Population Genomic Analyses of Wild and Farmed Striped Catfish Pangasianodon Hypophthalmus in the Lower Mekong River

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Tissue Collection
2.2. DNA Extraction, Library Preparation and Sequencing
2.3. Data Analysis
2.3.1. SNP Quality-Control and Outlier Detection
2.3.2. Basic Genetic Analysis
2.3.3. Cluster Analysis
2.3.4. Phylogenetic and Molecular Relative Times
2.3.5. Migration Rate
2.3.6. Effective Population Size
3. Results
3.1. Within Population Diversity
3.2. Genetic Distance
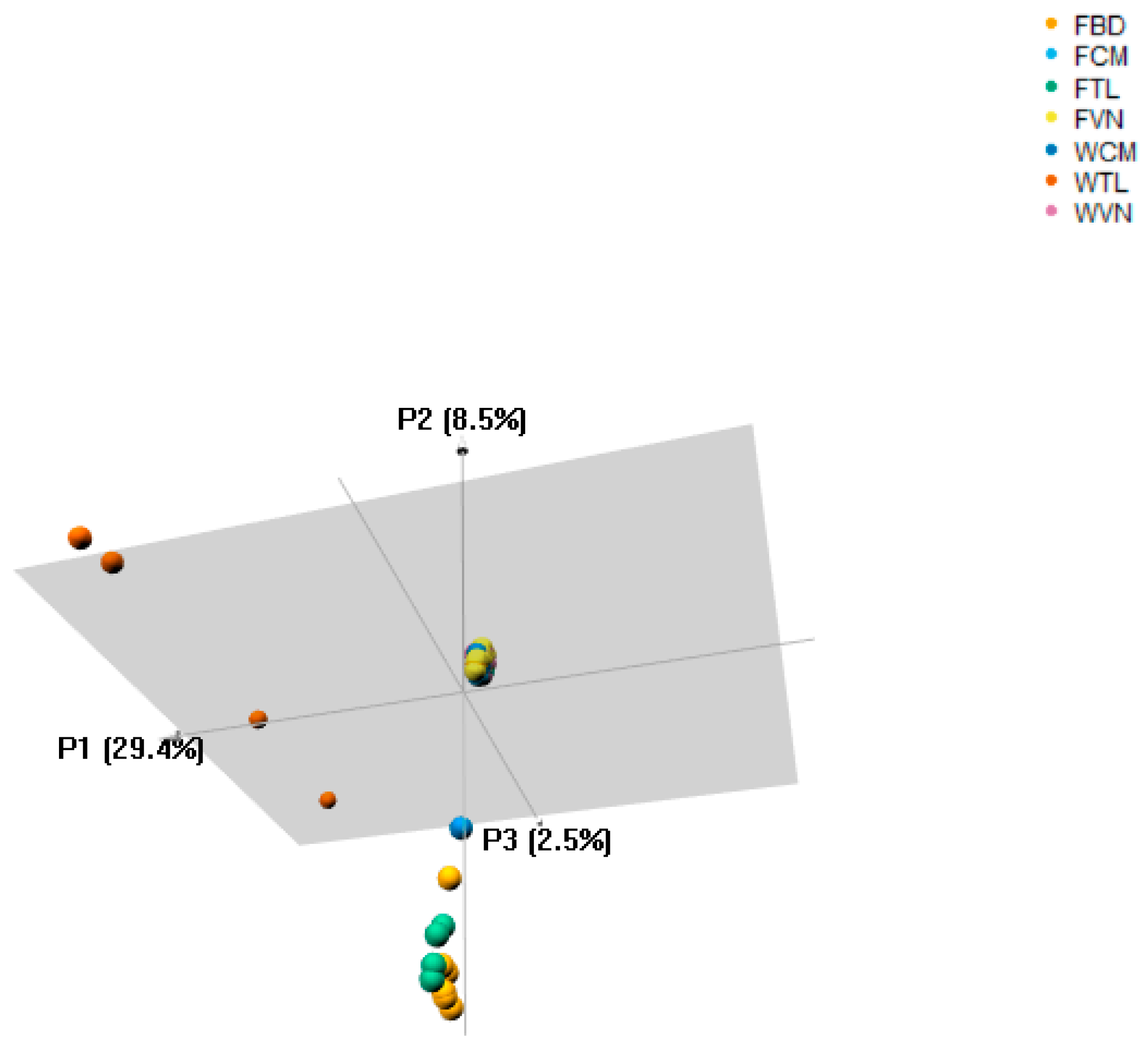
3.3. Population Structure
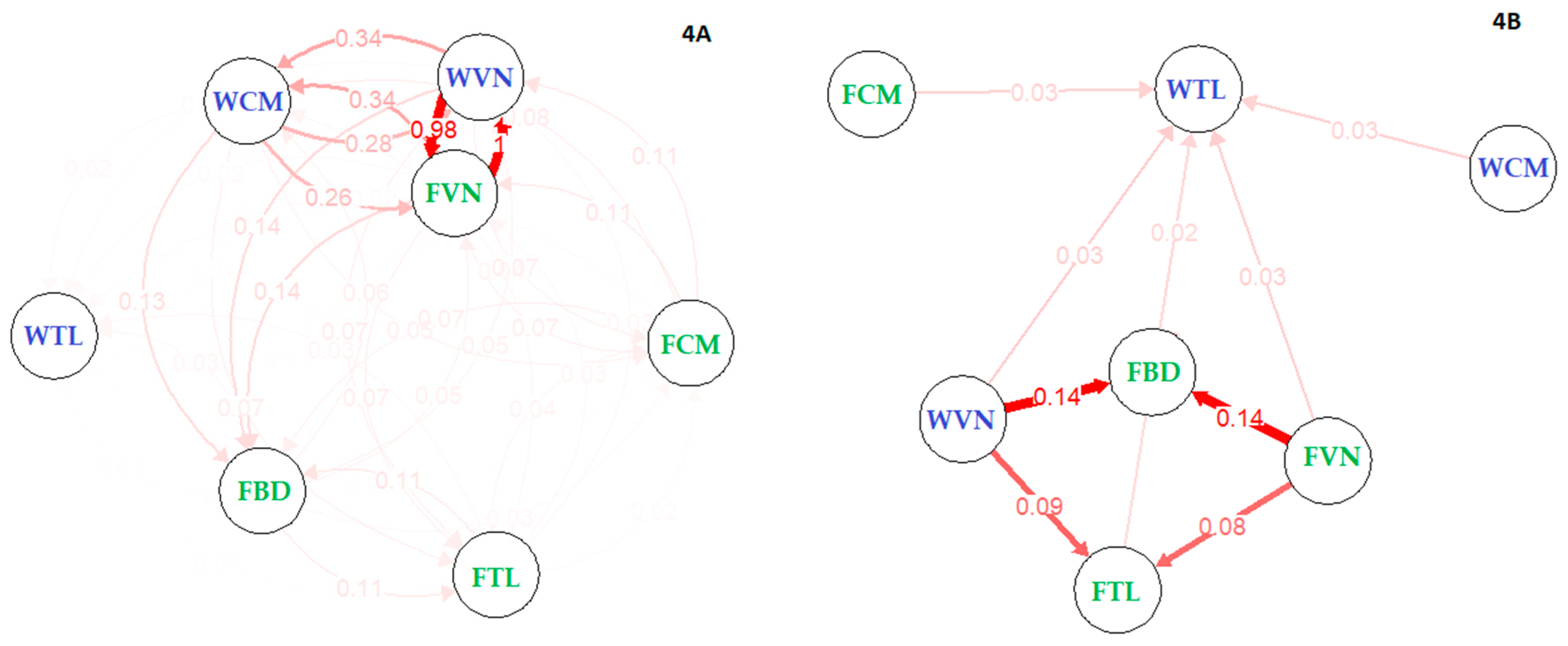
3.4. Migrations between Stocks
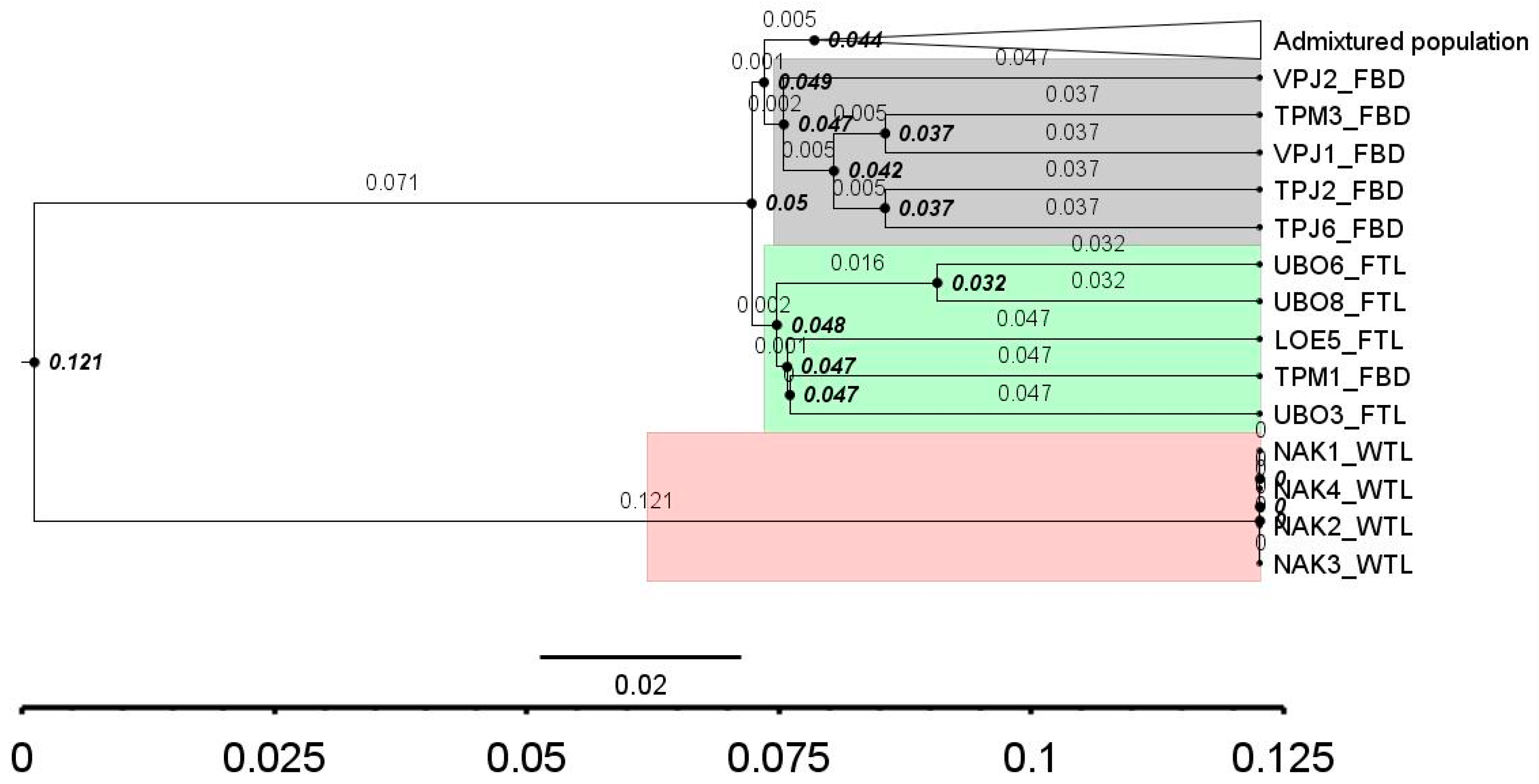
3.5. Molecular Relative Time
3.6. Effective Population Sizes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Touch, S. Life cycle of Pangasianodon hypophthalmus and the impact of catch and culture. In Proceedings of the Catfish Asia Conference, Bogor, Indonesia, 15–20 May 2000. [Google Scholar]
- Van Zalinge, N. Update on the status of the Cambodian inland capture fisheries sector with special reference to the Tonle Sap Great Lake. Mekong Fish. Catch Cult. 2002, 8, 1–9. [Google Scholar]
- Trong, T.Q.; Hao, N.V.; Griffiths, D. Status of Pangasiid Aquaculture in Viet Nam; MRC Technical Paper No. 2; Mekong River Commission: Phnom Penh, Cambodia, 2002; 16p, ISSN 1683-1489. [Google Scholar]
- Vũ, N.T.; Trọng, T.Q.; Đỉnh, L.T.; Nga, H.T.N.; Hiệp, N.V. Draft report: Survey results from Project “Investigation for develop national standard regulation: Freshwater fish-striped catfish broodtsock and fingerlings-quality requirements”. Draft. Rep. Fish. Dep. Res. Inst. Aquac. 2017, 25. [Google Scholar]
- Rainboth, W.J. Fishes of the Cambodian Mekong; Food & Agriculture Organization: Quebec City, QC, Canada, 1996. [Google Scholar]
- Roberts, T.R.; Vidthayanon, C. Systematic revision of the Asian catfish family Pangasiidae, with biological observations and descriptions of three new species. Proc. Acad. Nat. Sci. Phila. 1991, 143, 97–143. [Google Scholar]
- Na-Nakorn, U.; Moeikum, T. Genetic diversity of domesticated stocks of striped catfish, Pangasianodon hypophthalmus (Sauvage 1878), in Thailand: Relevance to broodstock management regimes. Aquaculture 2009, 297, 70–77. [Google Scholar] [CrossRef]
