrou, M.; Lapouméroulie, C.; Alberti, C.; Ducrocq, R.; Jacqz-Aigrain, E.; Elion, J.; Cartron, J.-P. Modulation of erythroid adhesion receptor expression by hydroxyurea in children with sickle cell disease. Haematologica 2008, 93, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Huang, W.; Ma, L.T.; Jiang, J.L.; Chen, Z.N. Importance of N-glycosylation on CD147 for its biological functions. Int. J. Mol. Sci. 2014, 15, 6356–6377. [Google Scholar] [CrossRef] [Green Version]
- Silva-Filho, J.C.; de Melo, C.G.F.; de Oliveira, J.L. The influence of ABO blood groups on COVID-19 susceptibility and severity: A molecular hypothesis based on carbohydrate-carbohydrate interactions. Med. Hypotheses 2020, 144, 110155. [Google Scholar] [CrossRef]
- Modrof, J.; Kerschbaum, A.; Farcet, M.R.; Niemeyer, D.; Corman, V.M.; Kreil, T.R. SARS-CoV-2 and the safety margins of cell-based biological medicinal products. Biologicals 2020, 68, 122–124. [Google Scholar] [CrossRef]
- Lam, L.M.; Murphy, S.J.; Kuri-Cervantes, L.; Weisman, A.R.; Ittner, C.A.G.; Reilly, J.P.; Pampena, M.B.; Betts, M.R.; Wherry, E.J.; Song, W.-C.; et al. Erythrocytes Reveal Complement Activation in Patients with COVID-19. MedRxiv 2020. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Zheng, Z.-H.; Wei, D.; Wen, A.; Zhang, Z.; Lian, J.-Q.; Kang, W.-Z.; Hao, C.-Q.; Wang, J.; Xie, R.-H.; et al. Safety and efficacy of meplazumab in healthy volunteers and COVID-19 patients: A randomized phase 1 and an exploratory phase 2 trial. Signal Transduct. Target. Ther. 2021, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Hulswit, R.J.G.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.J.; Bosch, B.J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, U.; Bauer, J.; Stehle, T. Viruses and sialic acids: Rules of engagement. Curr. Opin. Struct. Biol. 2011, 21, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Qing, E.; Hantak, M.; Perlman, S.; Gallagher, T. Distinct Roles for Sialoside and Protein Receptors in Coronavirus Infection. MBio 2020, 11, e02764-19. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Dong, W.; Milewska, A.; Golda, A.; Qi, Y.; Zhu, Q.K.; Marasco, W.A.; Baric, R.S.; Sims, A.C.; Pyrc, K.; et al. Human Coronavirus HKU1 Spike Protein Uses O-Acetylated Sialic Acid as an Attachment Receptor Determinant and Employs Hemagglutinin-Esterase Protein as a Receptor-Destroying Enzyme. J. Virol. 2015, 89, 7202–7213. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Hulswit, R.J.G.; Widjaja, I.; Raj, V.S.; McBride, R.; Peng, W.; Widagdo, W.; Tortorici, M.A.; van Dieren, B.; Lang, Y.; et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. USA 2017, 114, E8508–E8517. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhang, X.; Ostrikov, K.; Abrahamyan, L. Host receptors: The key to establishing cells with broad viral tropism for vaccine production. Crit. Rev. Microbiol. 2020, 46, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Koehler, M.; Delguste, M.; Sieben, C.; Gillet, L.; Alsteens, D. Initial Step of Virus Entry: Virion Binding to Cell-Surface Glycans. Annu. Rev. Virol. 2020, 7, 143–165. [Google Scholar] [CrossRef]
