
Naphthenic Acids Aggregation: The Role of Salinity
 , , , and
, , , and 
Abstract
:1. Introduction
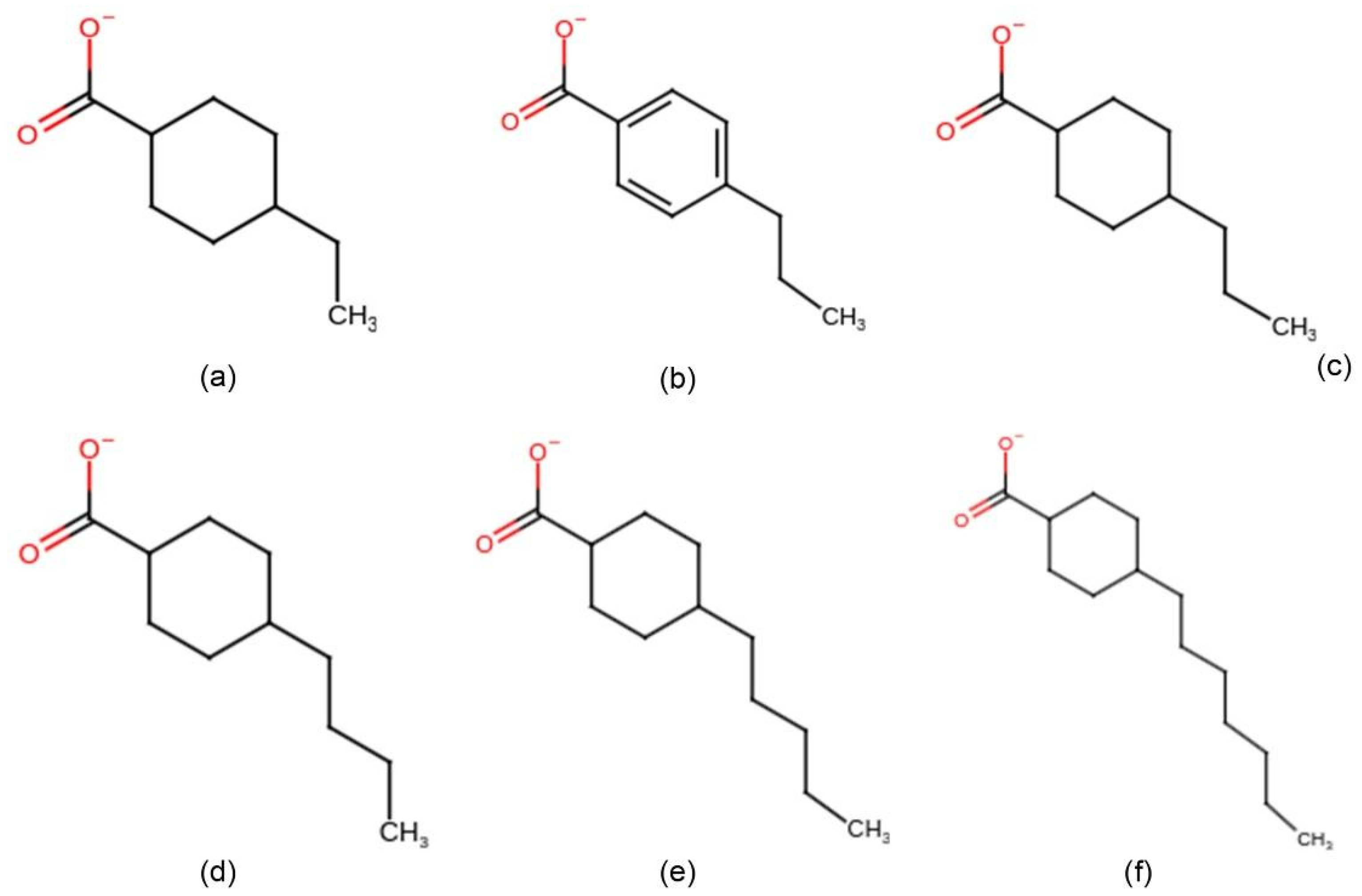
2. Materials and Methods
2.1. Quantum Chemical Continuum Solvent Calculations
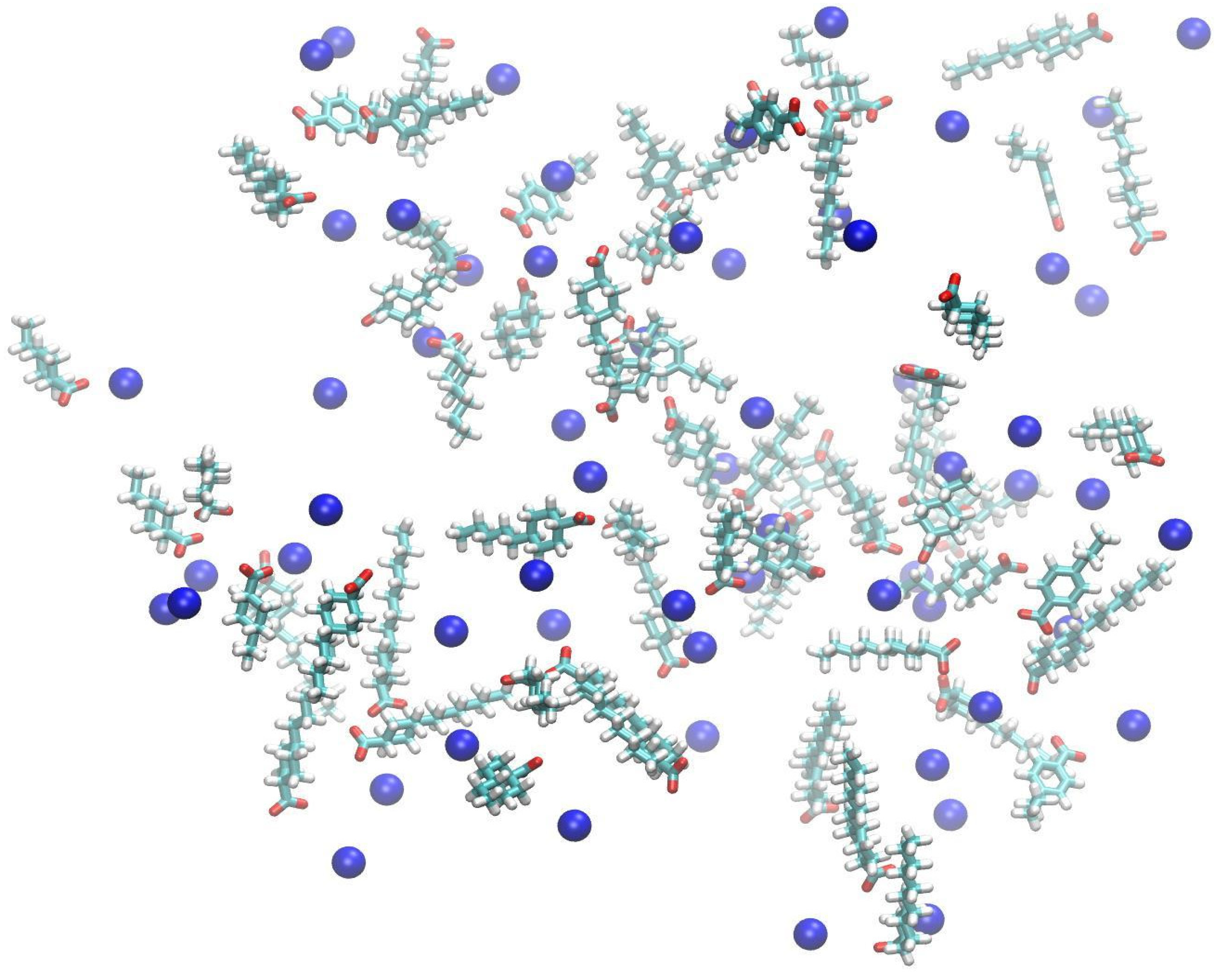
2.2. Molecular Dynamics Simulations
- 35 g/L—called here “low salinity” solution, comparable to injection water salinity; and
- 90 g/L—called here “high salinity” solution, comparable to the salinity of some pre-salt regions.