- VASEP. Available online: http://m.vasep.com.vn/Tin-Tuc/1018_52802/Hieu-qua-va-nang-suat-san-xuat-ca-tra-so-voi-ca-hoi-ca-chem-va-ca-trap-chau-Au.htm (accessed on 15 December 2019).
- Griffiths, D.; Van Khanh, P.; Trong, T.Q. Cultured Aquatic Species Information Programme: Pangasius Hypophthalmus. Fish. Aquac. Dep. Rome. Available online: http://www.fao.org/fishery/culturedspecies/Pangasius_hypophthalmus/en (accessed on 28 April 2015).
- Vu, N.T.; Van Sang, N.; Phuc, T.H.; Vuong, N.T.; Nguyen, N.H. Genetic evaluation of a 15-year selection program for high growth in striped catfish Pangasianodon hypophthalmus. Aquaculture 2019, 509, 221–226. [Google Scholar] [CrossRef]
- So, N.; Maes, G.; Volckaert, F. High genetic diversity in cryptic populations of the migratory sutchi catfish Pangasianodon hypophthalmus in the Mekong River. Heredity 2006, 96, 166–174. [Google Scholar] [CrossRef] [PubMed]
- So, N.; Van Houdt, J.K.; Volckaert, F.A. Genetic diversity and population history of the migratory catfishes Pangasianodon hypophthalmus and Pangasius bocourti in the Cambodian Mekong River. Fish. Sci. 2006, 72, 469–476. [Google Scholar] [CrossRef]
- Poulsen, A.F.; Hortle, K.; Valbo-Jorgensen, J.; Chan, S.; Chhuon, C.; Viravong, S.; Bouakhamvongsa, K.; Suntornratana, U.; Yoorong, N.; Nguyen, T.J.M. Distribution and ecology of some important riverine fish species of the Mekong River Basin. MRC Tech. Pap. 2004, 10, 116. [Google Scholar]
- Ha, H.P.; Nguyen, T.T.T.; Poompuang, S.; Na-Nakorn, U. Microsatellites revealed no genetic differentiation between hatchery and contemporary wild populations of striped catfish, Pangasianodon hypophthalmus (Sauvage 1878) in Vietnam. Aquaculture 2009, 291, 154–160. [Google Scholar] [CrossRef]
- Vu, N.T.; Sang, N.V.; Trong, T.Q.; Duy, N.H.; Dang, N.T.; Nguyen, N.H. Breeding for improved resistance to Edwardsiella ictaluri in striped catfish (Pangasianodon hypophthalmus): Quantitative genetic parameters. J. Fish. Dis. 2019, 42, 1409–1417. [Google Scholar] [CrossRef]
