
Chromosome-Level Genome Assembly of the Rough-Toothed Dolphin (Steno bredanensis)

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Genome Sequencing
2.2. Genome Assembly
2.3. Genome Annotation
2.4. Assessment of the Assembly and Annotation
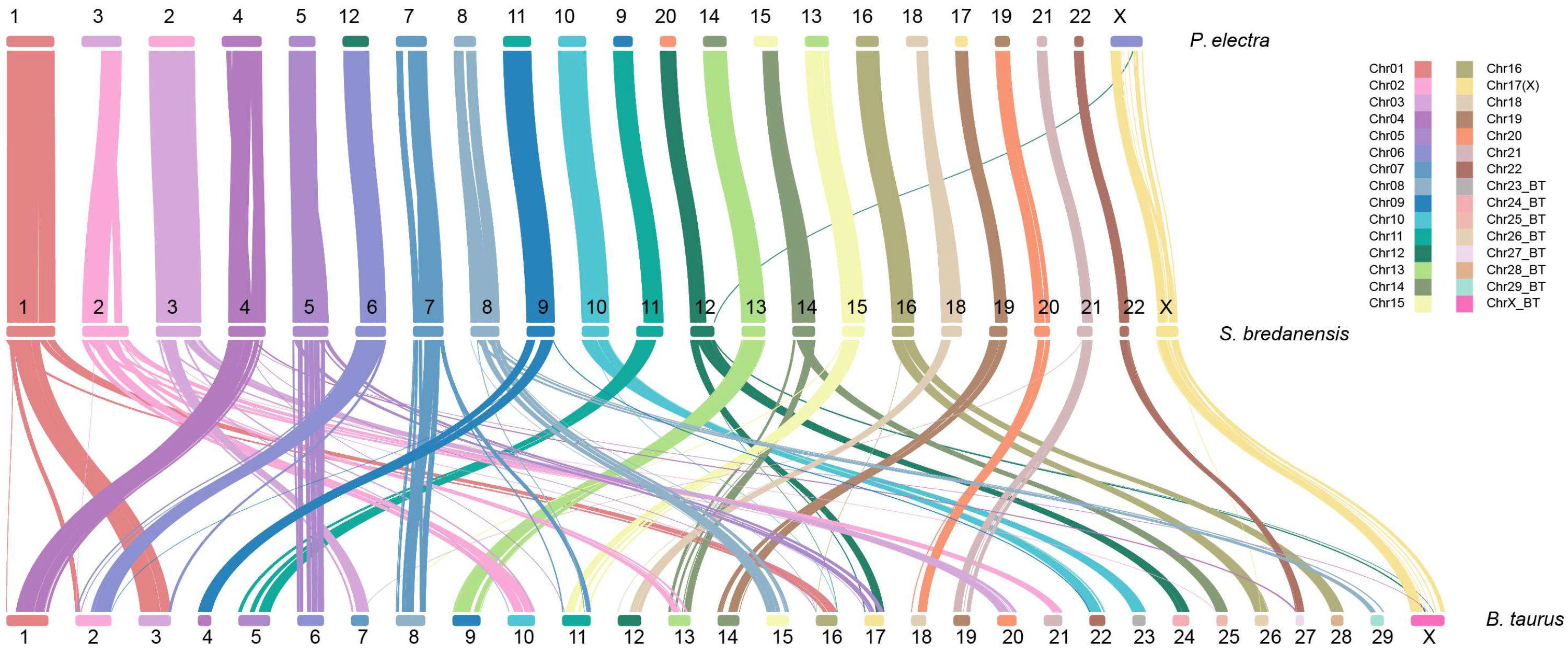
2.5. Evolution Analysis
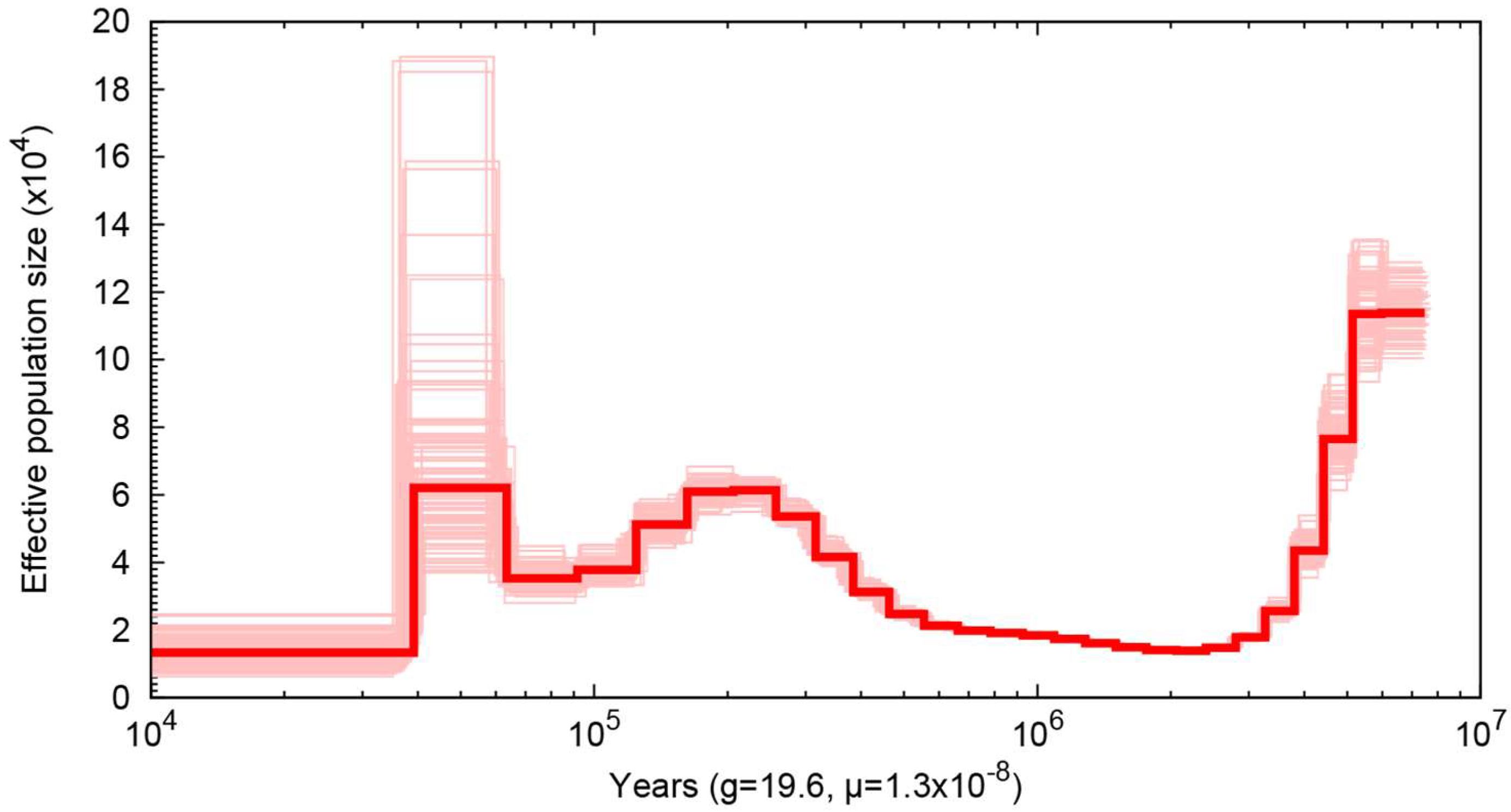
2.6. Demographic History
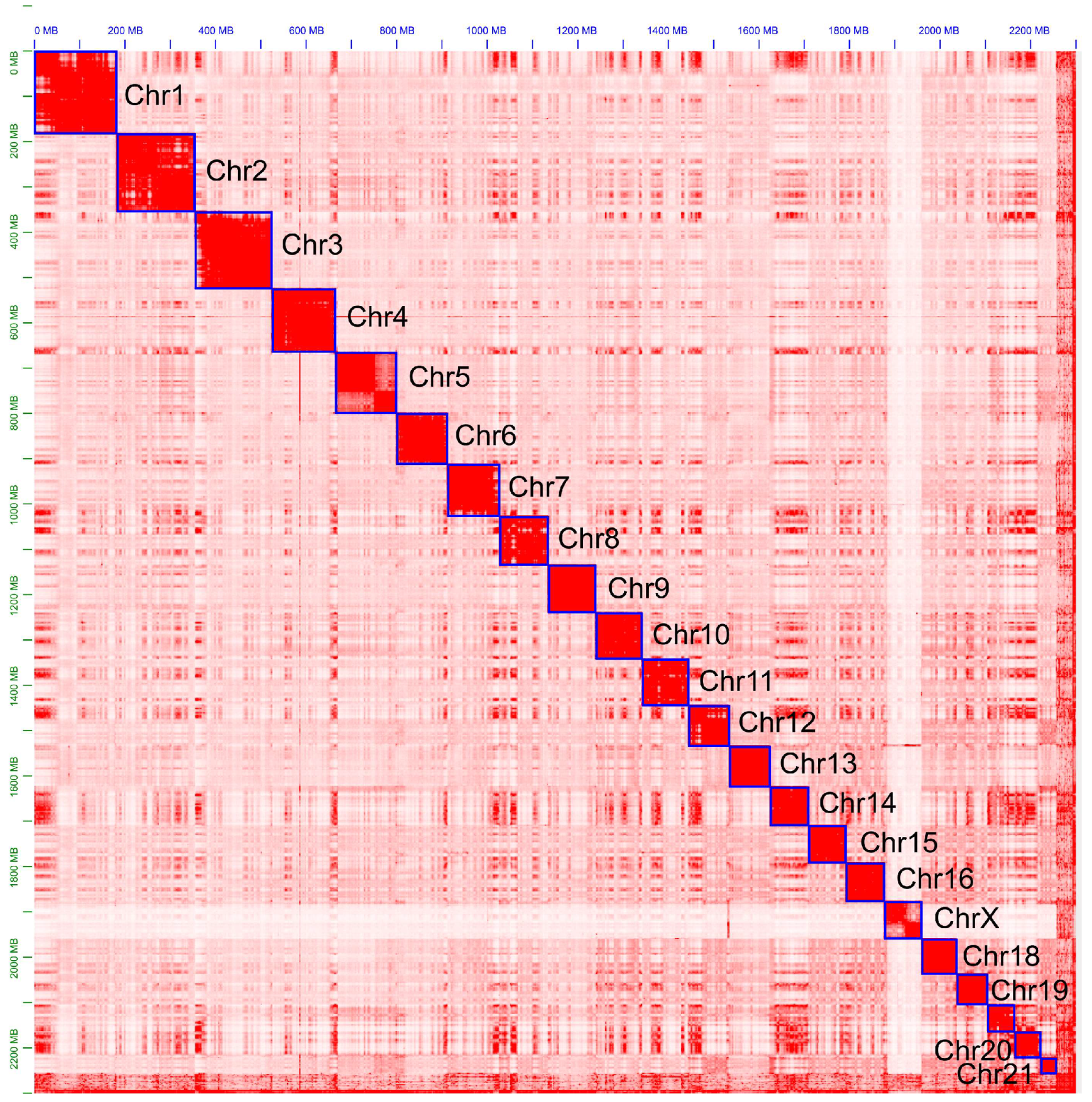
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baird, R.W.; Webster, D.L.; Mahaffy, S.D.; McSweeney, D.J.; Schorr, G.S.; Ligon, A.D. Site fidelity and association patterns in a deep-water dolphin: Rough-toothed dolphins (Steno bredanensis) in the Hawaiian Archipelago. Mar. Mammal Sci. 2008, 24, 535–553. [Google Scholar] [CrossRef]
- West, K.L.; Mead, J.G.; White, W. Steno bredanensis (Cetacea: Delphinidae). Mamm. Species 2011, 43, 177–189. [Google Scholar] [CrossRef]
- Jefferson, T.A. Rough-Toothed Dolphin: STENO Bredanensis, in Encyclopedia of Marine Mammals; Elsevier: Amsterdam, The Netherlands, 2009; pp. 990–992. [Google Scholar]