- Baum, J.; Ward, R.H.; Conway, D.J. Natural selection on the erythrocyte surface. Mol. Biol. Evol. 2002, 19, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Varki, A.; Gagneux, P. Multifarious roles of sialic acids in immunity. Ann. N. Y. Acad. Sci. 2012, 1253, 16–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Langereis, M.A.; van Vliet, A.L.W.; Huizinga, E.G.; de Groot, R.J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 9065–9069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, R.J. Structure, function and evolution of the hemagglutinin-esterase proteins of corona- and toroviruses. Glycoconj. J. 2006, 23, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, M.J.G.; Lang, Y.; Feitsma, L.J.; Hulswit, R.J.G.; de Poot, S.A.H.; van Vliet, A.L.W.; Margine, I.; de Groot-Mijnes, J.D.F.; van Kuppeveld, F.J.M.; Langereis, M.A.; et al. Betacoronavirus Adaptation to Humans Involved Progressive Loss of Hemagglutinin-Esterase Lectin Activity. Cell Host Microbe 2017, 21, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.; Herrler, G.; Klenk, H.D. Sialic Acid Receptors of Viruses. In SialoGlyco Chemistry and Biology II: Tools and Techniques to Identify and Capture Sialoglycans; Gerardy-Schahn, R., Delannoy, P., von Itzstein, M., Eds.; Springer International Publishing: New York, NY, USA, 2015; pp. 1–28. [Google Scholar]
- Miyagi, T.; Yamaguchi, K. 3.17—Sialic Acids. In Comprehensive Glycoscience; Kamerling, H., Ed.; Elsevier: Oxford, UK, 2007; pp. 297–323. [Google Scholar]
- Wagner, R.; Matrosovich, M.; Klenk, H.D. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 2002, 12, 159–166. [Google Scholar] [CrossRef]
- Lang, Y.; Li, W.; Li, Z.; Koerhuis, D.; van den Burg, A.C.S.; Rozemuller, E.; Bosch, B.-J.; van Kuppeveld, F.J.M.; Boons, G.-J.; Huizinga, E.G.; et al. Coronavirus hemagglutinin-esterase and spike proteins coevolve for functional balance and optimal virion avidity. Proc. Natl. Acad. Sci. USA 2020, 117, 25759–25770. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. The mystery of the pandemic’s ‘happy hypoxia’. Science 2020, 368, 455–456. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Lapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.D.; Sacco, C.; Alexia, B.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Price, L.C.; McCabe, C.; Garfield, B.; Wort, S.J. Thrombosis and COVID-19 pneumonia: The clot thickens! Eur. Respir. J. 2020, 56, 2001608. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329–2330. [Google Scholar] [CrossRef]
- Liao, C.G.; Kong, L.M.; Song, F.; Xing, J.L.; Wang, L.X.; Sun, Z.J.; Tang, H.; Yao, H.; Zhang, Y.; Wang, L.; et al. Characterization of basigin isoforms and the inhibitory function of basigin-3 in human hepatocellular carcinoma proliferation and invasion. Mol. Cell. Biol. 2011, 31, 2591–2604. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.; Staffler, G.; Hüttinger, R.; Hilgert, I.; Prager, E.; Cerný, J.; Steinlein, P.; Majdic, O.; Horejsí, V.; Stockinger, H. T cell activation-associated epitopes of CD147 in regulation of the T cell response, and their definition by antibody affinity and antigen density. Int. Immunol. 1999, 11, 777–786. [Google Scholar] [CrossRef]
- Lv, M.; Miao, J.; Zhao, P.; Luo, X.; Han, Q.; Wu, Z.; Zhang, K.; Zhu, P. CD147-mediated chemotaxis of CD4(+)CD161(+) T cells may contribute to local inflammation in rheumatoid arthritis. Clin. Rheumatol. 2018, 37, 59–66. [Google Scholar] [CrossRef]
- Schmidt, R.; Bültmann, A.; Fischel, S.; Gillitzer, A.; Cullen, P.; Walch, A.; Jost, P.; Ungerer, M.; Tolley, N.D.; Lindemann, S.; et al. Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ. Res. 2008, 102, 302–309. [Google Scholar] [CrossRef]
- Loh, D. The potential of melatonin in the prevention and attenuation of oxidative hemolysis and myocardial injury from cd147 SARS-CoV-2 spike protein receptor binding. Melatonin Res. 2020, 3, 380–416. [Google Scholar] [CrossRef]