- Lind, C.; Ponzoni, R.; Nguyen, N.; Khaw, H.J.R. Selective breeding in fish and conservation of genetic resources for aquaculture. Reprod. Domest. Anim. 2012, 47, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Robledo, D.; Palaiokostas, C.; Bargelloni, L.; Martínez, P.; Houston, R. Applications of genotyping by sequencing in aquaculture breeding and genetics. Rev. Aquac. 2018, 10, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Jasielczuk, I.; Piestrzyńska-Kajtoch, A.; Fornal, A.; Semik-Gurgul, E.; Bugno-Poniewierska, M.J.G. Genotyping-by-sequencing performance in selected livestock species. Genomics 2019, 111, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genomics 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Eltaher, S.; Sallam, A.; Belamkar, V.; Emara, H.A.; Nower, A.A.; Salem, K.F.; Poland, J.; Baenziger, P.S. Genetic diversity and population structure of F3: 6 nebraska winter wheat genotypes using genotyping-by-sequencing. Front. Genet. 2018, 9, 76. [Google Scholar] [CrossRef]
- Vignal, A.; Milan, D.; SanCristobal, M.; Eggen, A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 2002, 34, 275–305. [Google Scholar] [CrossRef]
- Morin, P.A.; Luikart, G.; Wayne, R.K. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Karlsson, S.; Moen, T.; Lien, S.; Glover, K.A.; Hindar, K. Generic genetic differences between farmed and wild Atlantic salmon identified from a 7K SNP-chip. Mol. Ecol. Resour. 2011, 11, 247–253. [Google Scholar] [CrossRef]
- Glover, K.A.; Pertoldi, C.; Besnier, F.; Wennevik, V.; Kent, M.; Skaala, Ø. Atlantic salmon populations invaded by farmed escapees: Quantifying genetic introgression with a Bayesian approach and SNPs. BMC Genet. 2013, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, C.; Hargrove, J.S.; Bowen, B.R.; Thongda, W.; Zhang, D.; Mohammed, H.; Beck, B.H.; Austin, J.D.; Peatman, E. SNP marker panels for parentage assignment and traceability in the Florida bass (Micropterus floridanus). Aquaculture 2018, 485, 30–38. [Google Scholar] [CrossRef]
- Puckett, E.E.; Eggert, L.S. Comparison of SNP and microsatellite genotyping panels for spatial assignment of individuals to natal range: A case study using the American black bear (Ursus americanus). Biol. Conserv. 2016, 193, 86–93. [Google Scholar] [CrossRef]