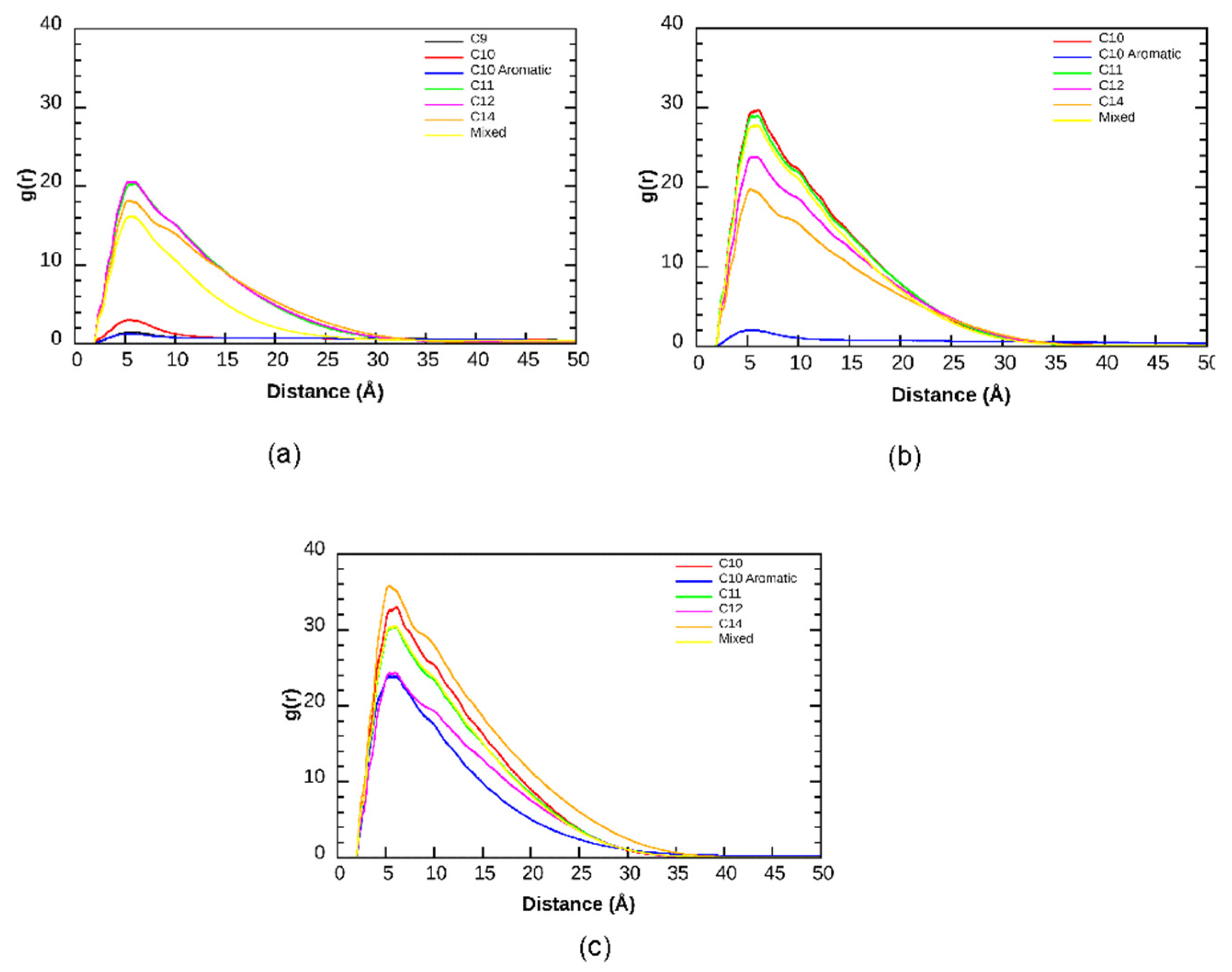
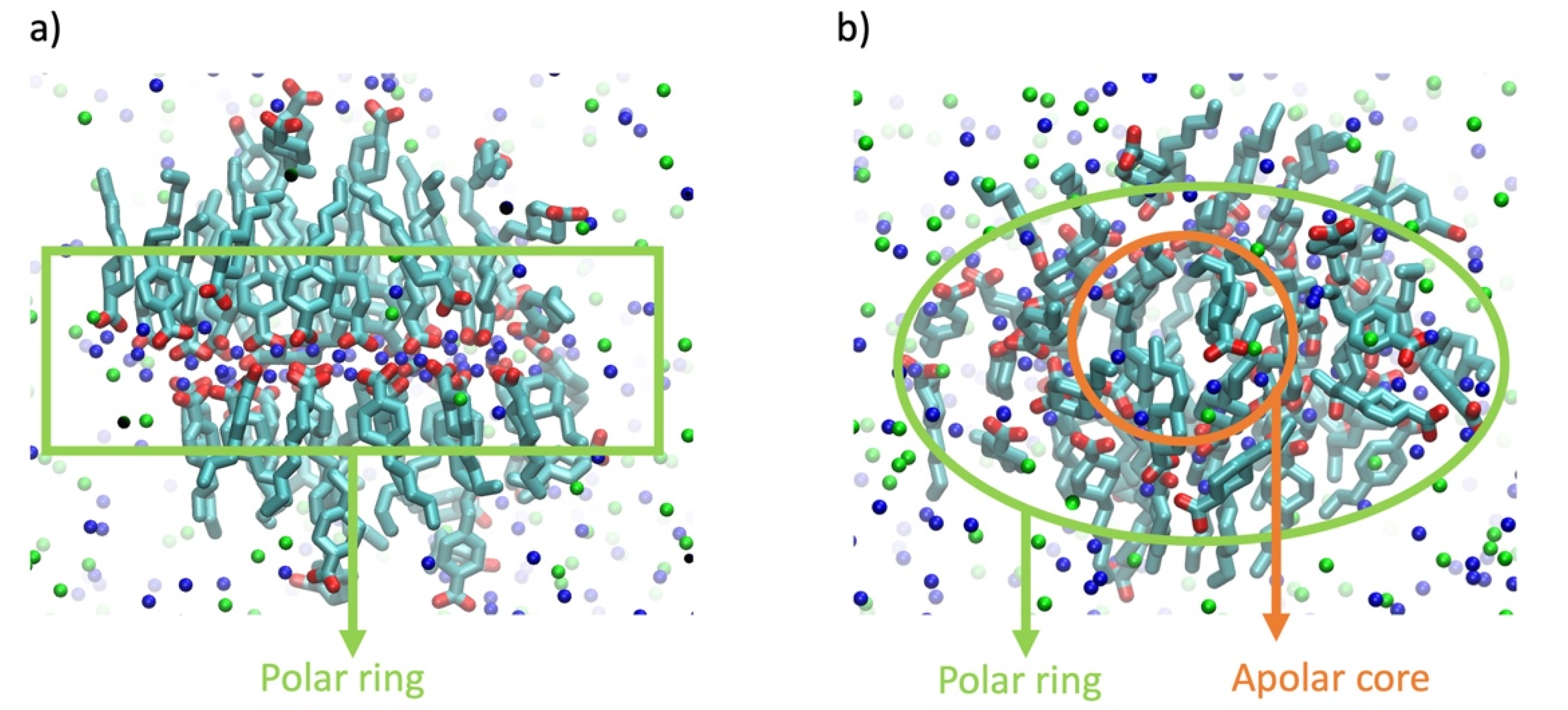
3. Results and Discussion
3.1. Hydrophobicity of NAs
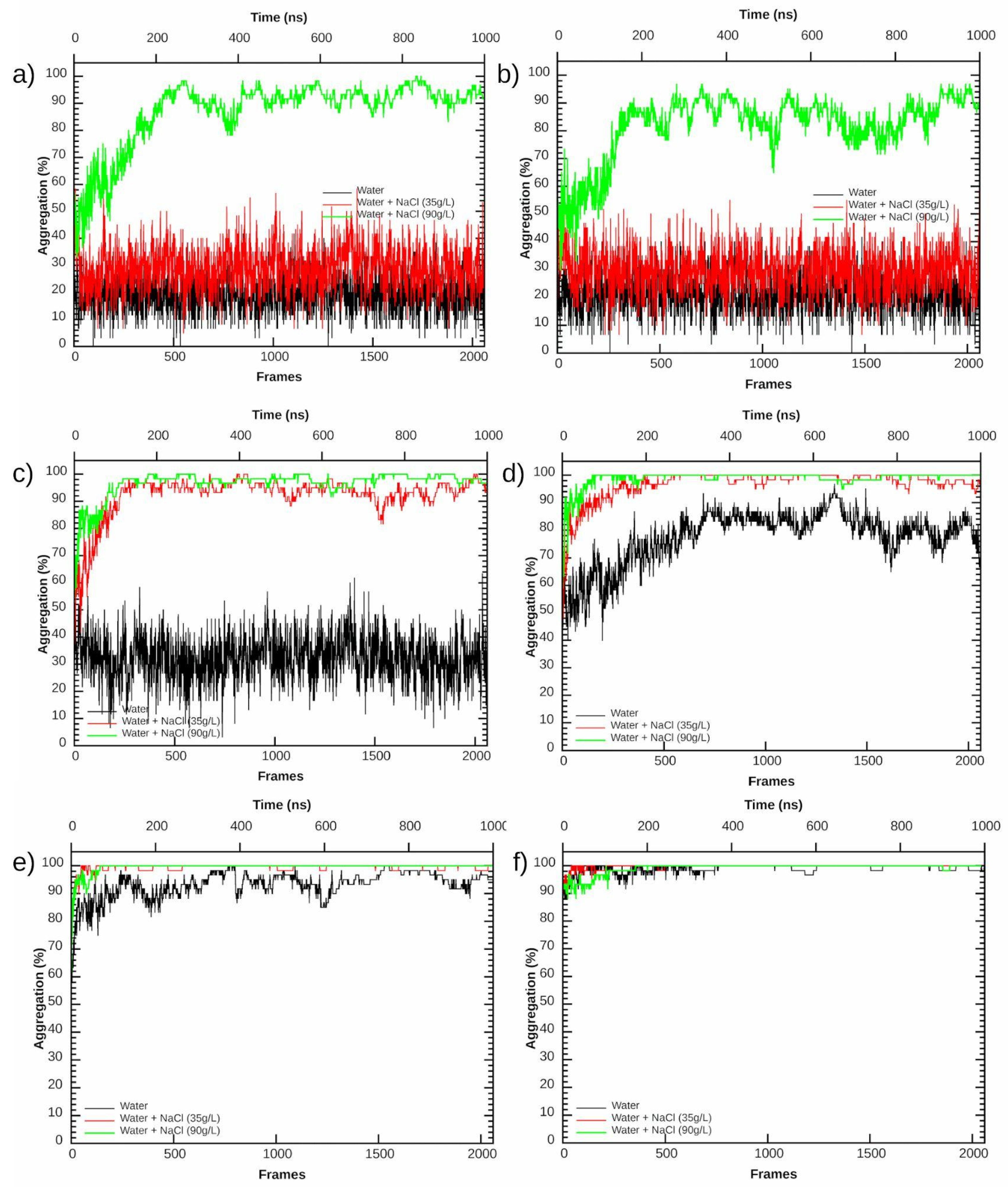
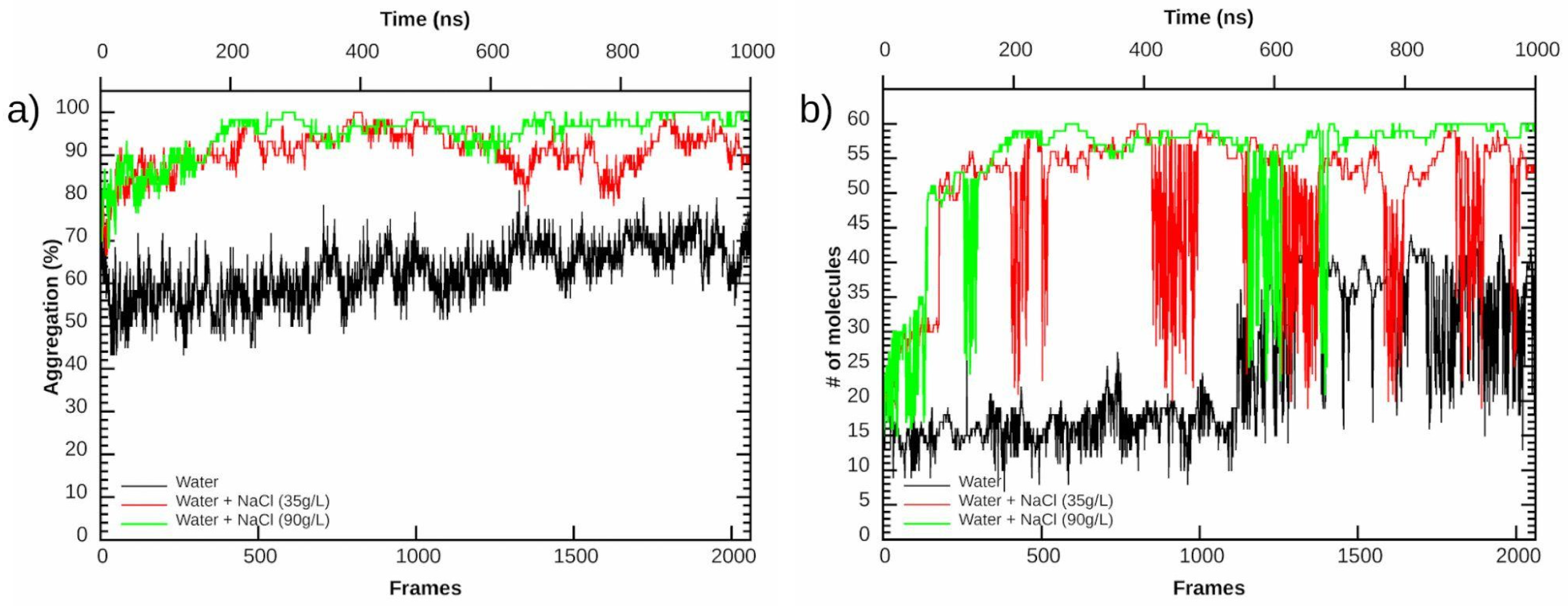
3.2. Aggregation Propensity of NAs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Clemente, J.S.; Fedorak, P.M. A Review of the Occurrence, Analyses, Toxicity, and Biodegradation of Naphthenic Acids. Chemosphere 2005, 60, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, A.J.; Frank, R.A.; Gillis, P.L.; Parrott, J.L.; Marentette, J.R.; Brown, L.R.; Hooey, T.; Vanderveen, R.; McInnis, R.; Brunswick, P.; et al. Toxicity of Naphthenic Acids to Invertebrates: Extracts from Oil Sands Process-Affected Water versus Commercial Mixtures. Environ. Pollut. 2017, 227, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Barros, E.V.; Dias, H.P.; Gomes, A.O.; Rodrigues, R.R.T.; Moura, R.R.; Sad, C.M.S.; Freitas, J.C.C.; Neto, A.C.; Aquije, G.M.F.V.; Romão, W. Study of Degradation of Acid Crude Oil by High Resolution Analytical Techniques. J. Pet. Sci. Eng. 2017, 154, 194–203. [Google Scholar] [CrossRef]
- Meriem-Benziane, M.; Bou-Saïd, B.; Boudouani, N. The Effect of Crude Oil in the Pipeline Corrosion by the Naphthenic Acid and the Sulfur: A Numerical Approach. J. Pet. Sci. Eng. 2017, 158, 672–679. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; Barrow, M.P. Mass Spectrometric Characterization of Naphthenic Acids in Environmental Samples: A Review. Mass Spectrom. Rev. 2009, 28, 121–134. [Google Scholar] [CrossRef]
- Kovalchik, K.A.; MacLennan, M.S.; Peru, K.M.; Headley, J.V.; Chen, D.D.Y. Standard Method Design Considerations for Semi-Quantification of Total Naphthenic Acids in Oil Sands Process Affected Water by Mass Spectrometry: A Review. Front. Chem. Sci. Eng. 2017, 11, 497–507. [Google Scholar] [CrossRef]
- Samanipour, S.; Reid, M.J.; Rundberget, J.T.; Frost, T.K.; Thomas, K.V. Concentration and Distribution of Naphthenic Acids in the Produced Water from Offshore Norwegian North Sea Oilfields. Environ. Sci. Technol. 2020, 54, 2707–2714. [Google Scholar] [CrossRef]