- Joseph, J.; Knobler, R.L.; Lublin, F.D.; Burns, F.R. Regulation of the expression of intercellular adhesion molecule-1 (ICAM-1) and the putative adhesion molecule Basigin on murine cerebral endothelial cells by MHV-4 (JHM). Adv. Exp. Med. Biol. 1993, 342, 389–391. [Google Scholar] [PubMed]
- De Back, D.Z.; Kostova, E.; Klei, T.; Beuger, B.; van Zwieten, R.; Kuijpers, T.; Juffermans, N.; van den Berg, T.; Korte, D.; van Kraaij, M.; et al. RBC Adhesive Capacity Is Essential for Efficient ‘Immune Adherence Clearance’ and Provide a Generic Target to Deplete Pathogens from Septic Patients. Blood 2016, 128, 1031. [Google Scholar] [CrossRef]
- Telen, M.J. Red blood cell surface adhesion molecules: Their possible roles in normal human physiology and disease. Semin. Hematol. 2000, 37, 130–142. [Google Scholar] [CrossRef]
- Yurchenko, V.; Constant, S.; Bukrinsky, M. Dealing with the family: CD147 interactions with cyclophilins. Immunology 2006, 117, 301–309. [Google Scholar] [CrossRef]
- Schulz, C.; von Brühl, M.L.; Barocke, V.; Cullen, P.; Mayer, K.; Okrojek, R.; Steinhart, A.; Ahmad, Z.; Kremmer, E.; Nieswandt, B.; et al. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J. Thromb. Haemost. 2011, 9, 1007–1019. [Google Scholar] [CrossRef]
- Von Ungern-Sternberg, S.N.I.; Zernecke, A.; Seizer, P. Extracellular Matrix Metalloproteinase Inducer EMMPRIN (CD147) in Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 507. [Google Scholar] [CrossRef] [Green Version]
- Yee, C.; Main, N.M.; Terry, A.; Stevanovski, I.; Maczurek, A.; Morgan, A.J.; Calabro, S.; Potter, A.J.; Iemma, T.L.; Bowen, D.G.; et al. CD147 mediates intrahepatic leukocyte aggregation and determines the extent of liver injury. PLoS ONE 2019, 14, e0215557. [Google Scholar] [CrossRef] [Green Version]
- Pennings, G.J.; Kritharides, L. CD147 in cardiovascular disease and thrombosis. Semin. Thromb. Hemost. 2014, 40, 747–755. [Google Scholar]
- Carbajo-Lozoya, J.; Ma-Lauer, Y.; Malešević, M.; Theuerkorn, M.; Kahlert, V.; Prell, E.; von Brunn, B.; Muth, D.; Baumert, T.F.; Drosten, C.; et al. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res. 2014, 184, 44–53. [Google Scholar] [CrossRef]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y.; et al. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc. Natl. Acad. Sci. USA 2001, 98, 6360–6365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Song, Z.; Zhang, S.; Nanda, A.; Li, G. CD147: A Novel Modulator of Inflammatory and Immune Disorders. Curr. Med. Chem. 2014, 21, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Dayer, M. Coronavirus (2019-nCoV) Deactivation via Spike Glycoprotein Shielding by Old Drugs, Bioinformatic Study. Preprints.Org. 2020. [Google Scholar] [CrossRef]
- Nallusamy, S.; Mannu, J.; Ravikumar, C.; Angamuthu, K.; Nathan, B.; Nachimuthu, K.; Ramasamy, G.; Muthurajan, R.; Subbarayalu, M.; Neelakandan, K. Shortlisting Phytochemicals Exhibiting Inhibitory Activity against Major Proteins of SARS-CoV-2 through Virtual Screening. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Suravajhala, R.; Parashar, A.; Malik, B.; Nagaraj, V.A.; Padmanaban, G.; Kavi Kishor, P.B.; Polavarapu, R.; Suravajhala, P. Comparative Docking Studies on Curcumin with COVID-19 Proteins. Preprints.Org. 2020. [Google Scholar] [CrossRef]