- Schaid, D.J.; Guenther, J.C.; Christensen, G.B.; Hebbring, S.; Rosenow, C.; Hilker, C.A.; McDonnell, S.K.; Cunningham, J.M.; Slager, S.L.; Blute, M.L.; et al. Comparison of microsatellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer–susceptibility loci. Am. J. Hum. Genet. 2004, 75, 948–965. [Google Scholar] [CrossRef]
- Fernández, M.E.; Goszczynski, D.E.; Lirón, J.P.; Villegas-Castagnasso, E.E.; Carino, M.H.; Ripoli, M.V.; Rogberg-Muñoz, A.; Posik, D.M.; Peral-García, P.; Giovambattista, G.J.G.; et al. Comparison of the effectiveness of microsatellites and SNP panels for genetic identification, traceability and assessment of parentage in an inbred Angus herd. Genet. Mol. Biol. 2013, 36, 185–191. [Google Scholar] [CrossRef]
- Trọng, T.Q.; van Bers, N.; Crooijmans, R.; Dibbits, B.; Komen, H.J.A. A comparison of microsatellites and SNPs in parental assignment in the GIFT strain of Nile tilapia (Oreochromis niloticus): The power of exclusion. Aquaculture 2013, 388, 14–23. [Google Scholar] [CrossRef]
- Dang, B.; Vu, Q.; Biesack, E.; Doan, T.; Truong, O.; Tran, T.; Ackiss, A.; Stockwell, B.; Carpenter, K.E. Population genomics of the peripheral freshwater fish Polynemus melanochir (Perciformes, Polynemidae) in a changing Mekong Delta. Conserv. Genet. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Dang, B.T.; Rahman, M.A.; Tran, S.Q.; Glenner, H. Genome-wide SNP analyses reveal population structure of Portunus pelagicus along Vietnam coastline. PLoS ONE 2019, 14, e0224473. [Google Scholar] [CrossRef]
- Tibihika, P.D.; Curto, M.; Dornstauder-Schrammel, E.; Winter, S.; Alemayehu, E.; Waidbacher, H.; Meimberg, H. Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv. Genet. 2019, 20, 357–372. [Google Scholar] [CrossRef]
- Ashton, D.T.; Hilario, E.; Jaksons, P.; Ritchie, P.A.; Wellenreuther, M. Genetic diversity and heritability of economically important traits in captive Australasian snapper (Chrysophrys auratus). Aquaculture 2019, 505, 190–198. [Google Scholar] [CrossRef]
- Thongda, W.; Zhao, H.; Zhang, D.; Jescovitch, L.N.; Liu, M.; Guo, X.; Schrandt, M.; Powers, S.P.; Peatman, E. Development of SNP panels as a new tool to assess the genetic diversity, population structure, and parentage analysis of the eastern oyster (Crassostrea virginica). Mar. Biotechnol. 