- Stanford, L.A.; Kim, S.; Klein, G.C.; Smith, D.F.; Rodgers, R.P.; Marshall, A.G. Identification of Water-Soluble Heavy Crude Oil Organic-Acids, Bases, and Neutrals by Electrospray Ionization and Field Desorption Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Environ. Sci. Technol. 2007, 41, 2696–2702. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, F.C.; Gruber, L.D.A.; Geller, A.M.; Vaz de Campos, M.C.; Gomes, A.O.; Guimarães, R.C.L.; Péres, V.F.; Jacques, R.A.; Caramão, E.B. Characterization of Naphthenic Acids Using Mass Spectroscopy and Chromatographic Techniques: Study of Technical Mixtures. Anal. Methods 2014, 6, 807–816. [Google Scholar] [CrossRef]
- Porto, N.d.A.; Crucello, J.; Facanali, R.; Junior, I.M.; Carvalho, R.M.; Hantao, L.W. Profiling Naphthenic Acids in Produced Water Using Hollow Fiber Liquid-Phase Microextraction Combined with Gas Chromatography Coupled to Fourier Transform Orbitrap Mass Spectrometry. J. Chromatogr. A 2021, 1655, 462485. [Google Scholar] [CrossRef]
- Vander Meulen, I.J.; Schock, D.M.; Parrott, J.L.; Mundy, L.J.; Pauli, B.D.; Peru, K.M.; McMartin, D.W.; Headley, J.V. Characterization of Naphthenic Acid Fraction Compounds in Water from Athabasca Oil Sands Wetlands by Orbitrap High-Resolution Mass Spectrometry. Sci. Total Environ. 2021, 780, 146342. [Google Scholar] [CrossRef] [PubMed]
- Santos Silva, H.; Alfarra, A.; Vallverdu, G.; Bégué, D.; Bouyssiere, B.; Baraille, I. Role of the Porphyrins and Demulsifiers in the Aggregation Process of Asphaltenes at Water/Oil Interfaces under Desalting Conditions: A Molecular Dynamics Study. Pet. Sci. 2020, 17, 797–810. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, X.; Shang, J.; Zhao, X.; Qu, T.; Wang, Y. The Effect of Naphthenic Acids on Physiological Characteristics of the Microalgae Phaeodactylum Tricornutum and Platymonas Helgolandica Var. Tsingtaoensis. Environ. Pollut. 