- de Moura, J.F.; da Rodrigues, É.S.; Siciliano, S. Epimeletic behaviour in rough-toothed dolphins (Steno bredanensis) on the east coast of Rio de Janeiro State, Brazil. Mar. Biodivers. Rec. 2009, 2, e12. [Google Scholar] [CrossRef]
- Kiszka, J.; Baird, R.; Braulik, G. Steno Bredanensis; The IUCN Red List of Threatened Species: Gland, Switzerland, 2019; p. e.T20738A50376703. [Google Scholar]
- Lodi, L.; Maricato, G. Rough-toothed dolphins (Cetartiodactyla: Delphinidae) habitat use in coastal urban waters of the South-western Atlantic. J. Mar. Biol. Assoc. UK 2020, 100, 471–479. [Google Scholar] [CrossRef]
- Garner, B.A.; Hand, B.K.; Amish, S.J.; Bernatchez, L.; Foster, J.T.; Miller, K.M.; Morin, P.A.; Narum, S.R.; O’Brien, S.J.; Roffler, G. Genomics in conservation: Case studies and bridging the gap between data and application. Trends Ecol. Evol. 2016, 31, 81–83. [Google Scholar] [CrossRef]
- Harrisson, K.A.; Pavlova, A.; Telonis-Scott, M.; Sunnucks, P. Using genomics to characterize evolutionary potential for conservation of wild populations. Evol. Appl. 2014, 7, 1008–1025. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Johansson, M.L.; Heath, D.D. Role of genomics and transcriptomics in selection of reintroduction source populations. Conserv. Biol. 2016, 30, 1010–1018. [Google Scholar] [CrossRef]
- Albertson, G.R.; Alexander, A.; Archer, F.I.; Caballero, S.; Martien, K.K.; Hemery, L.G.; Baird, R.W.; Oremus, M.; Poole, M.M.; Duffield, D.A. Worldwide phylogeography of rough-toothed dolphins (Steno bredanensis) provides evidence for subspecies delimitation. Mar. Mammal Sci. 2022, 38, 1371–1397. [Google Scholar] [CrossRef]
- Donato, A.; Siciliano, S.; Weksler, M.; Silva, D.; Carvalho, E.; Loiola, S.; Amaral, C. Population structure and the conservation status of the rough-toothed dolphins based on the analysis of the mitochondrial control region. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 294–295. [Google Scholar] [CrossRef]
- Albertson, G.R. Worldwide Phylogeography and Local Population Structure of the Rough-Toothed Dolphin (Steno bredanensis). Ph.D. Thesis, Oregon State University, Corvallis, OR, USA, 2014. [Google Scholar]
- Thomas, J.C.; Khoury, R.; Neeley, C.K.; Akroush, A.M.; Davies, E.C. A fast CTAB method of human DNA isolation for polymerase chain reaction applications. Biochem. Educ. 1997, 25, 233–235. [Google Scholar] [CrossRef]
- Wang, O.; Chin, R.; Cheng, X.; Wu, M.K.Y.; Mao, Q.; Tang, J.; Sun, Y.; Anderson, E.; Lam, H.K.; Chen, D. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 2019, 29, 798–808. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Pryszcz, L.P.; Gabaldón, T. Redundans: An assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016, 44, e113. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- RepeatMasker. Available online: http://www.repeatmasker.org/ (accessed on 18 September 2021).