2018, 20, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.; Nguyen, H.; Nguyen, T.; Tran, H.T.T. Identification and Analysis of SNPs in Population of Vietnamese Catfish (Pangasianodon hypophthalmus), using Next Generation Sequencing and SNP Validation. MOJ Curr. Res. Rev. 2018, 1, 12–19. [Google Scholar]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C. Diversity arrays technology: A generic genome profiling technology on open platforms. In Data Production and Analysis in Population Genomics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 67–89. [Google Scholar]
- Nguyen, N.H.; Premachandra, H.; Kilian, A.; Knibb, W. Genomic prediction using DArT-Seq technology for yellowtail kingfish Seriola lalandi. BMC Genom. 2018, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Team R Core. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Whitlock, M.C.; Lotterhos, K.E. Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of F ST. Am. Nat. 2015, 186, S24–S36. [Google Scholar] [CrossRef]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2011, 28, 298–299. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Wen, W.; Falush, D. Documentation for STRUCTURE software: Version 2. 2003. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.323.9675&rep=rep1&type=pdf (accessed on 10 February 2020).
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Belkhir, K.; Bonhomme, F. Partitionml: A Maximum Likelihood Estimation of the Best Partition of a Sample into Panmictic Units; Université de Montpellier: Montpellier, France, 2002; Available online: http://www.genetix.univ-montp2.fr/partitionml.htm (accessed on 15 February 2020).
- Smouse, P.E.; Waples, R.S.; Tworek, J.A. A genetic mixture analysis for use with incomplete source population data. Can. J. Fish. Aquat. Sci. 1990, 47, 620–634. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Sundqvist, L.; Keenan, K.; Zackrisson, M.; Prodöhl, P.; Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016, 6, 3461–3475. [Google Scholar] [CrossRef]
- Alcala, N.; Goudet, J.; Vuilleumier, S. On the transition of genetic differentiation from isolation to panmixia: What we can learn from GST and D. Theor. Popul. Biol. 2014, 93, 75–84. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-Implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Marandel, F.; Charrier, G.; Lamy, J.-B.; Le Cam, S.; Lorance, P.; Trenkel, V.M. Estimating effective population size using RADseq: Effects of SNP selection and sample size. Ecol. Evol. 2020, 10, 1929–1937. [Google Scholar] [CrossRef]