- Kalhor, H.; Sadeghi, S.; Abolhasani, H.; Kalhor, R.; Rahimi, H. Repurposing of the approved small molecule drugs in order to inhibit SARS-CoV-2 S protein and human ACE2 interaction through virtual screening approaches. J. Biomol. Struct. Dyn. 2020, 40, 1299–1315. [Google Scholar] [CrossRef]
- Yagisawa, M.; Foster, P.J.; Hanaki, H.; Omura, S. Global Trends in Clinical Studies of Ivermectin in COVID-19. Jpn. J. Antibiot. 2021, 74, 44–95. [Google Scholar]
- Campbell, W.C. History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr. Pharm. Biotechnol. 2012, 13, 853–865. [Google Scholar] [CrossRef]
- Juarez, M.; Schcolnik-Cabrera, A.; Dueñas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Rizzo, E. Ivermectin, antiviral properties and COVID-19: A possible new mechanism of action. Naunyn Schmiedebergs Arch. Pharm. 2020, 393, 1153–1156. [Google Scholar]
- Lehrer, S.; Rheinstein, P.H. Ivermectin Docks to the SARS-CoV-2 Spike Receptor-binding Domain Attached to ACE2. Vivo 2020, 34, 3023–3026. [Google Scholar] [CrossRef] [PubMed]
- Maurya, D. A Combination of Ivermectin and Doxycycline Possibly Blocks the Viral Entry and Modulate the Innate Immune Response in COVID-19 Patients. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Dasgupta, J.; Sen, U.; Bakashi, A.; Dasgupta, A. Nsp7 and Spike Glycoprotein of SARS-CoV-2 Are Envisaged as Potential Targets of Vitamin D and Ivermectin. Preprints 2020. [Google Scholar] [CrossRef]
- Kaur, H.; Shekhar, N.; Sharma, S.; Sarma, P.; Prakash, A.; Medhi, B. Ivermectin as a potential drug for treatment of COVID-19: An in-sync review with clinical and computational attributes. Pharmacol. Rep. 2021, 73, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.K.; Raihan, J. The Binding mechanism of Ivermectin and levosalbutamol with spike protein of SARS-CoV-2. Struct. Chem. 2021, 32, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Hussien, M.A.; Abdelaziz, A.E.M. Molecular docking suggests repurposing of brincidofovir as a potential drug targeting SARS-CoV-2 ACE2 receptor and main protease. Netw. Modeling Anal. Health Inform. Bioinform. 2020, 9, 56. [Google Scholar] [CrossRef]
- Santin, A.D.; Scheim, D.E.; McCullough, P.A.; Yagisawa, M.; Borody, T.J. Ivermectin: A multifaceted drug of Nobel prize-honored distinction with indicated efficacy against a new global scourge, COVID-19. New Microbes New Infect. 2021, 43, 100924. [Google Scholar] [CrossRef]
- Kory, P.; Meduri, G.U.; Varon, J.; Iglesias, J.; Marik, P.E. Review of the Emerging Evidence Demonstrating the Efficacy of Ivermectin in the Prophylaxis and Treatment of COVID-19. Am. J. Ther. 2021, 28, e299–e318. [Google Scholar] [CrossRef]
- Crump, A.; Ōmura, S. Ivermectin, ‘wonder drug’ from Japan: The human use perspective. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, C.A.; Furtek, C.I.; Porras, A.G.; Chen, C.; Tipping, R.; Clineschmidt, C.M.; Sciberras, D.G.; Hsieh, J.Y.; Lasseter, K.C. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J. Clin. Pharm. 2002, 42, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Camprubí, D.; Requena-Méndez, A.; Buonfrate, D.; Giorli, G.; Kamgno, J.; Gardon, J.; Boussinesq, M.; Muñoz, J.; Krolewiecki, A. Safety of high-dose ivermectin: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2020, 75, 827–834. [Google Scholar] [CrossRef] [PubMed]
- The 2015 Nobel Prize in Physiology or Medicine—Press Release; The Nobel Assembly at Karolinska Institutet: Solna, Sweden, 2015; Available online: https://www.nobelprize.org/prizes/medicine/2015/press-release/ (accessed on 22 February 2022).