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Li, H.; Coghlan, A.; Ruan, J.; Coin, L.J.; Heriche, J.K.; Osmotherly, L.; Li, R.; Liu, T.; Zhang, Z.; Bolund, L. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006, 34, D572–D580. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML, in Bioinformatics for DNA Sequence Analysis; Springer: Berlin/Heidelberg, Germany, 2009; pp. 113–137. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Cabrera, A.A.; Bérubé, M.; Lopes, X.M.; Louis, M.; Oosting, T.; Rey-Iglesia, A.; Rivera-León, V.E.; Székely, D.; Lorenzen, E.D.; Palsbøll, P.J. A Genetic Perspective on Cetacean Evolution. Annu. Rev. Ecol. Evol. Syst. 2021, 52, 131–151. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, Y.; Zhang, P.; Liu, C.; Wang, J.; Gao, H.; Hoelzel, A.R.; Seim, I.; Lv, M.; Lin, M. Comparative genomics provides insights into the aquatic adaptations of mammals. Proc. Natl. Acad. Sci. USA 2021, 118, e2106080118. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef]
- Pacifici, M.; Santini, L.; Di Marco, M.; Baisero, D.; Francucci, L.; Marasini, G.G.; Visconti, P.; Rondinini, C. Generation length for mammals. Nat. Conserv. 2013, 5, 89. [Google Scholar]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Brookwell, R.; Finlayson, K.; van de Merwe, J.P. A comparative analysis of the karyotypes of three dolphins–Tursiops truncatus Montagu, 1821, Tursiops australis Charlton-Robb et al., 2011, and Grampus griseus Cuvier, 1812. Comp. Cytogenet. 2021, 15, 53. [Google Scholar] [CrossRef]
- Cunha, H.A.; Moraes, L.C.; Medeiros, B.V.; Lailson-Brito, J., Jr.; da Silva, V.M.; Sole-Cava, A.M.; Schrago, C.G. Phylogenetic status and timescale for the diversification of Steno and Sotalia dolphins. PLoS ONE 2011, 6, e28297. [Google Scholar] [CrossRef] [PubMed]
- McGowen, M.R.; Tsagkogeorga, G.; Álvarez-Carretero, S.; Dos Reis, M.; Struebig, M.; Deaville, R.; Jepson, P.D.; Jarman, S.; Polanowski, A.; Morin, P.A. Phylogenomic resolution of the cetacean tree of life using target sequence capture. Syst. Biol. 2020, 69, 479–501. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Berta, A.; Sumich, J.L.; Kovacs, K.M. Marine Mammals: Evolutionary Biology; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Forti, E.; Aksanov, O.; Birk, R.Z. Temporal expression pattern of Bardet-Biedl syndrome genes in adipogenesis. Int. J. Biochem. Cell Biol. 2007, 39, 1055–1062. [Google Scholar] [CrossRef]
- Cai, M.; Chen, J.; Yu, C.; Xi, L.; Jiang, Q.; Wang, Y.; Wang, X. FAM134B promotes adipogenesis by increasing vesicular activity in porcine and 3T3-L1 adipocytes. Biol. Chem. 2019, 400, 523–532. [Google Scholar] [CrossRef]
- Cai, M.; Zhao, J.; Liu, Q.; Wang, X.; Wang, Y. FAM134B improves preadipocytes differentiation by enhancing mitophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158508. [Google Scholar] [CrossRef]
- Yuan, Z.; Song, D.; Wang, Y. The novel gene pFAM134B positively regulates fat deposition in the subcutaneous fat of Sus scrofa. Biochem. Biophys. Res. Commun. 2014, 454, 554–559. [Google Scholar] [CrossRef]