- England, P.R.; Cornuet, J.-M.; Berthier, P.; Tallmon, D.A.; Luikart, G. Estimating effective population size from linkage disequilibrium: Severe bias in small samples. Conserv. Genet. 2006, 7, 303. [Google Scholar] [CrossRef]
- Waples, R.S. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. J. Conserv. Genet. 2006, 7, 167. [Google Scholar] [CrossRef]
- Beerli, P. How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use. Popul. Genet. Anim. Conserv. 2009, 17, 42–79. [Google Scholar]
- Felsenstein, J. Distance methods for inferring phylogenies: A justification. Evolution 1984, 38, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Haque, M.M.; Belton, B. Striped catfish (Pangasianodon hypophthalmus, Sauvage, 1878) aquaculture in Bangladesh: An overview. Aquac. Res. 2013, 44, 950–965. [Google Scholar] [CrossRef]
- Nguyen, P.; Oanh, D. Striped catfish (Pangasianodon hypophthalmus) aquaculture in Viet Nam: An unprecedented development within a decade. In Success Stories in Asian Aquaculture; Springer: Dordrecht, The Netherlands; NACA: Bangkok, Thailand; IDRC: Ottawa, ON, Canada, 2009; pp. 133–150. [Google Scholar]
- Nomura, T.; Honda, T.; Mukai, F. Inbreeding and effective population size of Japanese Black cattle. J. Anim. Sci. 2001, 79, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Blower, D.C.; Pandolfi, J.M.; Bruce, B.D.; Gomez-Cabrera, M.D.C.; Ovenden, J.R. Population genetics of Australian white sharks reveals fine-scale spatial structure, transoceanic dispersal events and low effective population sizes. Mar. Ecol. Prog. Ser. 2012, 455, 229–244. [Google Scholar] [CrossRef]
- Tave, D. Inbreeding and Brood Stock Management; Food & Agriculture Organization: Quebec City, QC, Canada, 1999. [Google Scholar]
- Franklin, I.; Frankham, R. How large must populations be to retain evolutionary potential? J. Anim. Conserv. 1998, 1, 69–70. [Google Scholar] [CrossRef]
- Poulsen, A.F.; Valbo-Jørgensen, J.J. Fish migrations and spawning habits in the Mekong mainstream: A survey using local knowledge (Basin-Wide). In Assessment of Mekong fisheries: Fish Migrations Spawning the Impact of Water Management Component; Internal report; Mekong River Commission: Vientiane, Laos, 2000. [Google Scholar]