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Momekov, G.; Momekova, D. Ivermectin as a potential COVID-19 treatment from the pharmacokinetic point of view: Antiviral levels are not likely attainable with known dosing regimens. Biotechnol. Biotechnol. Equip. 2020, 34, 469–474. [Google Scholar] [CrossRef]
- Schmith, V.D.; Zhou, J.J.; Lohmer, L.R.L. The Approved Dose of Ivermectin Alone is not the Ideal Dose for the Treatment of COVID-19. Clin. Pharm. 2020, 108, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.M.; Buisson, B.; Bertrand, S.; Corringer, P.J.; Galzi, J.L.; Changeux, J.P.; Bertrand, D. Ivermectin: A positive allosteric effector of the alpha7 neuronal nicotinic acetylcholine receptor. Mol. Pharm. 1998, 53, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Ren, C.; Tong, Y.L.; Li, J.C.; Lu, Z.Q.; Yao, Y.M. The Protective Effect of Alpha 7 Nicotinic Acetylcholine Receptor Activation on Critical Illness and Its Mechanism. Int. J. Biol. Sci. 2017, 13, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Rajter, J.C.; Sherman, M.S.; Fatteh, N.; Vogel, F.; Sacks, J.; Rajter, J.-J. Use of Ivermectin is Associated with Lower Mortality in Hospitalized Patients with COVID-19 (ICON study). Chest 2020, 159, 85–92. [Google Scholar] [CrossRef]
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396. [Google Scholar] [CrossRef] [PubMed]
- Chaccour, C.; Casellas, A.; Blanco-Di Matteo, A.; Pineda, I.; Fernandez-Montero, A.; Ruiz-Castillo, P.; Richardson, M.-A.; Rodríguez-Mateos, M.; Jordán-Iborra, C.; Brew, J.; et al. The effect of early treatment with ivermectin on viral load, symptoms and humoral response in patients with non-severe COVID-19: A pilot, double-blind, placebo-controlled, randomized clinical trial. EClinicalMedicine 2021, 32, 100720. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulswit, R.J.G.; de Haan, C.A.M.; Bosch, B.J. Chapter Two—Coronavirus Spike Protein and Tropism Changes. In Advances in Virus Research, Ziebuhr, J., Ed.; Academic Press: New York, NY, USA, 2016; Volume 96, pp. 29–57. [Google Scholar]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, M.; Gulati, S.; Sarkar, D.; Tiwari, S.; Kateriya, S.; Ranjan, P.; Verma, S.K. The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses 2020, 12, 909. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Milanetti, E.; Miotto, M.; Rienzo, L.D.; Monti, M.; Gosti, G.; Ruocco, G. In-Silico evidence for two receptors based strategy of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Morniroli, D.; Giannì, M.L.; Consales, A.; Pietrasanta, C.; Mosca, F. Human Sialome and Coronavirus Disease-2019 (COVID-19) Pandemic: An Understated Correlation? Front. Immunol. 2020, 11, 1480. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef] [PubMed]
- DrugBank Online Database, Ivermectin (DB00602). Available online: https://go.drugbank.com/structures/search/small_molecule_drugs/structure?database_id=DB00602&search_type=similarity#results (accessed on 21 February 2022).
- CHARMM-GUI Archive—COVID-19 Proteins Library. Available online: https://www.charmm-gui.org/?doc=archive&lib=covid19 (accessed on 21 February 2022).
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell 2021, 184, 2121–2134.e13. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); ULC, Chemical Computing Group: Montreal, QC, Canada, 2019.
- Behloul, N.; Baha, S.; Shi, R.; Meng, J. Role of the GTNGTKR motif in the N-terminal receptor-binding domain of the SARS-CoV-2 spike protein. Virus Res. 2020, 286, 198058. [Google Scholar] [CrossRef]
- Di Gaetano, S.; Capasso, D.; Delre, P.; Pirone, L.; Saviano, M.; Pedone, E.; Mangiatordi, G.F. More Is Always Better Than One: The N-Terminal Domain of the Spike Protein as Another Emerging Target for Hampering the SARS-CoV-2 Attachment to Host Cells. Int. J. Mol. Sci. 2021, 22, 6462. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-4: SiteMap; Schrödinger, LLC.: New York, NY, USA, 2019.
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D.; et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchiano, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 Receptor Interaction by Natural and Semi-synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front. Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, I.T.E.; Cisneros, G. Amber 2021. Available online: https://ambermd.org/index.php (accessed on 21 February 2022).