2018, 240, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.C.; da Silva, T.B.G.; Francisco, A.D.d.S.; Bandeira, L.; Cunha, R.D.; Coutinho-Neto, M.D.; Homem-de-Mello, P.; Moraes de Almeida, J.; Orestes, E.; Nascimento, R.S.V. Hyperbranched Polyglycerols Derivatives as Cetyltrimethylammonium Bromide Nanocarriers on Enhanced Oil Recovery Processes. J. Appl. Polym. Sci. 2022, 139, 51725. [Google Scholar] [CrossRef]
- Kirch, A.; Razmara, N.; Mamani, V.F.S.; Meneghini, J.R.; Miranda, C.R. Multiscale Molecular Modeling Applied to the Upstream Oil & Gas Industry Challenges. Polytechnica 2020, 3, 54–65. [Google Scholar] [CrossRef]
- Alvim, R.S.; Lima, F.C.D.A.; Sánchez, V.M.; Headen, T.F.; Boek, E.S.; Miranda, C.R. Adsorption of Asphaltenes on the Calcite (10.4) Surface by First-Principles Calculations. RSC Adv. 2016, 6, 95328–95336. [Google Scholar] [CrossRef]
- Martyushev, D.A. Modeling and Prediction of Asphaltene-Resin-Paraffinic Substances Deposits in Oil Production Wells. Georesursy 2020, 22, 86–92. [Google Scholar] [CrossRef]
- Da Costa, L.M.; Stoyanov, S.R.; Gusarov, S.; Tan, X.; Gray, M.R.; Stryker, J.M.; Tykwinski, R.; Carneiro, J.W.d.M.; Seidl, P.R.; Kovalenko, A. Density Functional Theory Investigation of the Contributions of π–π Stacking and Hydrogen-Bonding Interactions to the Aggregation of Model Asphaltene Compounds. Energy Fuels 2012, 26, 2727–2735. [Google Scholar] [CrossRef]
- Stoyanov, S.R.; Yin, C.-X.; Gray, M.R.; Stryker, J.M.; Gusarov, S.; Kovalenko, A. Computational and Experimental Study of the Structure, Binding Preferences, and Spectroscopy of Nickel(II) and Vanadyl Porphyrins in Petroleum. J. Phys. Chem. B 2010, 114, 2180–2188. [Google Scholar] [CrossRef]
- Stoyanov, S.R.; Gusarov, S.; Kuznicki, S.M.; Kovalenko, A. Theoretical Modeling of Zeolite Nanoparticle Surface Acidity for Heavy Oil Upgrading. J. Phys. Chem. C 2008, 112, 6794–6810. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Chawla, M.; Hassan, A.A.; Abdulazeez, I.; Salawu, O.A.; Siddiqui, M.N.; Pervez, S.; Cavallo, L. Adsorption of Industrial Dyes on Functionalized and Nonfunctionalized Asphaltene: A Combined Molecular Dynamics and Quantum Mechanics Study. J. Mol. Liq. 2021, 337, 116433. [Google Scholar] [CrossRef]