- Zhou, Y.; Robciuc, M.R.; Wabitsch, M.; Juuti, A.; Leivonen, M.; Ehnholm, C.; Yki-Järvinen, H.; Olkkonen, V.M. OSBP-related proteins (ORPs) in human adipose depots and cultured adipocytes: Evidence for impacts on the adipocyte phenotype. PLoS ONE 2012, 7, e45352. [Google Scholar] [CrossRef]
- Micallef, P.; Vujičić, M.; Wu, Y.; Peris, E.; Wang, Y.; Chanclón, B.; Ståhlberg, A.; Cardell, S.L.; Asterholm, I.W. C1QTNF3 is upregulated during subcutaneous adipose tissue remodeling and stimulates macrophage chemotaxis and M1-like polarization. Front. Immunol. 2022, 13, 2679. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fan, M.; Zhang, S.; Qu, Y.; Zheng, S.; Song, J.; Miao, C. Intestinal Metrnl released into the gut lumen acts as a local regulator for gut antimicrobial peptides. Acta Pharmacol. Sin. 2016, 37, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zheng, S.; Wang, P.; Xu, T.; Guan, Y.; Zhang, Y.; Miao, C. Subfatin is a novel adipokine and unlike Meteorin in adipose and brain expression. CNS Neurosci. Ther. 2014, 20, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.R.; Long, J.Z.; White, J.P.; Svensson, K.J.; Lou, J.; Lokurkar, I.; Jedrychowski, M.P.; Ruas, J.L.; Wrann, C.D.; Lo, J.C. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 2014, 157, 1279–1291. [Google Scholar] [CrossRef]
- Rockel, J.S.; Yu, C.; Whetstone, H.; Craft, A.M.; Reilly, K.; Ma, H.; Tsushima, H.; Puviindran, V.; Al-Jazrawe, M.; Keller, G.M. Hedgehog inhibits β-catenin activity in synovial joint development and osteoarthritis. J. Clin. Investig. 2016, 126, 1649–1663. [Google Scholar] [CrossRef]
- Areal, H.; Abrantes, J.; Esteves, P.J. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol. Biol. 2011, 11, 368. [Google Scholar] [CrossRef]
- Ishengoma, E.; Agaba, M. Evolution of toll-like receptors in the context of terrestrial ungulates and cetaceans diversification. BMC Evol. Biol. 2017, 17, 54. [Google Scholar] [CrossRef]
- Borghese, F.; Clanchy, F.I. CD74: An emerging opportunity as a therapeutic target in cancer and autoimmune disease. Expert Opin. Ther. Targets 2011, 15, 237–251. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, W.; Zhou, R.; Gui, D.; Yu, X.; Wu, Y. Low major histocompatibility complex class II variation in the endangered Indo-Pacific humpback dolphin (Sousa chinensis): Inferences about the role of balancing selection. J. Hered. 2016, 107, 143–152. [Google Scholar] [CrossRef]
- Ratthe, C.; Girard, D. Interleukin-15 enhances human neutrophil phagocytosis by a Syk-dependent mechanism: Importance of the IL-15Rα chain. J. Leukoc. Biol. 2004, 76, 162–168. [Google Scholar] [CrossRef]
- Levin, M.; Romano, T.; Matassa, K.; De Guise, S. Validation of a commercial canine assay kit to measure pinniped cytokines. Vet. Immunol. Immunopathol. 2014, 160, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Ando, A.; Suzuki, R.; Takishita, K.; Kawato, M.; Katsumata, E.; Ohtsu, D.; Okutsu, K.; Tokutake, K.; Miyahara, H. Host–virus specificity of morbilliviruses predicted by structural modeling of the marine mammal SLAM, a receptor. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 227–241. [Google Scholar] [CrossRef]