- Preto, J.; Gentile, F. Assessing and improving the performance of consensus docking strategies using the DockBox package. J. Comput. Aided Mol. Des. 2019, 33, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.P.; Brenk, R.; Shoichet, B.K. Decoys for docking. J. Med. Chem. 2005, 48, 3714–3728. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [Green Version]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Toutain, P.L.; Upson, D.W.; Terhune, T.N.; McKenzie, M.E. Comparative pharmacokinetics of doramectin and ivermectin in cattle. Vet. Parasitol. 1997, 72, 3–8. [Google Scholar] [CrossRef]
- González Canga, A.; Sahagún Prieto, A.M.; Diez Liébana, M.J.; Fernández Martínez, N.; Sierra Vega, M.; García Vieitez, J.J. The Pharmacokinetics and Interactions of Ivermectin in Humans—A Mini-review. AAPS J. 2008, 10, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.; Ballester, M.R.; Antonijoan, R.M.; Gich, I.; Rodriguez, M.; Colli, E.; Gold, S.; Krolewiecki, A.J. Safety and pharmacokinetic profile of fixed-dose ivermectin with an innovative 18mg tablet in healthy adult volunteers. PLoS Negl. Trop. Dis. 2018, 12, e0006020. [Google Scholar] [CrossRef] [Green Version]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules; Springer: Berlin/Heidelberg, Germany, 2002; pp. 185–200. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Farsalinos, K.; Eliopoulos, E.; Leonidas, D.D.; Papadopoulos, G.E.; Tzartos, S.; Poulas, K. Nicotinic Cholinergic System and COVID-19: In Silico Identification of an Interaction between SARS-CoV-2 and Nicotinic Receptors with Potential Therapeutic Targeting Implications. Int. J. Mol. Sci. 2020, 21, 5807. [Google Scholar] [CrossRef]
- Gao, C.; Zeng, J.; Jia, N.; Stavenhagen, K.; Matsumoto, Y.; Zhang, H.; Li, J.; Hume, A.J.; Mühlberger, E.; van Die, I.; et al. SARS-CoV-2 Spike Protein Interacts with Multiple Innate Immune Receptors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cohen, M.; Varki, A. Chapter Three–Modulation of Glycan Recognition by Clustered Saccharide Patches. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: New York, NY, USA, 2014; Volume 308, pp. 75–125. [Google Scholar]
- Jaskiewicz, E.; Jodłowska, M.; Kaczmarek, R.; Zerka, A. Erythrocyte glycophorins as receptors for Plasmodium merozoites. Parasites Vectors 2019, 12, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, H.L.; Brodsky, I.E.; Mangalmurti, N.S. The Evolving Erythrocyte: Red Blood Cells as Modulators of Innate Immunity. J. Immunol. 2018, 201, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.W.; Lindstrom, W.; Olson, A.J.; Belew, R.K. Analysis of HIV Wild-Type and Mutant Structures via in Silico Docking against Diverse Ligand Libraries. J. Chem. Inf. Modeling 2007, 47, 1258–1262. [Google Scholar] [CrossRef] [Green Version]
- Mol-Instincts, Structure of IVERMECTIN (C48H74O14), Interactive 3-Dimensional (3D) Visualization. Available online: https://www.molinstincts.com/structure/IVERMECTIN-cstr-CT1079779157.html (accessed on 21 February 2022).