- Riahi, S.; Pourhossein, P.; Ganjali, M.R. Removal of Naphthenic Acids from Liquid Petroleum: Theoretical Study. Pet. Sci. Technol. 2010, 28, 68–78. [Google Scholar] [CrossRef]
- Olesen, K.B.; Pedersen, A.-S.D.; Nikolajsen, L.V.; Andersson, M.P.; Sølling, T.I.; Sauer, S.P.A.; Mikkelsen, K.V. Interfacial Tension in Water/n-Decane/Naphthenic Acid Systems Predicted by a Combined COSMO-RS Theory and Pendant Drop Experimental Study. Mol. Phys. 2020, 118, e1764645. [Google Scholar] [CrossRef]
- Kang, S.S.; Kang, J.W. Extraction of Naphthenic Acid from Low-Grade Crude Oil Using Diol Compounds. Fuel 2020, 275, 117828. [Google Scholar] [CrossRef]
- Alonso, G.; Gamallo, P.; Rincón, C.; Sayós, R. Interfacial Behavior of Binary, Ternary and Quaternary Oil/Water Mixtures Described from Molecular Dynamics Simulations. J. Mol. Liq. 2021, 324, 114661. [Google Scholar] [CrossRef]
- Teklebrhan, R.B.; Jian, C.; Choi, P.; Xu, Z.; Sjöblom, J. Role of Naphthenic Acids in Controlling Self-Aggregation of a Polyaromatic Compound in Toluene. J. Phys. Chem. B 2016, 120, 3516–3526. [Google Scholar] [CrossRef]
- Da Cunha, R.D.; Maciel, C.D.; Homem-de-Mello, P. The Cooperative Effect between Cyclic Naphthenic Acids and Surfactant Enhances the Separation Efficiency. J. Mol. Liq. 2021, 342, 117577. [Google Scholar] [CrossRef]
- Soteras, I.; Curutchet, C.; Bidon-Chanal, A.; Orozco, M.; Luque, F.J. Extension of the MST Model to the IEF Formalism: HF and B3LYP Parametrizations. J. Mol. Struct. Theochem. 2005, 727, 29–40. [Google Scholar] [CrossRef]
- Curutchet, C.; Orozco, M.; Luque, F.J. Solvation in Octanol: Parametrization of the Continuum MST Model. J. Comput. Chem. 2001, 22, 1180–1193. [Google Scholar] [CrossRef]