- Ohishi, K.; Maruyama, T.; Seki, F.; Takeda, M. Marine morbilliviruses: Diversity and interaction with signaling lymphocyte activation molecules. Viruses 2019, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Zhong, X.; McAlpine, W.; Liao, T.-C.; Zhang, D.; Fang, B.; Russell, J.; Ludwig, S.; Nair-Gill, E.; Zhang, Z. LMBR1L regulates lymphopoiesis through Wnt/β-catenin signaling. Science 2019, 364, eaau0812. [Google Scholar] [CrossRef] [PubMed]
- Parrish-Novak, J.; Dillon, S.R.; Nelson, A.; Hammond, A.; Sprecher, C.; Gross, J.A.; Johnston, J.; Madden, K.; Xu, W.; West, J. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 2000, 408, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Strengell, M.; Julkunen, I.; Matikainen, S. IFN-α regulates IL-21 and IL-21R expression in human NK and T cells. J. Leukoc. Biol. 2004, 76, 416–422. [Google Scholar] [CrossRef]
- Jia, K.; Bian, C.; Yi, Y.; Li, Y.; Jia, P.; Gui, D.; Zhang, X.; Lin, W.; Sun, X.; Lv, Y. Whole genome sequencing of Chinese white dolphin (Sousa chinensis) for high-throughput screening of antihypertensive peptides. Mar. Drugs 2019, 17, 504. [Google Scholar] [CrossRef]
- Warren, W.C.; Kuderna, L.; Alexander, A.; Catchen, J.; Pérez-Silva, J.G.; López-Otín, C.; Quesada, V.; Minx, P.; Tomlinson, C.; Montague, M.J. The novel evolution of the sperm whale genome. Genome Biol. Evol. 2017, 9, 3260–3264. [Google Scholar] [CrossRef]
- Amaral, A.R.; Beheregaray, L.B.; Bilgmann, K.; Freitas, L.; Robertson, K.M.; Sequeira, M.; Stockin, K.A.; Coelho, M.; Möller, L.M. Influences of past climatic changes on historical population structure and demography of a cosmopolitan marine predator, the common dolphin (genus Delphinus). Mol. Ecol. 2012, 21, 4854–4871. [Google Scholar] [CrossRef]
- Mace, G.M. The role of taxonomy in species conservation. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2004, 359, 711–719. [Google Scholar] [CrossRef]
- Fuentes-Pardo, A.P.; Ruzzante, D.E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Kang, H.; Zhang, Y.; Wang, J.; Lin, W.; Zhang, P.; Lin, M.; Liu, M.; Fan, G.; Li, S. Chromosome-Level Genome Assembly of the Rough-Toothed Dolphin (Steno bredanensis). J. Mar. Sci. Eng. 2023, 11, 418. https://doi.org/10.3390/jmse11020418
Gao H, Kang H, Zhang Y, Wang J, Lin W, Zhang P, Lin M, Liu M, Fan G, Li S. Chromosome-Level Genome Assembly of the Rough-Toothed Dolphin (Steno bredanensis). Journal of Marine Science and Engineering. 2023; 11(2):418. https://doi.org/10.3390/jmse11020418
Chicago/Turabian StyleGao, Haiyu, Hui Kang, Yaolei Zhang, Jiahao Wang, Wenzhi Lin, Peijun Zhang, Mingli Lin, Mingming Liu, Guangyi Fan, and Songhai Li. 2023. "Chromosome-Level Genome Assembly of the Rough-Toothed Dolphin (Steno bredanensis)" Journal of Marine Science and Engineering 11, no. 2: 418. https://doi.org/10.3390/jmse11020418
APA StyleGao, H., Kang, H., Zhang, Y., Wang, J., Lin, W., Zhang, P., Lin, M., Liu, M., Fan, G., & Li, S. (2023). Chromosome-Level Genome Assembly of the Rough-Toothed Dolphin (Steno bredanensis). Journal of Marine Science and Engineering, 11(2), 418. https://doi.org/10.3390/jmse11020418