- Hawkins, P.; Hortle, K.; Phommanivong, S.; Singsua, Y. Underwater video monitoring of fish passage in the Mekong River at Sadam Channel, Khone Falls, Laos. River Res. Appl. 2018, 34, 232–243. [Google Scholar] [CrossRef]
- Kang, B.; He, D.; Perrett, L.; Wang, H.; Hu, W.; Deng, W.; Wu, Y. Fisheries. Fish and fisheries in the Upper Mekong: Current assessment of the fish community, threats and conservation. Rev. Fish Biol. Fish. 2009, 19, 465. [Google Scholar] [CrossRef]
- Dugan, P.J.; Barlow, C.; Agostinho, A.A.; Baran, E.; Cada, G.F.; Chen, D.; Cowx, I.G.; Ferguson, J.W.; Jutagate, T.; Mallen-Cooper, M.; et al. Fish Migration, Dams, and Loss of Ecosystem Services in the Mekong Basin. AMBIO 2010, 39, 344–348. [Google Scholar] [CrossRef]
- Baran, E.; Myschowoda, C. Management. Dams and fisheries in the Mekong Basin. Aquat. Ecosyst. Health Manag. 2009, 12, 227–234. [Google Scholar] [CrossRef]
- Fukushima, M.; Jutagate, T.; Grudpan, C.; Phomikong, P.; Nohara, S. Potential Effects of Hydroelectric Dam Development in the Mekong River Basin on the Migration of Siamese Mud Carp (Henicorhynchus siamensis and H. lobatus) Elucidated by Otolith Microchemistry. PLoS ONE 2014, 9, e103722. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catfish Group | Observed Heterozygote * | Expected Heterozygote * | Inbreeding * | Diversity ** |
|---|---|---|---|---|
| FBD | 0.071 ± 0.141 | 0.083 ± 0.155 | 0.109 ± 0.330 | 0.082 ± 0.041 |
| FCM | 0.055 ± 0.182 | 0.056 ± 0.173 | 0.068 ± 0.512 | 0.055 ± 0.036 |
| FTL | 0.076 ± 0.161 | 0.090 ± 0.169 | 0.102 ± 0.398 | 0.088 ± 0.046 |
| FVN | 0.049 ± 0.123 | 0.049 ± 0.118 | 0.004 ± 0.178 | 0.049 ± 0.023 |
| WCM | 0.056 ± 0.138 | 0.059 ± 0.133 | 0.037 ± 0.322 | 0.060 ± 0.031 |
| WTL | 0.497 ± 0.459 | 0.333 ± 0.236 | −0.499 ± 0.712 | 0.363 ± 0.198 |
| WVN | 0.048 ± 0.119 | 0.049 ± 0.117 | 0.018 ± 0.196 | 0.049 ± 0.023 |
| Farmed catfish | 0.056 ± 0.114 | 0.064 ± 0.104 | 0.108 ± 0.249 | 0.064 ± 0.030 |
| Wild catfish | 0.092 ± 0.010 | 0.122 ± 0.105 | 0.096 ± 0.284 | 0.103 ± 0.049 |