- Zamora, W.J.; Pinheiro, S.; German, K.; Ràfols, C.; Curutchet, C.; Luque, F.J. Prediction of the N-Octanol/Water Partition Coefficients in the SAMPL6 Blind Challenge from MST Continuum Solvation Calculations. J. Comput. Aided. Mol. Des. 2020, 34, 443–451. [Google Scholar] [CrossRef]
- Viayna, A.; Pinheiro, S.; Curutchet, C.; Luque, F.J.; Zamora, W.J. Prediction of N-Octanol/Water Partition Coefficients and Acidity Constants (PKa) in the SAMPL7 Blind Challenge with the IEFPCM-MST Model. J. Comput. Aided. Mol. Des. 2021, 35, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zamora, W.J.; Curutchet, C.; Campanera, J.M.; Luque, F.J. Prediction of PH-Dependent Hydrophobic Profiles of Small Molecules from Miertus–Scrocco–Tomasi Continuum Solvation Calculations. J. Phys. Chem. B 2017, 121, 9868–9880. [Google Scholar] [CrossRef] [PubMed]
- Havre, T.E.; Sjöblom, J.; Vindstad, J.E. Oil/Water-Partitioning and Interfacial Behavior of Naphthenic Acids. J. Dispers. Sci. Technol. 2003, 24, 789–801. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A Fast and High-Quality Charge Model for the next Generation General AMBER Force Field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kunieda, M.; Nakaoka, K.; Liang, Y.; Miranda, C.R.; Ueda, A.; Takahashi, S.; Okabe, H.; Matsuoka, T. Self-Accumulation of Aromatics at the Oil−Water Interface through Weak Hydrogen Bonding. J. Am. Chem. Soc. 2010, 132, 18281–18286. [Google Scholar] [CrossRef]
- Da Silva, G.D.; de Freitas Martins, E.; Salvador, M.A.; Baptista, A.D.T.; de Almeida, J.M.; Miranda, C.R. From Atoms to Pre-Salt Reservoirs: Multiscale Simulations of the Low-Salinity Enhanced Oil Recovery Mechanisms. Polytechnica 2019, 2, 30–50. [Google Scholar] [CrossRef]
- Katende, A.; Sagala, F. A Critical Review of Low Salinity Water Flooding: Mechanism, Laboratory and Field Application. J. Mol. Liq. 2019, 278, 627–649. [Google Scholar] [CrossRef]