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Kiss, B.; Kis, Z.; Pályi, B.; Kellermayer, M.S.Z. Topography, Spike Dynamics, and Nanomechanics of Individual Native SARS-CoV-2 Virions. Nano Lett. 2021, 21, 2675–2680. [Google Scholar] [CrossRef]
- Xu, H.; Shaw, D.E. A Simple Model of Multivalent Adhesion and Its Application to Influenza Infection. Biophys. J. 2016, 110, 218–233. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, A.; Nastrucci, C.; Cesario, A.; Russo, P. Nicotine: Specific role in angiogenesis, proliferation and apoptosis. Crit. Rev. Toxicol. 2012, 42, 68–89. [Google Scholar] [CrossRef]
- Macklin, K.D.; Maus, A.D.; Pereira, E.F.; Albuquerque, E.X.; Conti-Fine, B.M. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J. Pharm. Exp. 1998, 287, 435–439. [Google Scholar]
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Reference | Binding Site Type | Residues | NTD/RBD |
|---|---|---|---|---|
| Site 1 | Milanetti et al. [103] | sialoside | L18-Q23, H66-T78 and G252-S254 | NTD |
| Site 2 | Behloul et al. [113] | sialoside | E154, F157, Y160 and the so-called stabilizing loop (N122-N125) | NTD |
| Site 3 | Baker et al. [12] | sialoside | (R21, Q23, L24, H69, F79, P82 and R246) | NTD |
| Site 4 (P1) | Di Gaetano et al. [114] | sialoside | R21, T22, Q23, L24, P26, R78, P82, V83, L110, F135, C136, N137 and R237 | NTD |
| Site 5 (P2) | Di Gaetano et al. [114] | sialoside | F92, S94, E96, K97, S98, R102, N121, V126, I128, M177, D178, K182, N188, R190, F192, I203, L226, V227 and L229. | NTD |
| Site 6–14 | Watanabe et al. [11] | glycosylation | N122, N149, N165, N17, N61, N74, N234, N282 | NTD |
| Site 15 | Fantini et al. [101] | ganglioside | Domain (111–158)- core Q-134 to D-138 | NTD |
| Site 16 | Carino et al. [117] | - | F342 N343 A343 T345 R346–W436 N437 S438–L441 D442 S443–G446–N448–Y451 L452 | RBD |
| Site 17 | Carino et al. [117] | - | S375–G404 D405–V502 G503–Q506–Y508 | RBD |
| Site 18 | Carino et al. [117] | - | E340 V341–F347 A348–N354 R355 K356–S399 F400 V401–V512 | RBD |
| Site 19 | Carino et al. [117] | - | F374–N388–Y495 G496 F497 | RBD |
| Site 20 | Carino et al. [117] | - | T376 F377 K378 C379 Y380–V407 R408–I410–V433 I444 A445 | RBD |
| Site 21–22 | Watanabe et al. [11] | glycosylation | N331–N3443 | RBD |
| Compound Name | Maximum |Score| | Open | Closed | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NTD | RDB | NTD | RBD | |||||||
| Score (kcal/mol) | At Site | Score (kcal/mol) | Site | Score (kcal/mol) | Site | Score (kcal/mol) | Site | Score (kcal/mol) | Site | |
| Ivermectin | −8.948 | NTD-open site 10 | −8.948 | site 10 | −8.256 | site 17 | −8.205 | site 4 | −7.735 | site 22 |
| Moxidectin | −8.902 | NTD-open site 2 | −8.902 | site 2 | −8.218 | site 21 | −7.659 | site 2 | −7.989 | site 18 |
| Doramectin | −8.885 | NTD-open site 2 | −8.885 | site 2 | −8.144 | site 21 | −8.867 | site 9 | −8.216 | site 19 |
| Oleandrin | −8.787 | RBD-closed site 19 | −7.787 | site 10 | −8.051 | site 22 | −8.083 | site 14 | −8.787 | site 19 |
| Selamectin | −8.774 | NTD-closed site 10 | −8.476 | site 15 | −7.432 | site 19 | −8.774 | site 10 | −8.142 | site 16 |
| Okadaic acid | −8.716 | NTD-open site 10 | −8.716 | site 10 | −8.067 | site 21 | −7.937 | site 4 | −8.25 | site 18 |
| Gitoformate | −8.514 | NTD-open site 10 | −8.514 | site 10 | −7.669 | site 21 | −7.88 | site 10 | −7.992 | site 19 |