| Source of Variance | Degree of Freedom | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among population (wild vs. farmed fish) | 1 | 743.4 | 4.61 (Va) | 1.53 |
| Among individuals and population | 87 | 29,098.9 | 33.34 (Vb) | 11.04 |
| Within individuals | 89 | 23,759.5 | 263.99 (Vc) | 87.43 |
| Stock | FBD | FCM | FTL | FVN | WCM | WTL | WVN |
|---|---|---|---|---|---|---|---|
| FBD | 6.2 | 7.3 | 1.4 | 3.0 | 0.3 | 1.4 | |
| FCM | 0.039 *** | 3.2 | 13.6 | 22.4 | 0.4 | 16.4 | |
| FTL | 0.033 *** | 0.072 *** | 0.9 | 1.8 | 0.4 | 0.9 | |
| FVN | 0.148 *** | 0.018 ** | 0.220 *** | 14.1 | 0.1 | 49.8 | |
| WCM | 0.076 *** | 0.011NS | 0.125 *** | 0.014 *** | 0.3 | 20.6 | |
| WTL | 0.468 *** | 0.363 *** | 0.407 *** | 0.666 *** | 0.487 *** | 0.1 | |
| WVN | 0.147 *** | 0.015 * | 0.217 *** | 0.005 *** | 0.012 *** | 0.664 *** |
| Stock | N | cNe | CI | hNe | CI* |
|---|---|---|---|---|---|
| FBD | 9 | 10.8 | 2.9 to 511.4 | 1043.9 | 746.5 to 1341.3 |
| FVN | 31 | 52.2 | 18.7 to infinite | 1239.6 | 1020.5 to 1458.7 |
| WCM | 7 | 14.9 | 2.9 to infinite | 1210.1 | 927.7 to 1492.4 |
| WVN | 32 | 73.6 | 32.1 to infinite | 1258.4 | 998.2 to 1518.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, N.T.; Ha, T.T.T.; Thuy, V.T.B.; Trang, V.T.; Nguyen, N.H. Population Genomic Analyses of Wild and Farmed Striped Catfish Pangasianodon Hypophthalmus in the Lower Mekong River. J. Mar. Sci. Eng. 2020, 8, 471. https://doi.org/10.3390/jmse8060471
Vu NT, Ha TTT, Thuy VTB, Trang VT, Nguyen NH. Population Genomic Analyses of Wild and Farmed Striped Catfish Pangasianodon Hypophthalmus in the Lower Mekong River. Journal of Marine Science and Engineering. 2020; 8(6):471. https://doi.org/10.3390/jmse8060471
Chicago/Turabian StyleVu, Nguyen Thanh, Tran Thi Thuy Ha, Vo Thi Bich Thuy, Vu Thi Trang, and Nguyen Hong Nguyen. 2020. "Population Genomic Analyses of Wild and Farmed Striped Catfish Pangasianodon Hypophthalmus in the Lower Mekong River" Journal of Marine Science and Engineering 8, no. 6: 471. https://doi.org/10.3390/jmse8060471
APA StyleVu, N. T., Ha, T. T. T., Thuy, V. T. B., Trang, V. T., & Nguyen, N. H. (2020). Population Genomic Analyses of Wild and Farmed Striped Catfish Pangasianodon Hypophthalmus in the Lower Mekong River. Journal of Marine Science and Engineering, 8(6), 471. https://doi.org/10.3390/jmse8060471