- Aghaeifar, Z.; Strand, S.; Austad, T.; Puntervold, T.; Aksulu, H.; Navratil, K.; Storås, S.; Håmsø, D. Influence of Formation Water Salinity/Composition on the Low-Salinity Enhanced Oil Recovery Effect in High-Temperature Sandstone Reservoirs. Energy Fuels 2015, 29, 4747–4754. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NA Type | log PN | log PI | log D (pH = 7) | log D (pH = 8) |
|---|---|---|---|---|
| c9 | 2.26 | −2.14 | 0.16 | −0.82 |
| c10 aromatic | 2.41 | −2.02 | 0.31 | −0.67 |
| c10 | 2.78 | −1.62 | 0.68 | −0.30 |
| c11 | 3.26 | −1.16 | 1.16 | 0.18 |
| c12 | 3.81 | −0.63 | 1.71 | 0.73 |
| c14 | 4.81 | 0.41 | 2.71 | 1.73 |
| NA Type | Water | Low Salinity Solution | High Salinity Solution |
|---|---|---|---|
| c9 | 19.5% | 29.4% | 93.0% |
| c10 aromatic | 21.0% | 29.7% | 85.0% |
| c10 | 32.7% | 93.5% | 98.1% |
| c11 | 81.3% | 98.5% | 99.4% |
| c12 | 94.1% | 99.6% | 100% |
| c14 | 99.4% | 100% | 100% |
| mixture | 66.3% | 91.1% | 97.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, R.D.; Ferreira, L.J.; Orestes, E.; Coutinho-Neto, M.D.; de Almeida, J.M.; Carvalho, R.M.; Maciel, C.D.; Curutchet, C.; Homem-de-Mello, P. Naphthenic Acids Aggregation: The Role of Salinity. Computation 2022, 10, 170. https://doi.org/10.3390/computation10100170
Cunha RD, Ferreira LJ, Orestes E, Coutinho-Neto MD, de Almeida JM, Carvalho RM, Maciel CD, Curutchet C, Homem-de-Mello P. Naphthenic Acids Aggregation: The Role of Salinity. Computation. 2022; 10(10):170. https://doi.org/10.3390/computation10100170
Chicago/Turabian StyleCunha, Renato D., Livia J. Ferreira, Ednilsom Orestes, Mauricio D. Coutinho-Neto, James M. de Almeida, Rogério M. Carvalho, Cleiton D. Maciel, Carles Curutchet, and Paula Homem-de-Mello. 2022. "Naphthenic Acids Aggregation: The Role of Salinity" Computation 10, no. 10: 170. https://doi.org/10.3390/computation10100170
APA StyleCunha, R. D., Ferreira, L. J., Orestes, E., Coutinho-Neto, M. D., de Almeida, J. M., Carvalho, R. M., Maciel, C. D., Curutchet, C., & Homem-de-Mello, P. (2022). Naphthenic Acids Aggregation: The Role of Salinity. Computation, 10(10), 170. https://doi.org/10.3390/computation10100170