| Amphotericin_B | −8.304 | NTD-open site 15 | −8.304 | site 15 | −7.516 | site 21 | −7.931 | site 4 | −7.332 | site 21 |
| P-57AS3 | −8.045 | NTD-open site 4 | −8.045 | site 4 | −7.663 | site 22 | −7.704 | site 5 | −7.627 | site 19 |
| Eprinomectin | −7.646 | NTD-open site 6 | −7.646 | site 6 | −7.584 | site 21 | −7.088 | site 6 | −7.302 | site 21 |
| Concanamycin A | −7.564 | NTD-open site 10 | −7.564 | site 10 | −7.335 | site 19 | −7.347 | site 3 | −7.302 | site 21 |
| Natamycin | −7.529 | RBD-open site 21 | −7.388 | site 13 | −7.529 | site 21 | −7.359 | site 4 | −6.87 | site 18 |
| Nystatin | −7.333 | RBD-open site 21 | −7.226 | site 6 | −6.845 | site 21 | −6.867 | site 14 | −6.773 | site 19 |
| beta-Escin | −7.324 | NTD-open site 10 | −7.324 | site 10 | −7.333 | site 21 | −7.264 | site 4 | −7.296 | site 19 |
| Fusicoccin | −6.705 | NTD-open site 2 | −6.705 | site 2 | −6.123 | site 22 | −6.353 | site 10 | −6.381 | site 18 |
| CD147 | α7nAChr | ||||
|---|---|---|---|---|---|
| Compound Name | Score (kcal/mol) | Site | Compound Name | Score (kcal/mol) | Site |
| Okadaic acid | −8.578 | site 5 | Ivermectin | −10.636 | Activated site 2 |
| Doramectin | −8.253 | site 1 | Doramectin | −10.243 | Activated site 2 |
| Selamectin | −8.082 | site 5 | Okadaic acid | −10.240 | Activated site 2 |
| P-57AS3 | −8.010 | site 1 | Moxidectin | −10.142 | Resting site 1 |
| Concanamycin A | −7.847 | site 9 | Concanamycin A | −9.932 | Activated site 2 |
| Ivermectin | −7.527 | site 5 | P-57AS3 | −9.799 | Desensitized site 3 |
| Amphotericin_B | −7.481 | site 1 | Gitoformate | −9.794 | Resting site 1 |
| Moxidectin | −7.469 | site 1 | beta-Escin | −9.711 | Resting site 3 |
| Oleandrin | −7.434 | site 4 | Natamycin | −9.611 | Activated site 1 |
| Gitoformate | −7.297 | site 8 | Oleandrin | −9.465 | Activated site 2 |
| Nystatin | −7.038 | site 9 | Selamectin | −9.397 | Activated site 2 |
| Eprinomectin | −6.827 | site 9 | Nystatin | −9.214 | Resting site 3 |
| beta-Escin | −6.755 | site 1 | Eprinomectin | −8.968 | Resting site 3 |
| Natamycin | −6.739 | site 7 | Fusicoccin | −8.814 | Resting site 3 |
| Fusicoccin | −5.872 | site 1 | Amphotericin_B | −8.811 | Resting site 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aminpour, M.; Cannariato, M.; Preto, J.; Safaeeardebili, M.E.; Moracchiato, A.; Doria, D.; Donato, F.; Zizzi, E.A.; Deriu, M.A.; Scheim, D.E.; et al. In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds. Computation 2022, 10, 51. https://doi.org/10.3390/computation10040051
Aminpour M, Cannariato M, Preto J, Safaeeardebili ME, Moracchiato A, Doria D, Donato F, Zizzi EA, Deriu MA, Scheim DE, et al. In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds. Computation. 2022; 10(4):51. https://doi.org/10.3390/computation10040051
Chicago/Turabian StyleAminpour, Maral, Marco Cannariato, Jordane Preto, M. Ehsan Safaeeardebili, Alexia Moracchiato, Domiziano Doria, Francesca Donato, Eric Adriano Zizzi, Marco Agostino Deriu, David E. Scheim, and et al. 2022. "In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds" Computation 10, no. 4: 51. https://doi.org/10.3390/computation10040051
APA StyleAminpour, M., Cannariato, M., Preto, J., Safaeeardebili, M. E., Moracchiato, A., Doria, D., Donato, F., Zizzi, E. A., Deriu, M. A., Scheim, D. E., Santin, A. D., & Tuszynski, J. A. (2022). In Silico Analysis of the Multi-Targeted Mode of Action of Ivermectin and Related Compounds. Computation, 10(4), 51. https://doi.org/10.3390/computation10040051